

Abstract
p97 (also referred to as DAP5, NAT1 or eIF4G2) has been proposed to act as a repressor of protein synthesis. However, we found that p97 is abundantly expressed in proliferating cells and p97 is recruited to ribosomes following growth factor stimulation. We also report that p97 binds eIF2β through its C-terminal domain and localizes to ribosome through its N-terminal MIF4G domain. When overexpressed, p97 increases reporter luciferase activity. In contrast, overexpression of the C-terminal two-thirds of eukaryotic initiation factor 4GI (eIF4GI), a region that shares significant homology with p97, or the N-terminal MIF4G domain of p97 markedly inhibits reporter activity, the rate of global translation and cell proliferation. Conversely, downregulation of p97 levels by RNA interference also decreases the rate of global translation and inhibits cell proliferation. This coincides with an increase in p27/Kip1 protein levels and a marked decrease in CDK2 kinase activity. Taken together, our results demonstrate that p97 is functionally different from the closely related C-terminal two-thirds of eIF4GI and it can positively promote protein synthesis and cell proliferation.
Keywords: eIF4GI, eIF2, p27/KIP1, p97/DAP5
Introduction
Eukaryotic initiation factor 4GI (eIF4GI) acts as a scaffold protein during translation initiation and binds several other translation initiation factors (reviewed by Kapp and Lorsch, 2004). Structurally, eIF4GI is thought to have four domains: an N-terminal domain, a central domain called MIF4G and two C-terminal domains called MA3 and W2. The N-terminal domain is the binding site for eIF4E, the cap-binding protein. The central MIF4G domain is the binding site for eIF4AI, which has a RNA helicase activity. It also interacts with eIF3, which binds with high affinity to 40S ribosomal subunits. The C-terminal MA3 domain has another eIF4AI binding site whereas the role of W2 domain is yet to be defined (Imataka and Sonenberg, 1997; Morino et al, 2000). During picornavirus infection, viral proteases cleave eIF4GI into the N-terminal one-third fragment and the C-terminal two-thirds fragment, called p100, which separates the cap-binding function of eIF4E from eIF4AI helicase and the eIF3 (Lamphear et al, 1995). This is presumed to be the cause of translational inhibition of capped cellular mRNAs in infected cells.
The eIF4G family consists of eIF4GI, p97 and eIF4G3/eIF4GII. Of these, p97 (also referred to as DAP5, NAT1 or eIF4G2) was isolated in 1997 by several groups independently and is only expressed in mammalian cells (Imataka et al, 1997; Levy-Strumpf et al, 1997; Shaughnessy et al, 1997; Yamanaka et al, 1997). p97 is highly homologous to eIF4GI, with the interesting exceptions that it lacks the N-terminal domain of eIF4GI necessary for eIF4E binding and that p97 has one eIF4AI-binding region in the N-terminal domain (Imataka et al, 1997). Consistent with this, p97 binds eIF3 and eIF4AI, but fails to interact with eIF4E (Imataka et al, 1997; Henis-Korenblit et al, 2002), suggesting that p97 may act as a natural counterpart to the p100 fragment of eIF4GI and that it acts as a general repressor of translation that sequesters eIF3 and eIF4AI into inactive complexes. Consistent with this, overexpression of p97 decreases both cap-dependent translation as well as cap-independent translation from the internal ribosome entry site (IRES) for encephalomyocarditis virus (EMCV) (Imataka et al, 1997; Yamanaka et al, 1997). However, the relative amounts of eIF4GI and eIF3 in HeLa cells have been reported to be 1:2.5 and eIF4AI is thought be the most abundant translation initiation factor (Duncan and Hershey, 1983; Mengod and Trachsel, 1985). Thus, in contrast to the condition where p97 is ectopically overexpressed, it is uncertain if endogenous p97 effectively competes with eIF4GI for eIF3 and eIF4AI binding since there is an excess pool of eIF3 proteins.
p97 has also been implicated in stress responses; during cell death triggered by Fas receptors or by p53 activation, p97 is rapidly converted into a truncated form called p86 through loss of the C-terminal domain (Henis-Korenblit et al, 2000). Furthermore, overexpression of p86, but not p97, induces preferential translation of cellular mRNAs that contain IRES elements including XIAP, Apaf-1, HIAP2 and p97/DAP5 itself (Henis-Korenblit et al, 2002; Nevins et al, 2003; Warnakulasuriyarachchi et al, 2004). However, the role of p97 in stress responses and in translation remains unclear.
In this study, we have explored the molecular functions of p97 in mammalian cells. We show that p97 directly binds eIF2β and can positively promote translation through its C-terminal domain. In contrast, the p100 fragment of eIF4GI, a region that shares significant homology with p97, or the N-terminal MIF4G domain of p97 represses global translation and inhibits cell proliferation. Conversely, downregulation of p97 protein levels by RNA interference also decreases the rate of global translation and inhibits cell proliferation. This effect coincides with an increase in p27/Kip1 protein levels and a decrease in CDK2 kinase activity. Taken together, our results demonstrate that p97 is functionally distinct from the C-terminal two-thirds of eIF4GI and it can positively promote protein synthesis as well as cell proliferation.
Results
p97 binds eIF2β through its C-terminal domain
A search with NCBI-BLAST revealed that the C-terminus of p97 is highly homologous to the C-terminal W2 domains of eIF2Bɛ and eIF5 (Figure 1A and C, listed in the NCBI Conserved Domains site) (Koonin, 1995). This domain contains two conserved tryptophans (hence the name W2) and participates in the interaction of eIF5 and eIF2Bɛ with eIF2β in yeast and mammalian cells (Asano et al, 1999; Das and Maitra, 2000). To test whether the C-terminal domain of p97 also binds eIF2β, we expressed GST-tagged full-length p97 (GST-p97wt), a GST-tagged C-terminal W2 domain deletion mutant of p97 (GST-p971−791) and a GST-tagged N-terminal deletion mutant of p97 (GST-p97349−907) proteins in Escherichia coli (Figure 1A, left panel). The ability of these proteins to bind eIF2β was determined by GST-pulldown assays using 293T total cell lysates as a source of eIF2 subunits. Both GST-p97wt and GST-p97349−907 proteins efficiently pulled down eIF2β and eIF2α, but GST-p971−791 did not (Figure 1A, right panel). Using recombinant proteins from E. coli, we observed that p97 directly bound to eIF2β through its C-terminal domain indicating eIF2β mediates an interaction of p97 with eIF2 complexes (data not shown). To determine the minimal region of p97 required for eIF2 binding, a series of C-terminal deletion mutants of GST-tagged p97 were generated and GST-pulldown assays were performed. GST-p97 mutants lacking the last 17 amino acids at the C-terminal end (GST-p971−890 and GST-p97349−890) were unable to pull down eIF2β and eIF2α (Figure 1B). To further define amino-acid residues necessary for eIF2β interaction, a co-immunoprecipitation assay was preformed. For this purpose, CMV-derived expression vectors encoding Myc-epitope tagged p97 harboring three different alanine scanning mutations in the C-terminal 17 amino-acid residues were prepared by site-directed mutagenesis, and co-transfected with a CMV-derived vector encoding HA-epitope tagged eIF2β (HA-eIF2β) into 293T cells (Figure 1C, top panel). At 48 h after transfection, cells were lysed and co-immunoprecipitated with anti-Myc antibody. The p97 mutant harboring a substitution of four consecutive glutamate residues with alanines (Myc-p97898−901A4) failed to bind HA-eIF2β whereas Myc-p97893−895A3 and Myc-p97903−905A3 bound HA-eIF2β (Figure 1C, bottom panels). In dependent of binding to eIF2β through its C-terminal region, the N-terminal MIF4G domain in p97 was required to bind eIF3 and eIF4AI (Figure 1D). To exclude the possibility that p97 might indirectly interact with eIF2β through a preinitiation complex (PIC), Myc-tagged wild type or mutants of p97 proteins were ectopically expressed in 293T cells, S100 supernatants were prepared to eliminate PIC, and subjected to immunoprecipitation analysis using anti-Myc antibody. Myc-tagged full-length p97 were able to bind eIF2β and eIF4AI in S100 supernatants. In contrast, the C-terminal deletion mutants of p97 or Myc-p97898−901A4 failed to bind eIF2β whereas these mutants of p97 bound eIF4AI (Figure 1E). These results suggest that the C-terminal domain in p97 mediates the eIF2 complex binding independent of PIC and that the N-terminal MIF4G domain mediates eIF4AI and eIF3-binding in vivo. However, It was also notable that the ability of Myc-p971−791, a p86 fragment of p97, to bind eIF4AI was markedly decreased (Figure 1E, lane 8).
Figure 1.
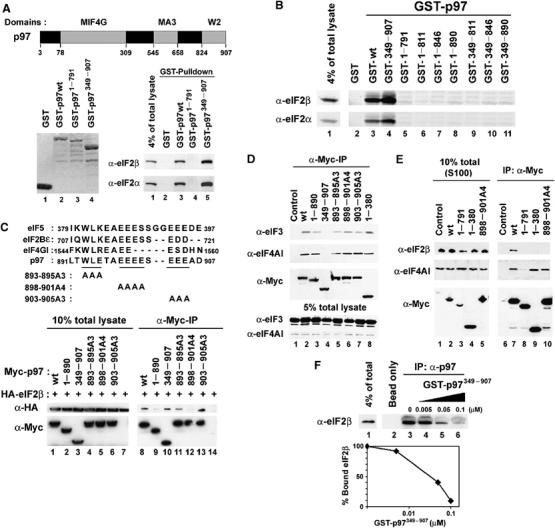
The C-terminal domain of p97 binds to eIF2β in vitro and in vivo. (A) (Top) The domains of p97 are shown schematically. The amino acids at the boundaries of these regions are indicated by numbers. (Bottom left) Coomassie staining of purified recombinant GST-tagged p97 proteins (1 μg each) used for in vitro GST-pulldown assay. (Bottom right) 293T total cell lysates were incubated with the indicated purified recombinant GST-tagged p97 proteins and glutathione-agarose beads. The beads were extensively washed and the bound eIF2β and eIF2α proteins were determined by immunoblot analysis. (B) In vitro GST-pulldown assay to identify the minimal region in p97 for eIF2β binding. (C) (Top) Amino-acid sequences of the minimal eIF2β binding region in p97 were aligned with corresponding amino-acid sequences in eIF5, eIF2Bɛ and eIF4GI. The positions of alanine scanning mutations in the minimal eIF2β binding region in p97 used for co-immunoprecipitation assay are shown. (Bottom left) Myc-tagged full-length or mutants of p97 were co-expressed in 293T cells with HA-tagged eIF2β. The cells were lysed and a 1/10 of total lysates used for co-immunoprecipitation was visualized with immunoblot analysis. (Bottom right) anti-Myc rabbit polyclonal antibody and protein A-agarose beads were added to an indicated total cell lysates. The beads were washed and the bound proteins were visualized with immunoblot analysis. (D, E) Myc-tagged full-length or mutants of p97 were expressed in 293T cells. Total cell lysates (D) or S100 supernatants (E) were prepared and subjected to immunoprecipitation analysis using anti-Myc antibody. Bound proteins were visualized with immunoblot analysis. (F) p97 binds eIF2β in vivo. (Top) 293T total cell lysates were incubated with goat polyclonal antibody raised against the N-terminal end of p97 and protein A-agarose beads. To determine the relative affinity of p97 to eIF2β, increasing amounts of purified GST-tagged N-terminal truncated p97 (GST-p97349−907) that cannot be recognized by the anti-p97 antibody were added during incubation. The beads were washed and bound eIF2β was visualized with immunoblot analysis. (Bottom) eIF2β bound to p97 was quantified with a densitometer. The amount of bound eIF2β proteins was plotted against the concentration of GST-p97349−907 protein.
To determine if the endogenous p97 proteins could bind eIF2β in vivo, a co-immunoprecipitation assay of endogenous proteins was performed using 293T cell lysates and an antibody directed to the N-terminus of p97. To demonstrate specificity and to determine relative binding affinity, increasing amount of a purified N-terminal deletion mutant of p97 (GST-p97349−907) was included in the cell lysates to compete with endogenous p97 binding to eIF2β. Inclusion of 50 nM GST-p97349−907 reduced the amount of eIF2β bound to the endogenous p97 protein by approximately 50% (Figure 1F). This result demonstrates that p97 binds to eIF2β in vivo with physiologically relevant affinity in the submicromolar range.
p97 co-sediments with eIF2 proteins in the 40S ribosomal subunit fraction in a growth factor dependent manner
Our findings that p97 binds the eIF2 complex prompted us to investigate the role of p97 at the ribosomes. First, to determine if p97 is a ribosome-associated protein, T98G glioblastoma cell extracts were fractionated into a hypotonic lysis fraction (cytoplasmic) and a ribosomal salt wash (RSW) fraction. The presence of p97 in each fraction was determined by immunoblot analysis. MEK1 was used as a marker of the cytoplasmic fraction and eIF4GI for RSW fraction. As with eIF4GI, ∼50% of total p97 was detected in the RSW fraction, whereas MEK1 protein was only detected in the soluble cytoplasmic fraction (Figure 2A).
Figure 2.
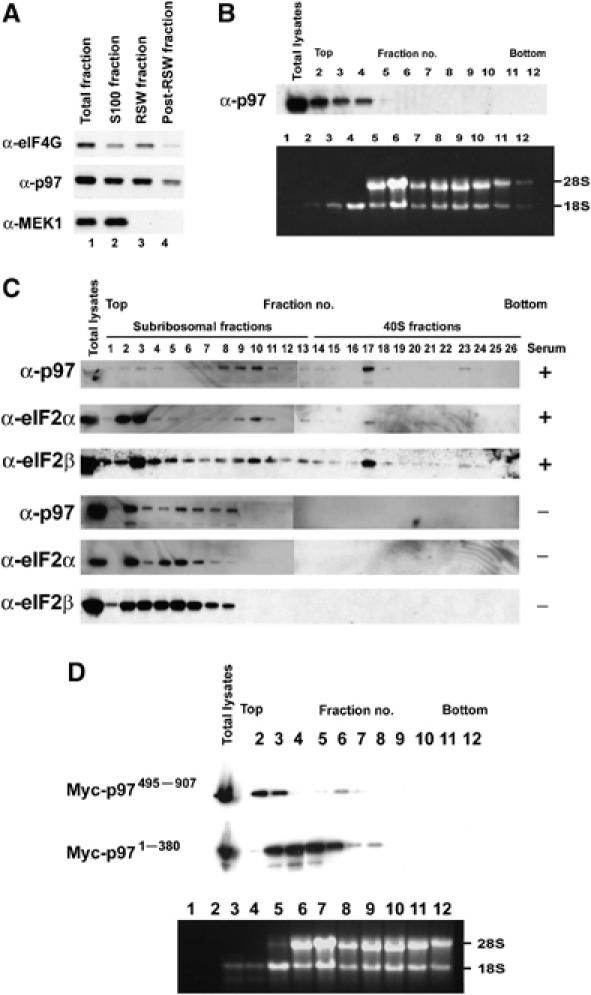
p97 is a ribosome-associated protein which co-sediments with eIF2β and eIF2α in 40S ribosomal fractions in a growth factor dependent manner and the N-terminal MIF4G domain of p97 is required for ribosomal localization of p97. (A) Exponentially growing T98G cells were lysed in hypotonic lysis buffer. Fraction containing ribosomes was pelleted with centrifugation. The resulting pellets were washed with RSW buffer. An equal volume of lysates from each fraction was separated and analyzed by immunoblot. MEK1 was used as a marker for soluble cytoplasmic fraction and eIF4GI was used as a marker for pellet fractions contains ribosome-associated proteins. (B) T98G whole-cell extracts prepared using a lysis buffer containing ∼1.2% Triton X-100 and ∼1.2% deoxycholate were resolved on sucrose density gradient by centrifugation. Gradients were separated into 12 fractions. (Top) A half of each fraction was precipitated with TCA. p97 protein was identified with immunoblot analysis. (Bottom) The remaining half of each fraction was used to exact total RNA and subjected to gel analysis to determine the presences of 18S and 28S rRNAs. rRNAs were detected by ethidium bromide staining. (C) p97 co-sediments with eIF2β and eIF2α in ribosomal fraction in a growth factor dependent manner. Whole-cell extracts were prepared from either U87 cells grown exponentially in the presence of serum or U87 cells serum starved for 48 h. Cell extracts were resolved on sucrose density gradient. Top one-third of the gradient was separated into 26 fractions. Each fraction was precipitated with TCA and the presence of p97, eIF2β and eIF2α was determined by immunoblot analysis. (D) Myc-tagged MIF4G domain (Myc-p971−380) or Myc-tagged MIF4G deletion mutant of p97 (Myc-p97495−907) was expressed in HEK293 cells. Whole-cell extracts were prepared and resolved on sucrose density gradient. Gradients were separated into 12 fractions. A half of each fraction was precipitated with TCA and Myc-tagged proteins were identified with immunoblot analysis using anti-Myc polyclonal antibody.
To determine which ribosomal subunits associate with p97, extracts of T98G cells were resolved by sucrose density gradient velocity sedimentation and one half of each fraction was analyzed for the presence of p97 protein by immunoblot analysis (Figure 2B, top panel). Total RNA in the other half of each fraction was extracted and subjected to gel analysis to determine the presence of 18S and 28S rRNA (Figure 2B, bottom panel). The majority of p97 was detected in fractions 1–2 that are lighter than 40S subunit fraction. The p97 was also detected in fractions 3–4 that corresponded to 40S subunit fraction only containing 18S rRNA (Figure 2B). The discrepancy for the p97 distribution between Figure 2A and B most likely resulted from differences in lysis conditions that a hypotonic lysis buffer was used for Figure 2A (see Materials and methods).
To determine if recruitment of p97 to 40S ribosomal subunit is regulated in a growth factor dependent manner and to confirm the co-sedimentation of p97 with eIF2β in other cell line, sucrose density gradient velocity sedimentation was performed with whole-cell lysates from U87 glioblastoma cell extracts. In an effort to determine where p97 and eIF2 proteins were present under conditions of serum starvation, the top one third of the gradient was separated into 26 fractions and subjected to immunoblot analysis. p97, eIF2α and eIF2β proteins were detected in fractions 1–24 (Figure 2C, top panels). In contrast, under condition of serum starvations, the majority of p97, eIF2α and eIF2β proteins sedimented in fractions 2–8 (Figure 2C, bottom panels). Taken together, these results suggest that p97 is a ribosome-associated factor that is recruited to 40S subunits in a growth factor dependent manner.
The N-terminal MIF4G domain of p97 is required for ribosomal localization of p97
To identify the p97 domain that is required for ribosome association, CMV-derived mammalian expression vector encoding either the Myc-tagged N-terminus MIF4G domain of p97 (Myc-p971−380) or the Myc-tagged C-terminal domain of p97 lacking the MIF4G domain (Myc-p97495−907) were transfected into HEK293 cells because of their high transfection efficiency and subjected to sucrose density gradient velocity sedimentation analysis (Figure 2D). Total RNA in one half of each fraction was extracted and subjected to gel analysis to determine the presence of 18S and 28S rRNA (Figure 2D, bottom panel). The majority of Myc-p97495−907 sedimented in fraction 2, whereas a large amount of Myc-p971−380 sedimented in 40S subunit fractions 3–5 (Figure 2D, top panels). Thus, the MIF4G domain in p97 mediates ribosomal association of p97, most likely by binding to eIF3 (Figure 1D, lane 8) that bound to the 40S ribosome subunit.
Ectopic expression of p97 induces luciferase activity through its C-terminal domain and the N-terminal MIF4G domain of p97 is a dominant negative form of p97
Our finding that the C-terminal domain of p97 binds the eIF2 complex prompted us to determine if the C-terminal domain of p97 affects rates of translation. To determine this, a monocistronic translational reporter assay was performed using pCDNA3 vector inserted with cDNA encoding firefly luciferase. We used 293T cells because of their high transfection efficiency and protein production. pCDNA3 vector encoding firefly luciferase was co-transfected with an increasing amount of CMV-derived mammalian expression vector encoding Myc-tagged full-length p97 or Myc-tagged C-terminal deletion mutants of p97. At 20 h after transfection, luciferase activity was measured. This activity increased with increasing amount of Myc-p97wt (Figure 3A). Conversely, a deletion of the C-terminal domain in p97 failed to increase luciferase activity (Figure 3A and B). Furthermore, increasing amount of the MIF4G domain of p97 (Myc-p971−380) markedly decreased luciferase activity indicating that Myc-p971−380 is a dominant negative form of p97 (Figure 3A and B). These changes of luciferase activities were most likely due to translation rather than transcription since there was no measurable difference in luciferase mRNA levels determined by Northern blot analysis (Figure 3C). Ectopic expression of Myc-p97898−901A4 that failed to bind eIF2β (Figure 1C, lane 12) was also unable to increase luciferase activity compared to Myc-p97wt (Figure 3D and E). Taken together, these results suggest that p97 can positively mediate translation through its C-terminal domain.
Figure 3.
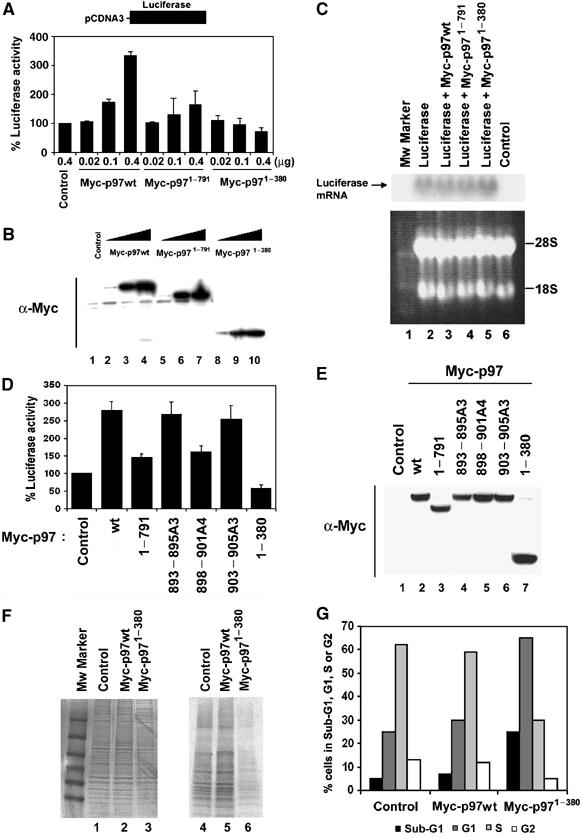
Ectopic expression of p97 induces luciferase activity through its C-terminal domain and the MIF4G domain of p97 inhibits general protein synthesis and induces cell cycle arrest. (A, D) CMV-derived mammalian expression vectors encoding Myc-tagged full-length or the indicated mutants of p97 were co-transfected with pCDNA3 vector encoding firefly luciferase into 293T cells. At 20 h after transfection, cells were lysed and analyzed for luciferase activity. Average of luciferase activity from five independent experiments is shown with standard deviation. (B, E) The same amount of each lysates were separated and subjected to immunoblot analysis. (C) Northern blot analysis of luciferase mRNA in 293T cells. Total cellular RNA was isolated and 20 μg of total cellular RNA was subjected to Northern blot analysis for luciferase mRNA (Top). To ensure equal loading of RNAs, 28S and 18S rRNAs were visualized by staining with Ethidium Bromide (Bottom). Control refers to the cells without transfection (lane 6). (F) Myc-tagged wild type p97 or Myc-tagged MIF4G domain of p97 (Myc-p971−380) proteins were expressed in HEK293 cells. Cells were pulse-labeled with [35S]methionine and [35S]cysteine mix for 1 h, and total cell extracts were prepared and separated through SDS polyacrylamide gel. (Left) The loading of total protein was visualized with Coomassie staining. (Right) The same gel was dried and subjected to auto-radiography. (G) An aliquot of transfected HEK293 cells with an indicated DNA plasmid construct was stained with PI (PI) and subjected to FACS analysis.
Ectopic expression of the N-terminal MIF4G domain of p97 inhibits general protein synthesis and induces cell cycle arrest
The marked inhibition of reporter activity following overexpression of the MIF4G domain of p97 prompted us to determine its effect on the rate of global protein synthesis. To address the question, HEK293 cells were transiently transfected with CMV-derived mammalian expression vectors encoding Myc-tagged full-length p97 or the Myc-tagged N-terminal MIF4G domain of p97 (Myc-p971−380). HEK293 cells were used because of their high transfection efficiency. At 48 h after transfection, cells were pulse-labeled with [35S]methionine and [35S]cysteine mix, and we determined the accumulation of newly synthesized proteins. Despite equal amounts of protein loaded, ectopic expression of Myc-p971−380 showed a decrease in the global translation (Figure 3F). Overexpression of Myc-p971−380 decreased a rate of global translation by ∼50% and markedly induced cell cycle arrest at G1 and sub-G1 phases compared to either control transfectants or Myc-p97wt transfectants (Figure 3F and G).
p97 is functionally different from the closely related C-terminal two-thirds of eIF4GI
p97 is homologous to the C-terminal two-thirds of eIF4GI (Figure 3F), but lacks the N-terminal eIF4E binding domain that is present in eIF4GI (Imataka et al, 1997). This difference is thought to account for the inhibitory effects of p97 on global translation by sequestering eIF3 from the eIF4F complex. We wished to compare the ability of p97 and eIF4GI to regulate translation. A vector encoding HA-tagged full-length eIF4GI was transfected into 293T cells and luciferase activity was measured 20 h later. Ectopic expression of eIF4GI increased luciferase activity by ∼4 fold compared to empty vector control (Figure 4B and C), as expected. To determine if the C-terminal two-thirds of eIF4GI could increase luciferase activity in a comparable manner to p97, a CMV-derived mammalian expression vector encoding a Myc-tagged N-terminal deletion mutant of eIF4GI (Myc-eIF4GI621−1560) was transfected into 293T cells and luciferase activity was measured 20 h later. The fragment of eIF4GI encoding the C-terminal two-thirds of the protein failed to increase luciferase activity even though levels of expression were comparable to levels of Myc-p97, suggesting that these proteins are not functionally equivalent in vivo despite significant homology between p97 and the C-terminal two-thirds of eIF4GI (Figure 4B and C).
Figure 4.
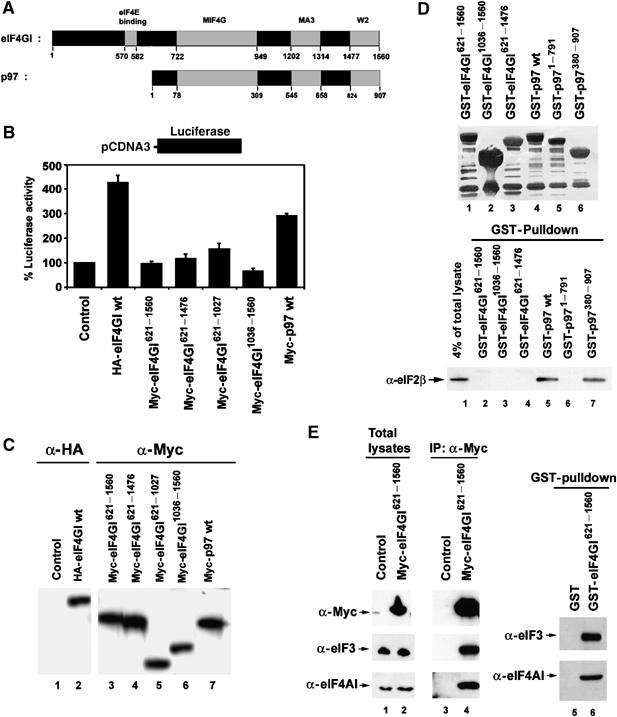
p97 is functionally different from the closely related C-terminal two-thirds of eIF4GI. (A) The domains of eIF4GI and p97 are shown schematically. The amino acids at the boundaries of these regions are indicated by numbers. (B) CMV-derived mammalian expression vectors encoding eIF4GI or p97 were co-transfected with pCDNA3 encoding firefly luciferase into 293T cells. At 20 h after transfection, cells were lysed and 10 μl of each lysate was analyzed for luciferase activity. The average of luciferase activity from five independent experiments is shown with standard deviations. (C) The same amount of each lysates were separated and subjected to immunoblot analysis. (D) (Top) Coomassie staining of purified recombinant GST-eIF4GI and GST-p97 proteins (1 μg each) used for in vitro GST-pulldown assay. (Bottom) 293T total cell lysates were incubated with the indicated purified recombinant GST-tagged eIF4GI and GST-tagged p97 proteins and glutathione-agarose beads. The beads were extensively washed and the bound eIF2β was determined by immunoblot analysis. (E) Myc-tagged eIF4GI621−1560 proteins were expressed in 293T cells and subjected to immunoprecipitation and immunoblot analysis (lanes 1–4). 293T total cell lysates were incubated with GST-tagged eIF4GI621−1560 proteins and the bound proteins were determined with immunoblot analysis (lanes 5 and 6).
Like p97, the C-terminal region of eIF4GI contains a W2 domain, which is expected to bind eIF2β (Figure 4A). To determine if eIF4GI binds eIF2β, several deletion mutant forms of recombinant GST-tagged eIF4GI proteins were prepared from E. coli (Figure 4D, top panel). None of the GST-eIF4GI proteins bound eIF2β whereas both Myc-eIF4GI621−1560 and GST-eIF4GI621−1560 proteins efficiently pulled down eIF4AI and eIF3 in vivo and in vitro (Figure 4E). These results further suggest that p97 and eIF4GI are functionally distinct despite their sequence homology.
Downregulation of p97 protein levels by RNA interference inhibits cell proliferation and decreases a rate of general protein synthesis
p97 mRNA are highly expressed in transformed and nontransformed breast cells and we found that p97 makes up ∼0.1–0.3% of total cellular proteins in ectopically growing transformed and non-transformed breast carcinoma cells (data not shown). The constitutive and abundant expression of p97 protein in proliferating cells and recruitment of p97 to the ribosomes under growth factor stimulation (Figure 2C) are not compatible with a role for p97 in suppressing translation and cell growth. Thus, we asked if downregulation of endogenous p97 protein levels would have an effect on cell proliferation and a rate of general protein synthesis. For this purpose, a small interfering RNA (siRNA) against p97 was designed and transfected into T98G cells because of their high efficiency of siRNA transfection. siRNA against p97 efficiently induced a decrease in p97 protein levels (Figure 5A, top panel). p97 protein levels were efficiently downregulated up to 90 h after p97 siRNA transfection (data not shown). At 40 h after p97 siRNA transfections, the density of T98G cells was substantially decreased (Figure 5B). A decrease in cell number by downregulation of p97 protein levels was also observed in other cell lines including MCF7, U87, SF188, U20S, HEK293, 293T and wild-type human fibroblast WI38 cells (data not shown). To test if this decrease in cell number was due to growth arrest, T98G cells were stained with 5-bromo-2-deoxyuridine (Br-dU) at 40 h after p97 siRNA transfection. The percentage of Br-dU positive cells in p97 siRNA transfectants was reduced from ∼25 to ∼16% compared to control non-silencing siRNA transfectants (Figure 5C).
Figure 5.
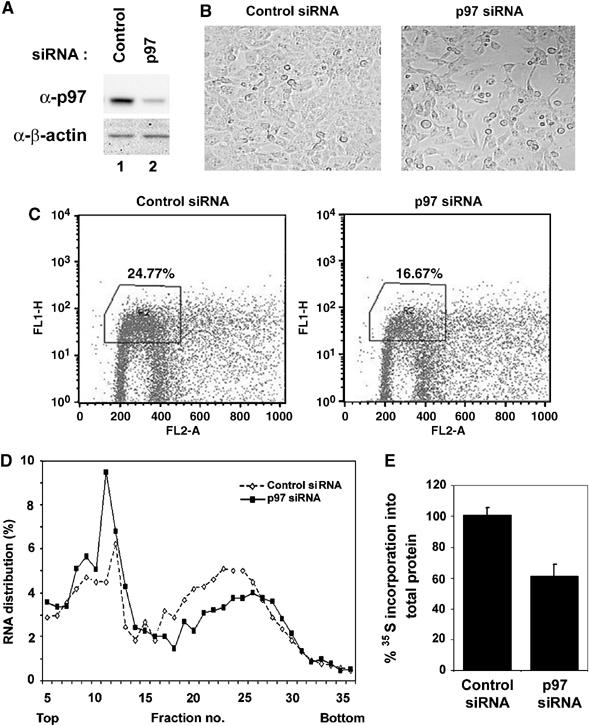
Downregulation of p97 protein levels by RNA interference inhibit cell proliferation and decreases a rate of general protein synthesis. (A) Control non-silencing or p97 siRNA was transfected into T98G cells. At 48 h after transfections, total lysates were prepared. Total lysates (20 μg) were separated and subjected to immunoblot analysis for p97 (Top) and β-actin for loading control (Bottom). (B) Control non-silencing (Left) or p97 siRNA (Right) were transfected into T98G cells and photographed 48 h after transfection. (C) T98G cells were labeled with Br-dU and subjected to FACS analysis. The percentages of Br-dU positive T98G cells from three independent experiments are shown. (D) Control non-silencing or p97 siRNA was transfected into T98G cells. At 48 h after transfection, cell extracts were prepared and resolved on sucrose density gradient. Gradients were separated into 36 fractions. RNA was isolated from each fraction quantified by absorbance at 260 nm. The amount of RNA in each fraction was measured at OD260 and plotted against fraction numbers. (E) Control non-silencing or p97 siRNA was transfected into T98G cells. At 48 h after transfection, cells were pulse-labeled with [35S]methionine and [35S]cysteine mix for 1 h and subjected to filter binding assay. The amount of [35S]methionine and [35S]cysteine incorporation into total protein was determined by liquid scintillation counting.
To determine if downregulation of p97 protein levels affected the polysome profile, sucrose density gradient velocity sedimentation and fractionation assays were performed after transfections with either control non-silencing or p97 siRNA. Compared to the polysome profile from lysate transfected with control non-silencing siRNA, the percent distributions of total RNA in polysome fractions compared to in sub-ribosomal and monosome fractions were decreased in lysates transfected with p97 siRNA (Figure 5D). Furthermore, compared to the control non-silencing siRNA, p97 siRNA caused a decrease in protein synthesis by ∼30–40% (Figure 5E).
To determine if the loss of polysomes and a decrease in protein synthesis by downregulation of p97 proteins levels arose from a decrease in mRNAs in polysomes, but not from mRNA degradation, sucrose density gradient velocity sedimentation and fractionation assays were performed. At 48 h after siRNA transfections, pCDNA3 vector encoding firefly luciferase was transfected and cell lysates were prepared 20 h later. Compared to control non-silencing siRNA, p97 siRNA caused a decrease in luciferase activity by ∼40% (Figure 6A, right). In agreement with this result, downregulation of p97 protein levels caused a shift of the majority of luciferase mRNA to fractions 2–5 likely where either monosomes or a small polysomes were present (Figure 6B–D). Taken together, these results suggest that endogenous p97 proteins can promote cell proliferation and protein synthesis.
Figure 6.
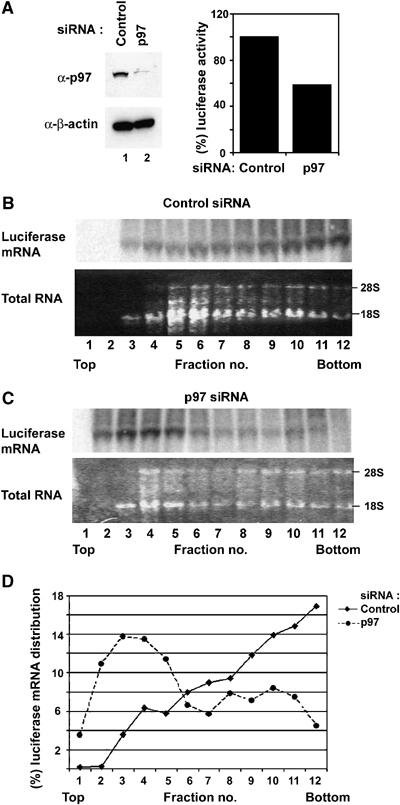
Downregulation of p97 proteins levels induces a reduction of luciferase mRNA in polysomes and luciferase activity. Control non-silencing or p97 siRNA was transfected into T98G cells. At 48 h after siRNA transfections, pCDNA3 vector encoding firefly luciferase was transfected. At 20 h after transfection, Cell extracts were prepared and resolved on sucrose density gradient. (A) Cell extracts (20 μg) was separated and subjected to immunoblot analysis for p97 and β-actin (Left). Cell extracts (20 μg) were analyzed for luciferase activity (Right). (B, C) Gradients were separated into 12 fractions and each fraction was used to exact total RNA and subjected to Northern blot analysis for luciferase mRNA and gel analysis to determine the presences of 18S and 28S rRNAs by ethidium bromide staining. (D) Intensity of bands corresponding to luciferase mRNA in each fraction was quantified by Storm-imager analysis using ImageQuant software and relative changes of luciferase mRNA were plotted against an indicated fraction.
Inhibition of cell proliferation by downregulation of p97 protein levels coincides with an increase in p27/Kip1 protein levels
Next, we asked if inhibition of cell proliferation by downregulation of p97 protein levels would correlate with expression of cell cycle inhibitory proteins, such as p27/Kip1. Figure 7A shows that indeed downregulation of p97 by siRNA resulted in accumulation of p27/Kip1 in T98G cells. Moreover, p97 downregulation in T98G cells led to cell cycle arrest at G1 (Figure 7B). Increased levels of p27/Kip1 correlated with marked inhibition of CDK2 kinase activity (Figure 7C). It is possible that upregulation of p27/Kip1 protein levels might be due to downregulation of Skp2, a substrate recognition subunit of the SCFSkp2 ubiquitin ligase complex, which targets p27/Kip1 and p21/Cip1 proteins for ubiquitination and subsequent degradation (Carrano et al, 1999; Bornstein et al, 2003). To test this possibility, Skp2 protein levels were monitored in response to p97 siRNA transfection. Skp2 protein levels did not change at 36 h after p97 siRNA transfection, although Skp2 protein levels gradually decreased 48 h after p97 siRNA transfection, probably due to a decrease in the rate of general protein synthesis (Figure 7A). To ensure that upregulation of p27/Kip1 was due to an increase in protein synthesis rather than inhibition of protein degradation, protein synthesis rate was measured in response to p97 downregulation. p97 siRNA was transfected into T98G cells. At 48 h after transfection, cells were pulse-labeled with [35S]methionine and [35S]cysteine mix for 1 h. Cells were harvested and immunoprecipitated with anti-p27/Kip1 antibody. As a negative control, immunoprecipitation with anti-Cyclin E antibody was included. Immunoprecipitated [35S] p27/Kip1 and Cyclin E protein were visualized by autoradiography and quantified. Downregulation of p97 increased p27/Kip1 protein synthesis rates by ∼2-fold compared to either control non-silencing siRNA whereas there was no measurable increase in Cyclin E protein synthesis rates (Figure 7D). p97 siRNA resulted in marked decrease of p97 mRNA levels, whereas there were no measurable changes of p27/Kip1 mRNA levels suggesting that an increase of protein synthesis rates were not due to increase of mRNA levels (data not shown). To further determine a half-life of p27/Kip1 protein, a pulse-chase analysis was performed after either control non-silencing or p97 siRNA transfections (Figure 7E). Downregulation of p97 by siRNA transfection increased p27/Kip1 protein synthesis rates by ∼2-fold compared to control non-silencing siRNA. However, the half-life of p27/Kip1 protein was not significantly different between control non-silencing siRNA transfectants and p97 siRNA transfectants (Figure 7E). Thus, increased p27/Kip1 protein levels were most likely due to an increase in protein synthesis rates rather than changes in protein stability. Taken together, these results indicate that p97 can positively promote proliferation of mammalian cells.
Figure 7.
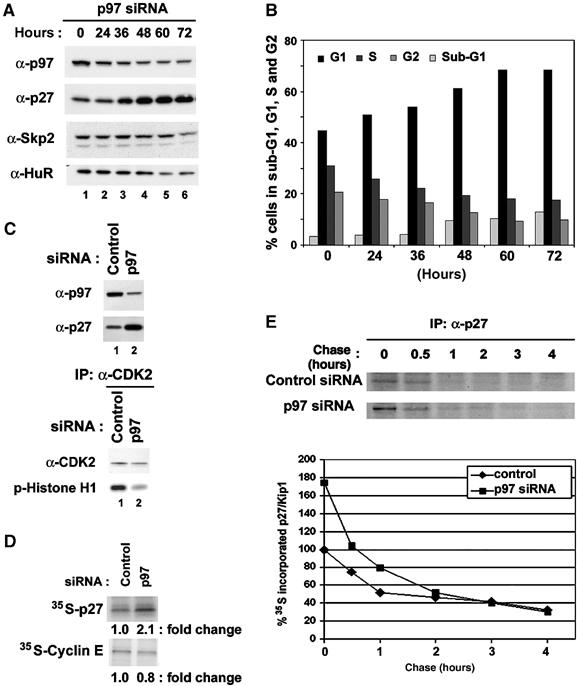
Downregulation of p97 protein levels increases in p27/Kip1 protein levels and inhibits CDK2 kinase activity. (A) Control non-silencing siRNA or p97 siRNA (sequences are shown in experimental procedures) was transfected into T98G cells. Cells were lysed at indicated time points and subjected to immunoblot analysis. (B) p97 siRNA transfected T98G cells were collected at indicated time point, stained with PI, and subjected to FACS analysis. (C) Immunoprecipitation and kinase assay. Total proteins were extracted from T98G cells 48 h after transfections with indicated siRNAs and immunoprecipitated with antibodies for CDK2 and were subjected to kinase assay with Histone H1 protein as a substrate and immunoprecipitated CDK2 protein levels were determined with immunoblot analysis. (D) Control non-silencing or p97 siRNA was transfected into T98G cells. Cells were pulse-labeled with [35S]methionine and [35S]cysteine mix for 1 h, cell extracts were prepared. [35S]-labeled cell extracts were immunoprecipitated with anti-p27/Kip1 or anti-Cyclin E antibodies together with protein A-agarose beads. Bound proteins were separated and subjected to autoradiography. Intensity of bands corresponding to an indicated protein was quantified by Storm-imager analysis using ImageQuant software and relative fold changes of indicated protein bands were shown. (E) Control non-silencing siRNA or p97 siRNA was transfected into T98G cells. Cells were pulse-labeled with [35S]methionine and [35S]cysteine mix for 1 h and chased for an indicated time. [35S]-labeled cell extracts were immunoprecipitated with anti-p27/Kip1 antibody together with protein A-agarose beads. Bound proteins were separated and subjected to autoradiography (Top). Intensity of bands corresponding to p27/Kip1 protein was quantified by Storm-imager analysis using ImageQuant software and relative changes of indicated protein bands were plotted against an indicated chase time (Bottom).
Discussion
In this paper, we show that p97 is a ribosome-associated factor that can positively promote cell proliferation and protein synthesis in some mRNAs. We found that expression of p97 is constitutive and makes up about 0.1–0.3% of total cellular proteins (data not shown), and that p97 is recruited to ribosomes following growth factor stimulation (Figure 2C). Downregulation of p97 protein levels by RNA interference decreased the rate of protein synthesis and cell growth (Figures 5E and 7B). Under these conditions, we observed a marked increase in p27/Kip1 protein levels and decrease in CDK2 kinase activity (Figure 7). In a monocistronic translational reporter system, we show a linear increase of reporter activity by titration of exogenous p97 (Figure 3A). In contrast, the C-terminal two-thirds of eIF4GI failed to increase luciferase activity, indicating that it is not equivalent in function to p97. We also found the N-terminal MIF4G domain of p97 is a dominant negative form of p97 as it markedly repressed reporter activity, a rate of global translation and cell proliferation (Figure 3) and it is required for ribosomal localization of p97 presumably through binding to eIF3 and eIF4AI (Figures 1 and 2). Furthermore, the MIF4G domain of p97, when it is ectopically overexpressed, can efficiently compete to eIF4GI and p97 for ribosome binding (data not shown) explaining its dominant negative functions. However, it is important to note that other truncations that remove or change the C-terminal W2 domain of p97 were less active, but did not repressed reporter activity (Figure 3A and D). We speculate that an inefficient recruitment of other translation factors including eIF2 and eIF4AI might cause a decrease in translation rate.
One prevailing model suggests that p97 represses translation, presumably because binds eIF3 and eIF4AI, but not the cap-binding protein eIF4E, and so sequesters eIF3 and eIF4AI into inactive complexes. Subsequent studies based on transient transfections with bicistronic reporter systems showed that overexpression of p97 in HeLa cells repressed both cap-dependent and cap-independent translation from EMCV IRESs (Imataka et al, 1997; Yamanaka et al, 1997). Furthermore, inducible expression of p97 in S2-6 cells reduced overall protein synthesis by ∼20% (Imataka et al, 1997), whereas we show an increase of reporter activity by exogenous p97 in S2-6 cells and ectopic expression of p97 did not reduce overall protein synthesis in these cells (data not shown). On the other hand, disruption of the mouse p97 gene does not change in global translation in embryonic stem cells, but causes embryonic lethality during gastrulation (Yamanaka et al, 2000).
How can this discrepancy be explained? One possible explanation is that the relative contribution of p97 and eIF4GI to global translation might vary in a cell type-dependent manner. Here, we show that the ability of p97 to promote translation using a monocistronic translational reporter system is lower than eIF4GI by ∼20–30% (Figure 3H). In addition, we found that p97, when it is only ectopically overexpressed, can compete to eIF4GI for ribosome binding (data not shown). Thus, in cells which global translation depends heavily on eIF4GI, overexpression of p97 may reduce the rate of global translation by competing with eIF4GI for relatively limiting amount of translation initiation factors such as eIF3. However, the levels of eIF3 protein in HeLa cells have been reported to be ∼0.8–1.5% of total protein (Mengod and Trachsel, 1985). We determined that the levels of p97 protein are ∼0.1–0.3% of total protein and the relative expression ratio of p97 to eIF4GI is 1–2:1 in exponentially growing breast carcinoma cells (data not shown). Thus, it is likely that endogenous p97, in contrast to ectopically overexpressed p97, does not compete with eIF4GI for eIF3 binding effectively since there is pool of excess eIF3 proteins. We attempted to overcome this potential complication using RNA interference. Downregulation of endogenous p97 protein levels by RNA interference induced a decrease in the rate of protein synthesis (Figure 5E) indicating that p97 does not function as a global translation repressor. However, it is important to note that overexpression of Myc-p97wt in HEK293 cells did not increase the rate of protein synthesis (Figure 3F) but increase our reporter activity (Figures 3 and 4). Furthermore, downregulation of p97 protein levels by RNA interference selectively induced a marked increase in p27/Kip1 protein levels whereas the rate of global translation was decreased (Figure 7). These results suggest that p97 might control translation of selective rather than bulk mRNAs, and also raise the interesting possibility that inactivation or modification of p97, such as a rapid conversion of p97 into the C-terminal truncated form called p86 during apoptosis, could be involved in switching general translation to a stress response related translation under an unfavorable condition for cell growth (Henis-Korenblit et al, 2002; Nevins et al, 2003; Warnakulasuriyarachchi et al, 2004). Notably, the ability of p86 to bind eIF4AI was markedly decreased (Figure 1E, lane 8) whereas Myc-p971−380, which contains only the MIF4G domain of p97, efficiently bound to eIF4AI (Figure 1E, lane 9), indicating a conformational change in the MIF4G domain of p86 by the C-terminal truncation. Further study is necessary to understand a biological significance of decrease in interaction between p86 and eIF4AI.
In summary, in contrast to a generally accepted view, we have demonstrated that p97 is functionally distinct from the C-terminal two-thirds of eIF4GI and that p97 is an abundantly expressed protein that requires for cell proliferation in mammalian cells. Understanding the contributions of p97 to general protein synthesis will require extensive characterizations of interactions with other eIFs and the relative effects on mRNA translation. During the manuscript preparation, it was reported that p97 promotes internal ribosome entry sequence (IRES)-driven translation and uncapped mRNA in vitro (Hundsdoerfer et al, 2005; Mikami et al, 2006). We also found adding an increasing amount of purified full-length p97 protein steadily promoted translation of uncapped mRNA in 7-methyl-GTP-Sepharose bead-depleted reticulocyte lysate where eIF4GI and eIF4E were removed (data not shown) whereas p971−380, a dominant-negative form of p97, inhibited translation in vitro.
Materials and methods
Cell lines, cell culture and reagents
Human glioblastoma T98G and U87 cells, human embryonic kidney HEK293 and 293T cells were maintained in Dullbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Gibco). Transient DNA transfections were performed with Lipofectamine Plus using the protocol provided by the manufacturer (Invitrogen).
The Myc-tagged full-length p97 was constructed by inserting p97/DAP5 cDNA (generous gift from Dr Holcik, University of Ottawa) into CMV-based mammalian expression vector pCAN1-Myc as EcoRI–XhoI fragments. Deletion mutants of p97 were generated using PCR with appropriate oligonucleotide primers and the PCR-derived DNAs were cloned into pCAN1-Myc as EcoRI–XhoI fragments. pCDNA3 encoding firefly luciferase gene constructed by inserting firefly luciferase fragment into pCDNA3 vector as HindIII and XbaI was a generous gift from Dr Tetsu (UCSF). The HA-tagged eIF2β was constructed by inserting PCR-derived eIF2β cDNA (Image ID, 3447202) into CMV-based mammalian expression vector pCAN1-HA as EcoRI–XhoI fragment. The vector encoding HA-tagged full-length eIF4GI was a generous gift from Dr Sonenberg (McGill University). The full-length eIF4GI used in this study represents eIF4GI isoform-b (Byrd et al, 2002). Deletion mutants of eIF4GI were generated using PCR with appropriate oligonucleotide primers and the PCR-derived DNAs were cloned into pCAN1-Myc as EcoRI–XhoI fragments. All GST expression vectors for p97 and eIF4GI were constructed using PCR with appropriate oligonucleotide primers and the PCR-derived DNAs were cloned into pET42a vector (Novagen) as EcoRI–XhoI fragments.
p97 small interfering (si) RNA oligonucleotide target sites were selected and generated as recommended (Qiagen). The target sequences are as p97 siRNA#1, 5′-AACAGATTCATCCTCTGCTCC (antisense direction of human p97 cDNA nucleotides 2370–2391). The DNA target sequence of non-silencing control siRNA was 5′-AATTCTCCGAACGTGTCACGT (Qiagen). siRNAs were transfected twice in 24 h intervals using Lipofectamine Plus (Invitrogen) at a final concentration of 240 nM.
Following monoclonal and polyclonal primary antibodies, horseradish peroxidase (HRP) conjugated secondary antibodies were used. p97 (G-20, Santa Cruz), eIF2α (Cell signaling), eIF2β (P-3, Santa Cruz), eIF4GI (N-20 and D-20, Santa Cruz), eIF4AI (N-19, Santa Cruz), eIF3 (p100) (L-18, Santa Cruz), Cyclin E (C-19, Santa Cruz), CDK2 (M2, Santa Cruz), p27/Kip1 and HuR (Transduction Laboratories), Skp2 (8D9, Zymed), β-actin (Sigma), sheep anti-mouse IgG HRP, donkey anti-rabbit IgG HRP (Amersham, Roche) and bovine anti-goat IgG HRP (Santa Cruz).
GST pulldown assay
Recombinant GST-p97 and GST-eIF4GI proteins were expressed in E. coli strain BL21(DE3)pLysS and purified with glutathione agarose beads (Amersham). To measure binding of eIF2 to immobilized GST-tagged p97 and GST-eIF4GI proteins, 1 μg of each GST-tagged protein was loaded onto glutathione agarose beads in binding buffer (10 mM sodium phosphate, pH 7.3, 150 mM NaCl, 0.1% Triton X-100 and 1 mM DTT) at 4°C for 30 min. The beads were extensively washed and incubated with 500 μg of 293T cell lysates overnight at 4°C. The matrix was extensively washed with binding buffer prior to addition of SDS–PAGE sample buffer. In all binding assays, immunoblot analysis was used to measure proteins that remained bound to the respective solid supports after extensive washing. Samples were electrophoresed through SDS polyacrylamide gels and transferred onto nitrocellulose. eIF2β was detected with anti-eIF2β mouse monoclonal antibody (P-3, SantaCruz) and eIF2α was detected with anti-eIF2α rabbit polyclonal antibody (Cell signaling).
Immunoprecipitation analysis of p97 and eIF2β proteins
In Figure 1C and D, 293T cells were co-transfected with an equal amount of CMV-derived mammalian expression vectors pCAN1 encoding an indicated epitope-tagged full-length or mutants p97 and eIF2β proteins. At 48 h after transfections, cells were lysed in NP-40 cell lysis buffer (50 mM Tris–HCl (pH 8.0), 120 mM NaCl, 0.1% NP-40) containing 1 mM DTT and Complete Mini (Roche). To inhibit phosphatase activity, 0.4 mM sodium orthovanadate and 1 mM sodium fluoride were included in the lysis buffer. In Figure 1E, S100 fractions were prepared as previously described (Deutscher, 1990) with a modification during dialysis. Briefly, cells were collected by centrifugation (2000 r.p.m.) and washed with cold PBS. The cells were then suspended in buffer A (10 mM HEPES–NaOH (pH 7.9 at 25°C), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.5 mM PMSF) and allowed to stand on ice for 10 min to swell. The cells were lysed by 10 strokes in a glass-glass Dounce homogenizer using a tight-fitting pestle and centrifuged (2000 r.p.m.) for 10 min. The supernatant fractions were added 0.11 vol of buffer B (0.3 mM HEPES–NaOH (pH 7.9 at 25°C), 30 mM MgCl2, 1.4 M KCl) with gentle stirring followed by centrifugation (100 000 g, 1 h). The supernatant fractions were dialyzed against 0.1% NP-40 cell lysis buffer and used for immunoprecipitation analysis.
To determine interaction of p97 with eIF2β in vivo, 293T cells lysates were prepared in 0.1% NP-40 cell lysis buffer. To compete with interaction of p97 to eIF2β, increase amounts of purified GST-tagged p97349−907 (amino acids 349–907) were included in the lysates. Immunoprecipitation of endogenous p97 proteins was performed with 2 μg of a goat polyclonal antibody directed to N-terminus of p97 (G-20, Santa Cruz) and 20 μl protein A beads per sample (Santa Cruz). The presence of endogenous eIF2β proteins in the immunoprecipitates was determined by immunoblot analysis with anti-eIF2β mouse monoclonal antibody (Santa Cruz).
Fractionation and RSW analysis
In Figure 2A, RSW analysis was performed as previously described (Chang et al, 1993). In brief, exponentially growing T98G cells were collected by centrifugation (2000 r.p.m.) and washed with cold PBS. The cells were then suspended in hypotonic lysis buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.1 mM PMSF). The cells were homogenized by 50 strokes in a glass-glass Dounce homogenizer using a tight-fitting pestle and centrifuged (2000 r.p.m.) for 10 min to remove nuclei. The supernatant was centrifuged at 100 000 g for 1 h, and the pellet was resuspended in RSW buffer (5 mM Tris–HCl (pH 7.6), 0.1 mM EDTA, 1 mM DTT, 0.25 M sucrose and a final concentration of 0.5 M KCl achieved by slowly adding 4 M KCl), agitated for 1 h and centrifuged at 100 000 g for 2 h. The remaining pellet was suspended in 1% NP40 cell lysis buffer.
Polyribosome and immunoblot analysis of ribosomal fractions
Polyribosome analysis was performed as previously described (Krieg, 1996). Briefly, cycloheximide (final concentration 90 μg/ml) was added to cells culture 10 min before harvesting and left the cell culture at 37°C. Cells were briefly trypsinized, washed two times with cold PBS and collected by spinning at 2000 r.p.m. for 5 min at 4°C. Cells were resuspended in 450 μl of cold reticulocyte standard buffer (RSB) (10 mM NaCl, 10 mM Tris–HCl (pH 7.4), 15 mM MgCl2), lysed by addition of 60 μl of lysis buffer (10% Triton X-100, 10% deoxycholate in RSB) and centrifuged for 3 min at 4°C. The resulting cytosol was diluted with an equal volume of polysomal buffer (25 mM Tris–HCl (pH 7.4 at 4°C), 10 mM MgCl2, 25 mM NaCl, 0.05% Triton X-100, 0.14 M Sucrose, 500 μg heparin/ml, 200 U of RNasin (Promega), 1 mM DTT) and applied to 10–45% sucrose density gradient in buffer A (25 mM Tris–HCl (pH 7.4 at 4°C), 25 mM NaCl, 5 mM MgCl2) and centrifuged at 4°C in SW40 Ti Beckman rotor for 2.5–3 h at 37 000 r.p.m. Gradient analysis was performed by manually collecting 12 fractions. In Figure 2C, top, one-third of the gradient was separated into 26 fractions. In Figure 5D, after centrifugation, the gradients were divided into 36 fractions. RNA was isolated from each fraction by using an RNeasy minikit (QIAGEN) and quantified by absorbance at 260 nm. For protein analysis, indicated fractions were pooled and precipitated with TCA to a final concentration of 10%, washed with ice-cold acetone. The resulting protein pellets were resuspended in SDS sample buffer. The samples were electrophoresed through SDS polyacrylamide gels and subjected to immunoblot analysis as described above.
Protein synthesis analysis
To determine a rate of general protein synthesis, 48 h after transfections with siRNAs, T98G cells were pulse-labeled with 0.1. mCi of [35S]methionine and [35S]cystein label mix (Perkin-Elmer) for 1 h, and cells were lysed in 1% NP-40 lysis buffer (50 mM Tris–HCl (pH 8.0), 120 mM NaCl, 1% NP-40) containing 1 mM DTT and Complete Mini (Roche). To measure the total uptake of radioactivity, an equal amount of proteins from the cell lysates was subjected to filter binding assay. In brief, the samples were filtered through 0.22 μm GS filter (Millipore). The filters were washed three times with cold PBS, and analyzed by liquid scintillation counting.
For reporter analysis as shown in Figures 3 and 4, 293T cells were co-transfected with pCDNA3 encoding firefly luciferase with indicated amount of CMV-based mammalian expression vectors encoding HA-tagged full-length eIF4GI, Myc-tagged full-length p97 or Myc-tagged deletion mutants of p97 or eIF4GI. At 20 h after transfection, firefly luciferase expression was determined using luciferase assay system (Promega).
Immunoprecipitation and protein kinase assay
In Figure 7C, total cell lysates were prepared in cell lysis buffer 50 mM Tris–HCl (pH 8.0), 120 mM NaCl, 0.2% NP-40 containing 1 mM DTT, the protease inhibitor cocktail, Complete Mini (Roche), and Phosphatase inhibitor mix I and II (Sigma). Extracts containing 100 μg proteins were immunoprecipitated with 2 μg rabbit antibodies to CDK2 at 4°C for 2 h and collected with 30 μl Protein G PLUS-Agarose (Santa Cruz) at 4°C for 1 h. After three washes in washing buffer 50 mM Tris–HCl (pH 8.0), 120 mM NaCl, 0.1% NP-40 containing 1 mM DTT, the protease inhibitor cocktail, Complete Mini (Roche), and Phosphatase inhibitor mix I and II (Sigma). Precipitated immune complex and washed once with kinase buffer and was used for protein kinase assays. Reactions were performed with 25 μl kinase buffer, 50 mM Tris–HCl (pH 8.0), 10 mM β-glycerophosphate, 1 mM DTT, 0.1 mM Na3VO4, 1 mM NaF, 10 mM MgCl2 containing 20 μM ATP, and 0.37 M Bq γ-32P ATP plus 2 μg histone H1 (Upstate) at 30°C for 30 min.
Isolation of total cellular RNA and Northern blot analysis
Total cellular RNA was isolated from cell plates using TRIzol reagent (Gibco). Total cellular RNA (20 μg) was resolved on a 1% agarose/formaldehyde gel and then transferred onto a Hybond-N nylon membrane (Amersham) by capillary transfer. Antisense probes were synthesized in vitro using a PCR-generated template complementary to luiferase (nt 215–715). PCR-generated DNA templates were gel purified and labeled with [32P]α-CTP using Rediprime II random prime labeling system (Amersham). The filter membranes were prehybridized for 30 min at 60°C, followed by probe hybridization at 60°C overnight. The membranes were washed with 2 × SSC and 0.2 × SSC followed by autoradiography.
Cell cycle analysis
After p97 siRNA-transfected cells and control non-silencing siRNA-transfected cells (1.0 × 106/plate) were incubated for 40 h in DMEM supplemented with 10% FBS at 37°C, Br-dU was added to the culture medium (10 μM), and the cultures were incubated for an additional 30 min at 37°C. Cells were fixed in 70% ethanol at −20°C. To denature the DNA, fixed cells were incubated for 30 min at room temperature in 2 N HCl with 0.5% Triton X-100. After neutralization with 0.1 M sodium tetraborate (pH 8.5), cells were incubated with anti-Br-dU-FITC antibody (Becton Dickinson, San Jose, CA) for 30 min at room temperature and resuspended in 5 μg/ml propidium iodide (PI). Cells were analyzed by using a FACSCaliber with Cellquest (Becton Dickinson) at the UCSF Cancer Center Cytometry Core. For cell cycle analysis, PI stained cells were pelleted and resuspended in 1 ml of 0.1% sodium citrate containing 0.3% NP-40, 0.0002 mg/ml RNase and 50 μg/ml PI, and incubated for 30 min on ice. Cell cycle profile was analyzed with a FACSCaliber with Cellquest (Becton Dickinson) and Modifit software.
Acknowledgments
We thank Dr Martin Holcik for the plasmid containing p97/DAP5 and Dr Nahum Sonenberg for the plasmid containing eIF4GI. We are grateful to Dr Osamu Tetsu, Dr Luika Timmerman, Dr Pablo Rodriguez-Viciana, Dr Madhu Macrae and Ms Suchitra Ananthnarayan for critical readings of this manuscript and sharing of reagents. We thank members of the McCormick laboratory, and the UCSF Comprehensive Cancer Center Cytometry Core and Genome core facilities, especially Dr Ginzinger for real-time Q-PCR Analysis.
References
- Asano K, Krishnamoorthy T, Phan L, Pavitt GD, Hinnebusch AG (1999) Conserved bipartite motifs in yeast eIF5 and eIF2Bepsilon, GTPase-activating and GDP–GTP exchange factors in translation initiation, mediate binding to their common substrate eIF2. EMBO J 18: 1673–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A (2003) Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem 278: 25752–25757 [DOI] [PubMed] [Google Scholar]
- Byrd MP, Zamora M, Lloyd RE (2002) Generation of multiple isoforms of eukaryotic translation initiation factor 4GI by use of alternate translation initiation codons. Mol Cell Biol 22: 4499–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M (1999) SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1: 193–199 [DOI] [PubMed] [Google Scholar]
- Chang KH, Brown EA, Lemon SM (1993) Cell type-specific proteins which interact with the 5′ nontranslated region of hepatitis A virus RNA. J Virol 67: 6716–6725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Maitra U (2000) Mutational analysis of mammalian translation initiation factor 5 (eIF5): role of interaction between the beta subunit of eIF2 and eIF5 in eIF5 function in vitro and in vivo. Mol Cell Biol 20: 3942–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher MP (ed) (1990) Guide to protein purification. New York: Academic press [Google Scholar]
- Duncan R, Hershey JW (1983) Identification and quantitation of levels of protein synthesis initiation factors in crude HeLa cell lysates by two-dimensional polyacrylamide gel electrophoresis. J Biol Chem 258: 7228–7235 [PubMed] [Google Scholar]
- Henis-Korenblit S, Shani G, Sines T, Marash L, Shohat G, Kimchi A (2002) The caspase-cleaved DAP5 protein supports internal ribosome entry site-mediated translation of death proteins. Proc Natl Acad Sci USA 99: 5400–5405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henis-Korenblit S, Strumpf NL, Goldstaub D, Kimchi A (2000) A novel form of DAP5 protein accumulates in apoptotic cells as a result of caspase cleavage and internal ribosome entry site-mediated translation. Mol Cell Biol 20: 496–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundsdoerfer P, Thoma C, Hentze MW (2005) Eukaryotic translation initiation factor 4GI and p97 promote cellular internal ribosome entry sequence-driven translation. Proc Natl Acad Sci USA 102: 13421–13426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imataka H, Olsen HS, Sonenberg NA (1997) New translational regulator with homology to eukaryotic translation initiation factor 4G. EMBO J 16: 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imataka H, Sonenberg N (1997) Human eukaryotic translation initiation factor 4G (eIF4G) possesses two separate and independent binding sites for eIF4AI. Mol Cell Biol 17: 6940–6947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapp LD, Lorsch JR (2004) The molecular mechanics of eukaryotic translation. Annu Rev Biochem 73: 657–704 [DOI] [PubMed] [Google Scholar]
- Koonin EV (1995) Multidomain organization of eukaryotic guanine nucleotide exchange translation initiation factor eIF-2B subunits revealed by analysis of conserved sequence motifs. Protein Sci 4: 1608–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg PA (ed) (1996) A Laboratory Guide to RNA: Isolation, Analysis, and Synthesis. New York, NY: Wiley-Liss, Inc [Google Scholar]
- Lamphear BJ, Kirchweger R, Skern T, Rhoads RE (1995) Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. J Biol Chem 270: 21975–21983 [DOI] [PubMed] [Google Scholar]
- Levy-Strumpf N, Deiss LP, Berissi H, Kimchi A (1997) DAP-5, a novel homolog of eukaryotic translation initiation factor 4G isolated as a putative modulator of gamma interferon-induced programmed cell death. Mol Cell Biol 17: 1615–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengod G, Trachsel H (1985) Eukaryotic protein synthesis initiation factor eIF-3: determination of concentration and association with ribosomes in rabbit reticulocyte and HeLa cell lysates. Biochim Biophys Acta 825: 169–174 [DOI] [PubMed] [Google Scholar]
- Mikami S, Masutani M, Sonenberg N, Yokoyama S, Imataka H (2006) An efficient mammalian cell-free translation system supplemented with translation factors. Protein Exp Purif 46: 348–357 [DOI] [PubMed] [Google Scholar]
- Morino S, Imataka H, Svitkin YV, Pestova TV, Sonenberg N (2000) Eukaryotic translation initiation factor 4E (eIF4E) binding site and the middle one-third of eIF4GI constitute the core domain for cap-dependent translation, and the C-terminal one-third functions as a modulatory region. Mol Cell Biol 20: 468–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins TA, Harder ZM, Korneluk RG, Holcik M (2003) Distinct regulation of internal ribosome entry site-mediated translation following cellular stress is mediated by apoptotic fragments of eIF4G translation initiation factor family members eIF4GI and p97/DAP5/NAT1. J Biol Chem 278: 3572–3579 [DOI] [PubMed] [Google Scholar]
- Shaughnessy JD Jr, Jenkins NA, Copeland NG (1997) cDNA cloning, expression analysis, and chromosomal localization of a gene with high homology to wheat eIF-(iso)4F and mammalian eIF-4G. Genomics 39: 192–197 [DOI] [PubMed] [Google Scholar]
- Warnakulasuriyarachchi D, Cerquezzi S, Cheung HH, Holcik M (2004) Translational induction of the Inhibitor of Apoptosis protein HIAP2 during endoplasmic reticulum stress attenuates cell death and is mediated via an inducible IRES element. J Biol Chem 279: 17148–17157 [DOI] [PubMed] [Google Scholar]
- Yamanaka S, Poksay KS, Arnold KS, Innerarity TL (1997) A novel translational repressor mRNA is edited extensively in livers containing tumors caused by the transgene expression of the apoB mRNA-editing enzyme. Genes Dev 11: 321–333 [DOI] [PubMed] [Google Scholar]
- Yamanaka S, Zhang XY, Maeda M, Miura K, Wang S, Farese RV Jr, Iwao H, Innerarity TL (2000) Essential role of NAT1/p97/DAP5 in embryonic differentiation and the retinoic acid pathway. EMBO J 19: 5533–5541 [DOI] [PMC free article] [PubMed] [Google Scholar]