


Abstract
Viral integrase (IN) and Vpr are both components of the human immunodeficiency virus type 1 (HIV-1) pre-integration complex. To investigate whether these proteins interact within this complex, we investigated the effects of Vpr and its subdomains on IN activity in vitro. When a 21mer oligonucleotide was used as a donor and acceptor, both Vpr and its C-terminal DNA-binding domain [(52–96)Vpr] inhibited the integration reaction, whereas the (1–51)Vpr domain did not affect IN activity. Steady-state fluorescence anisotropy showed that both full-length and (52–96)Vpr bind to the short oligonucleotide, thereby extending previous observations with long DNA. The concentrations of the two proteins required to inhibit IN activity were consistent with their affinities for the oligonucleotide. The use of a 492 bp mini-viral substrate confirmed that Vpr can inhibit the IN-mediated reaction. However, the activity of (52–96)Vpr differed notably since it stimulated specifically integration events involving two homologous mini-viral DNAs. Order of addition experiments indicated that the stimulation was maximal when IN, (50–96)Vpr and the mini-viral DNA were allowed to form a complex. Furthermore, in the presence of (50–96)Vpr, the binding of IN to the mini-viral DNA was dramatically enhanced. Taken together, these data suggest that (52–96)Vpr stimulates the formation of a specific complex between IN and the mini-viral DNA.
INTRODUCTION
The integration of a DNA copy of the retroviral RNA genome is a key step in the human immunodeficiency virus (HIV) replication cycle. This process is carried out by the viral integrase (IN), which directs two distinct reactions: (i) a 3′ processing reaction in which a dinucleotide is removed from the 3′ end of the proviral DNA; and (ii) a strand transfer reaction, resulting in the joining of processed 3′ ends to 5′ phosphates in the target DNA (1,2). In vivo, the integration of both extremities of the viral DNA is a concomitant process, referred to as concerted integration. Although recombinant integrase is capable of performing both 3′ processing and strand transfer reactions in vitro, it does not reproduce the in vivo process accurately. In particular, the number of concerted integration products generated remains low compared with the in vivo process (3–6). In vivo, the integration activity is carried out by a pre-integration complex (PIC). Thus, the enzyme may only be able to perform its function correctly when it is included in a complex that comprises at least the integrase and the viral DNA associated with viral and/or cellular factors. Defective IN activity may reflect the lack of these factors in in vitro assays. This hypothesis is supported by the finding that fidelity is improved when the reaction is performed either with IN purified from viral particles or with pre-integration complexes purified from HIV-infected cells (7–9). It has been proposed that the viral nucleocapside NCp7 is a viral cofactor given that it is present within purified PIC and that it can stimulate IN activity in vitro (3,10). Small, basic, cellular proteins, such as HMGI(Y), which are able to modulate the condensation of DNA, also stimulate IN activity (5,11,12). Although these proteins are believed to be part of the PICs, they are not thought to be tightly bound given the low stringency conditions necessary for their co-purification with the PIC (13,14). The viral protein R, Vpr, is also associated with the PIC and interacts with the nucleocapsid protein (15–17). Vpr is a 15 kDa accessory protein of HIV-1, packaged into the virion through its interaction with Gag (13,18,19). Vpr has a number of activities, including the induction of G2/M cell cycle arrest, the modulation of apoptosis and a weak transcriptional activity in infected cells (20–26). Vpr participates in the nuclear import of the HIV PIC (15,27). These functions are mediated by the interaction between Vpr and different cellular proteins [for a review see Bukrinsky and Adzhubel (28)]. Furthermore, Vpr binds DNA and RNA in a non-specific manner (29,30). The nucleic acid-binding activity, a property that has not yet been shown to be associated with any particular function, has been mapped to the (52–96) C-terminal domain of Vpr. Vpr, and in particular (52–96)Vpr, may act as a transfection agent due to its ability to induce DNA condensation, a phenomenon that is highly reminiscent of the properties of NCp7 (31,32). The analogy between Vpr and NCp7 DNA-binding properties prompted us to examine whether Vpr and its (52–96) subdomain affect the integration reaction in vitro. First, we showed that the full-length Vpr and (52–96)Vpr inhibit integration activity using a short oligonucleotide substrate (ODN). This effect was due to the strong binding of both proteins to the short ODN, as demonstrated by static fluorescence experiments. Secondly, we observed that the C-terminal domain (52–96) of Vpr can specifically stimulate the homologous strand transfer resulting from the integration of a 492 bp mini-viral DNA substrate into a homologous fragment. Pre-incubation assays suggested that this effect was caused by the stimulation of the formation of an integration-competent complex involving IN and the mini-viral substrate. This hypothesis was reinforced by the dramatic enhancement of IN binding to the mini-viral DNA in the presence of (52–96)Vpr.
MATERIALS AND METHODS
Proteins and peptides
Peptide (1–96)Vpr from HIV-1 strain LAI was used. The sequence of this peptide was QAPEDQGPQREPYNDWTLELLEELKNEAVRHFPRIWLHSLGQHIYETYGDTWT GVEALIRILQQLLFIHRIGCRHSRIGIIQQRRTRNGASKS. Peptides were synthesized as described previously (16). Electrospray mass spectrometry was used to confirm the identities of peptides (1–51)Vpr [theoretical molecular mass (MMth) = 6165.81; calculated molecular mass (MMcalc) = 6167], (52–96)Vpr (MMth = 5247.3; MMcalc = 5249), Vpr (1–96) (MMth = 11 394.9; MMcalc = 11 392.6), (70–96)Vpr (MMth = 3148.66; MMcalc = 3148) and (60–80)Vpr (MMth = 2577.18; MMcalc = 2578). Recombinant IN protein was purified as previously described (33).
Oligonucleotides
Oligonucleotides U5B 5′-GTGTGGAAAATCTCTAGC AGT-3′, U5-B-2 5′-GTGTGGAAAATCTCTAGCA-3′, U5A 5′-ACTGCTAGAGATTTTCCACAC-3′ and D 5′-TGC TAGTTCTAGCAGGCCCTTGGGCCGGCGCTTGCGCC-3′ were purchased from Eurogentec and purified further on an 18% denaturing acrylamide/urea gel. For processing assays, 100 pmol of U5B was radiolabeled using T4 polynucleotide kinase and 50 µCi of [γ-32P]ATP (3000 Ci/mmol). The T4 kinase was heat inactivated and unincorporated nucleotides were removed using a Sephadex G-25 column (Pharmacia). NaCl was added to a final concentration of 0.1 M and the complementary unlabeled strand U5A was added. The mixture was incubated at 90°C for 3 min and the DNA was annealed by slow cooling. Pre-processed U3U5 DNA substrate consists of a 492 bp DNA fragment generated by NdeI restriction of pU3U5 (10).
IN activity assays
Processing reactions were performed using U5 double-stranded oligonucleotide substrates in buffer containing 20 mM HEPES pH 7.0, 50 mM NaCl, 2 mM dithiothreitol (DTT) with 10 mM MnCl2. The reaction was initiated by adding substrate DNA. The reaction mixture was incubated for up to 1 h at 37°C. The reaction was stopped by phenol–chloroform extraction, and DNA products were precipitated with ethanol. The products were dissolved in TE buffer containing 7 M urea and subjected to electrophoresis on an 18% denaturing acrylamide/urea gel. Long fragment integration assays were carried out using 10 ng of [32P]U3U5 DNA fragment and 40 ng of pSP70 vector as a heterologous integration target in a buffer containing 20 mM HEPES pH 7.0, 50 mM NaCl, 2 mM DTT, 0.05% NP-40, 0.5 mM CHAPS and 10 mM MnCl2. The mixture was pre-incubated at 4°C for 10 min and then at 37°C for 1 h. After phenol–chloroform extraction and DNA precipitation, the products were separated by electrophoresis on a 1% agarose gel containing 0.1% SDS. Gels were dried and vizualized using a STORM™ Molecular Dynamics phosphorimager.
DNA gel electrophoretic mobility shift assay
Experiments were performed in the same buffers as the strand transfer assay. After 10 min at 4°C, DNA loading buffer was added and samples were loaded on a 1% agarose gel. Gel electrophoresis was carried out in 0.5× Tris/borate buffer. Gels were dried and visualized on a Molecular Dynamics STORM™ phosphoimager.
Fluorescence anisotropy study
Steady-state fluorescence anisotropy experiments were carried out using a Beacon 2000 Fluorescence Polarization System (Pan Vera P3200) to investigate the interaction between Vpr, or its fragments, and DNA. The target DNA used in the following studies was 21 bp long, representing the terminal sequence of the HIV-1 viral DNA, and 5′-labeled with fluorescein 5′-F-GTG TGG AAA ATC TCT AGC AGT-3′ (E21AF5). Oligonucleotides E21B (5′-ACT GCT AGA GAT TTT CCA CAC-3′) and E21AF5 were purchased from Cybergene. Fluorescein was attached to the ODN through a six-carbon linker to minimize perturbation of the DNA–Vpr interaction. The duplex was pre-formed in 50 mM NaCl, 20 mM HEPES pH 7.2. The solution was incubated at 85°C for 5 min and allowed to cool slowly on the bench. Concentrations of double-stranded DNA (dsDNA) were estimated by measuring the optical density at 260 nm (corrected for hypochromic effects). To follow a change in anisotropy, the concentration of dsDNA was set to 10 nM and a small aliquot (1 or 2 µl) of protein was added successively to 100 µl of total volume to avoid a dilution effect. The anisotropy value of labeled dsDNA alone in our conditions was approximately 0.04. This value was systematically subtracted from the anisotropy value of the DNA–protein complex (delta anisotropy). A theoretical binding curve following the Hill formulation was fitted to experimental data using the maximum delta anisotropy (δmax), Kapp and the Hill coefficient n as adjustable parameters.
δ = δmax(1 + (Kapp/[P])n)1
where [P] refers to the total peptide concentration and Kapp the apparent dissociation constant. The difference between experimental and theoretical values was minimized using the non-linear regression module of Prism 3.0 (Graphpad software).
RESULTS
Full-length Vpr and (52–96)Vpr inhibit IN activity on short ODNs
Both the DNA-binding properties of Vpr and its presence within the PIC prompted us to explore whether Vpr affects the integration process. First, we examined the effect of Vpr and its (52–96) DNA-binding domain by assaying HIV-1 IN activity in the presence of short oligonucleotide substrates. In this assay, a double-stranded 21mer ODN mimicking the HIV-1 U5 long terminal repeat (LTR) extremity acted as both a DNA donor and acceptor. In the presence of IN, both activities, i.e. 3′ processing and strand transfer, were observed. The incubation of the enzyme in the presence of the 21mer substrate led to the accumulation of a 19mer oligonucleotide product corresponding to the trimmed substrate (Fig. 1, lane 7). The larger bands were the products of the subsequent strand transfer reaction resulting from the covalent insertion of a donor into a homologous acceptor ODN. Vpr strongly inhibited both activities of IN (Fig. 1). This inhibition was dose dependent, with an EC50 of 0.2 µM.
Figure 1.

Inhibition of HIV-1 IN-mediated 3′ processing by Vpr and its subdomains. (A) Dose–response effect of full-length Vpr on 3′ processing of a 21mer ODN substrate. The 5′-end-labeled U5 substrate (10 nM) was incubated for 1 h at 37°C with 65 nM IN. Lanes 1–6, 50, 100, 200, 400 and 800 nM, and 1.6 µM Vpr, respectively; lane 7, no Vpr; lane 8, negative control in the absence of IN. The 21mer DNA substrate, 19mer 3′ processed product and the strand transfer products are indicated on the left. (B) Quantification of experimental data obtained in the presence of full-length Vpr (filled circles), (1–51) (open squares) and (52–96) (open diamonds) peptides of Vpr. Data from two independent experiments were averaged.
The same experiment was performed with the N-terminal (1–51) domain or with the C-terminal DNA-binding (52–96) domain of Vpr. IN activity was tested in the presence of these peptides and the results obtained were compared with those obtained with the full-length protein. The 3′ processing activity was quantified in the presence of both peptides (Fig. 1B). The (52–96) peptide strongly impaired the IN activity, whereas the (1–51) peptide did not affect IN activity at concentrations up to 5 µM. The inhibition due to (52–96)Vpr was comparable with that of the full-length protein, suggesting that IN inhibition was related to the DNA-binding property of Vpr.
Affinity of full-length and truncated Vpr for ODN
The affinity of full-length and truncated Vpr for nucleic acids was evaluated previously by measuring their capacity to aggregate long nucleic acids (30,31). This approach does not make it possible to estimate the apparent association constant for short oligonucleotides. To investigate the possibility that full-length and (52–96)Vpr bind to short ODNs and subsequently compete with IN for the 21mer oligonucleotide, we used steady-state fluorescence anisotropy to evaluate their affinity for the U5 oligonucleotide. This method makes it possible to quantify the association between a fluorescently labeled ODN and a putative ligand by monitoring the fluorescence anisotropy increase as a function of the amount of complex present in solution. The addition of increasing concentrations of Vpr and (52–96)Vpr to a solution of fluorescein-labeled U5 21mer induced a dramatic increase in the steady-state anisotropies of the ODN (Fig. 2). In the presence of (1–51)Vpr, no significant change of anisotropy was detected. We concluded that both full-length Vpr and (52–96)Vpr readily associated with the oligonucleotide, whereas no binding was detectable for (1–51)Vpr. The sigmoidal shape of the experimental binding curve hinted at a positive cooperative binding. Indeed, data fitted best a binding isotherm corresponding to a simple Hill model (see Materials and Methods). Apparent dissociation constants (Kapp) were derived from the binding isotherms, yielding values of 0.48 × 10–6 M for Vpr and 0.25 × 10–6 M for (52–96)Vpr. Hill coefficients were 1.5 and 2, respectively, for Vpr and (52–96)Vpr. Moreover, similar values were obtained with a non-viral oligonucleotide, thus indicating that binding of both Vpr and (52–96)Vpr to short ODNs was not sequence specific (data not shown).
Figure 2.

Binding of Vpr and (52–96)Vpr to the double-stranded 21mer U5 ODN. Change in steady-state fluorescence anisotropy of fluorescein-labeled U5 ODN (10 nM) was monitored as a function of increasing concentrations of either full-length Vpr (filled squares) or (52–96)Vpr (filled triangles). The anisotropy value of labeled dsDNA alone recorded in our condition was close to 0.04 and was systematically subtracted from the anisotropy value of the DNA–protein complex (δ anisotropy). Addition of (1–51)Vpr did not lead to a change in anisotropy, indicating the absence of complex formation. Isotherm binding curves were fitted with Prism3.0 software.
These values were consistent with the EC50 concentrations obtained for integrase inhibition using the U5 ODN as a DNA substrate, suggesting that these proteins actually compete with IN for the oligonucleotide.
(52–96)Vpr stimulates homologous integration of long DNA fragments
To avoid the possibility that an effect of Vpr and (52–96)Vpr might be masked by this competition for the short substrate, we decided to use longer substrates that could accommodate both proteins simultaneously. The 21mer oligonucleotide was substituted for a 492 bp dsDNA fragment containing both U5 and U3 extremities as a mini-viral donor DNA. pSP70 was used as the heterologous acceptor DNA. In the presence of IN, both homologous (bands b) and heterologous (bands c) integration products were detectable (Fig. 3A). Homologous integration refers to the integration of one 492 bp fragment into a second homologous fragment, whereas heterologous integration corresponds to the insertion of one or several LTR fragments into the pSP70 target (10). In the presence of increasing concentrations of Vpr, the strand transfer activity was inhibited (Fig. 3A, lanes 3–5, and B). Both homologous and heterologous strand transfer were equally impaired, confirming the effect previously observed with the short oligonucleotide substrate. Likewise, the (1–51) fragment did not affect IN activity (Fig. 3A, lanes 6–8). In contrast, (52–96)Vpr stimulated strand transfer (Fig. 3A, lanes 9–11, and B). At concentrations of 1.6 µM, which inhibit 90% of the oligonucleotide 3′ processing activity, the strand transfer activity was enhanced 4-fold. Furthermore, analysis of the integration products indicated that the overall stimulation resulted exclusively from an enhancement of the homologous strand transfer, whereas the heterologous strand transfer was not affected (Fig. 3B). Thus, unlike other DNA-binding proteins that stimulate the integration reaction in vitro, the (52–96) peptide specifically affected integration into the mini-viral DNA.
Figure 3.

Vpr effects on long DNA integration activity. (A) Phosphorimage of long DNA strand transfer assay. Reactions were performed by incubation for 1 h at 37°C of 2 pmol IN, 10 ng of U3U5 substrate and 40 ng of pSP70 target in the presence of increasing concentrations of Vpr and its subdomains: 0.4 µM (lanes 3, 6 and 9), 0.8 µM (lanes 4, 7 and 10), 1.6 µM (lanes 5, 8 and 11) of Vpr, (1–51)Vpr or (52–96)Vpr, respectively. Components were pre-incubated for 10 min on ice before incubation at 37°C, with the exception of the pSP70 target, which was added at the beginning of the reaction. The nature of the integration product is reported on the right: bold arrows indicate the main products corresponding to integration of one mini-viral DNA; fainter arrows indicate products corresponding to multiple integration events. (a) 32P-labeled U3U5 substrate; (b) homologous integration products; (c) heterologous integration products. (B) Quantification of homologous integration products using ImageQuant software. Homologous integration in the presence of (52–96)Vpr (filled circles); (1–51)Vpr (filled inverted triangles) and Vpr (filled squares).
To determine whether the properties of (52–96)Vpr could be mapped to one or several specific sequences, the overlapping (52–96), (52–70), (60–80) and (70–96) Vpr peptides were assayed in the long DNA strand transfer assay. Increasing concentrations of peptides were assayed. Figure 4 presents heterologous (Fig. 4A) and homologous (Fig. 4B) strand transfer products. Homologous integration was stimulated 10-fold in the presence of 1.6 µM of the (52–96) fragment (Fig. 4B, lanes 2–4). At higher concentrations, integration was strongly inhibited (Fig. 4A and B, lanes 4 and 5). The (52–70) and (70–96) peptides did not significantly affect the strand transfer activity (lanes 6–8 and 9–11). The (60–80) peptide strongly inhibited both homologous and heterologous strand transfer activity without significant specific changes in the homologous integration (lanes 12–14). Interestingly, the stimulating effect of (52–96)Vpr was restricted to a narrow concentration range. Increasing the concentration of (52–96)Vpr above 2 µM led to the inhibition of the whole reaction, showing that the stimulating effect only occurs for a given ratio of reaction components.
Figure 4.

Effects of the overlapping C-terminal peptides of Vpr on mini-viral DNA strand transfer activity. Reactions were performed by incubation for 1 h at 37°C of 2 pmol IN, 10 ng of U3U5 substrate and 40 ng of pSP70 target in the presence of increasing concentrations of Vpr peptides. Concentrations of 0.8, 1.6, 3.2 µM of (52–96) were used in lanes 3–5, of (52–70) in lanes 6–8, of (70–96) in lanes 9–11 and of (60–80) in lanes 12–14, without Vpr peptides in lane 2 or without IN in lane 1. Components were pre-incubated together for 10 min on ice before incubation at 37°C. (A) Phosphorimage of heterologous strand transfer; bands located above the main products correspond to multiple integration events. (B) Phosphorimage of homologous strand transfer products.
Stimulation by the (52–96)Vpr requires a viral extremity
Unlike the oligonucleotide assay, the use of long DNA fragments implicated the presence of two structurally different DNA molecules. The 492 bp donor DNA is a linear fragment containing LTR-derived sequences at both extremities, whereas the acceptor DNA is a circular DNA devoid of viral sequences. We considered the possibility that the specificity of homologous integration was due to these differences between donor and acceptor DNA. To address this issue, the experiment was repeated with various heterologous DNAs. First, a linear instead of circular pSP70 was used as the unspecific acceptor DNA. Homologous integration events again were stimulated specifically in the presence of 0.8 µM (52–96)Vpr (Fig. 5B). This result indicates that the specificity does not originate from a preference of IN for linear substrates. Secondly, we examined the possibility that IN and the viral sequence specifically interact in the context of the circular DNA. The pSP70 target DNA was substituted with pU3U5 DNA containing the 492 bp mini-viral fragment. Again, the homologous integration was stimulated specifically in the presence of (52–96) peptide (Fig. 5A). Taken together, these data demonstrated that stimulation occurred exclusively in the context of strand transfer between linear DNAs bearing LTR-derived sequences at their extremities.
Figure 5.
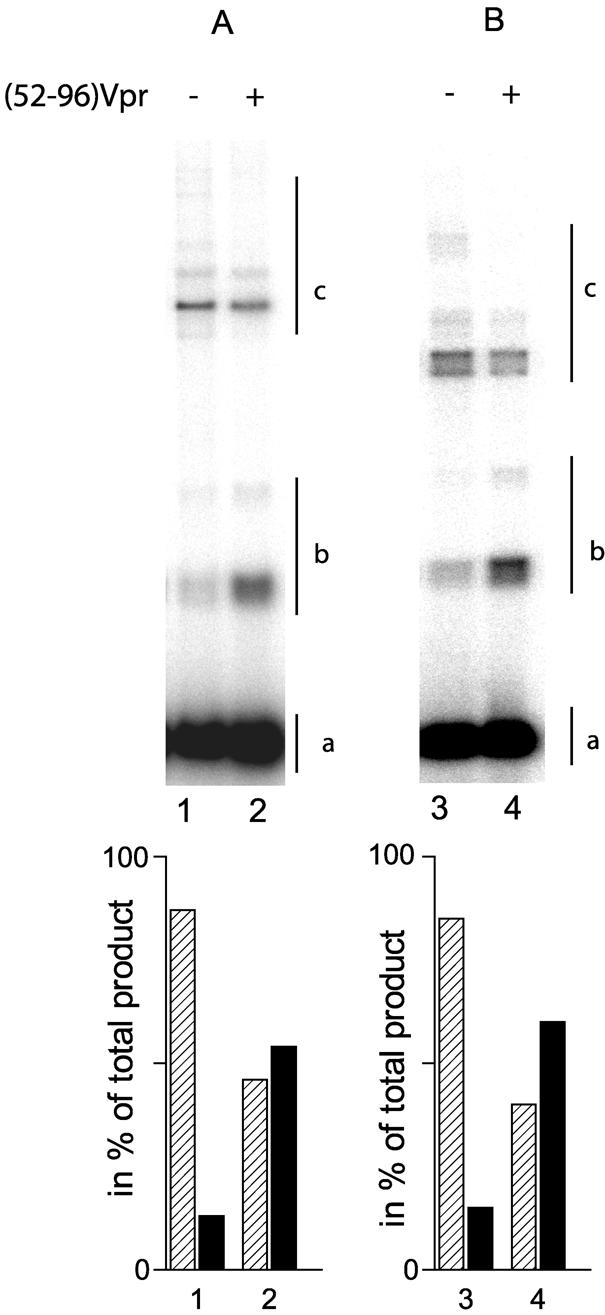
Effect of (52–96) peptide on mini-viral DNA strand transfer using a linear target. The reaction was performed as described in Figure 3, except that the circular pSP70 plasmid was exchanged with either the circular pU3U5 plasmid DNA (A) or with a linear 2500 bp target resulting from the digestion of the pU3U5 plasmid with NdeI (B). The assay was carried out in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of 0.8 µM of (52–96)Vpr. The nature of the integration product is reported on the right: 32P-labeled U3U5 substrate (a), homologous integration products (b) and heterologous integration products (c).
Stimulation of homologous strand transfer does not require commitment of IN on the mini-viral DNA
There are two hypotheses to explain this effect. First, the Vpr bound to DNA may compete for non-specific IN-binding sites but not for LTR-specific IN-binding sites, thereby impairing the binding of IN to the acceptor DNA. Alternatively, the simultaneous binding of Vpr and IN to the mini-viral donor DNA may stimulate the formation of a complex that can undergo integration. To discriminate between these two models, we tested whether the pre-incubation of (52–96)Vpr and IN with DNA would modulate the stimulating effect. IN and (52–96)Vpr were pre-incubated with both DNA molecules in a number of different conditions (Fig. 6). First, IN, the mini-viral DNA and the target DNA simultaneously were mixed and pre-incubated together. In the absence of the (52–96)Vpr, strand transfer into the plasmid DNA was the main result, as expected given the excess of plasmid DNA over the mini-viral fragment which would favor the commitment of IN to the plasmid DNA (Fig. 6, lane 1). The addition of (52–96)Vpr stimulated the homologous strand transfer products (Fig. 6, lane 2). IN was pre-incubated with the plasmid DNA. In the absence of (52–96)Vpr, the whole reaction yield decreased slightly, indicating that IN was trapped by the unspecific DNA upon the addition of the mini-viral DNA. Nevertheless, the presence of (52–96)Vpr shifted the integration pattern from heterologous to homologous integration (lanes 3 and 4). Thirdly, IN and the donor DNA were pre-incubated together (lanes 5 and 6). Both the overall activity yield and the homologous to heterologous ratio increased (52% of total integration product) (compare lane 5 with lanes 1 and 3), indicating the early commitment of IN onto the mini-viral DNA. In these conditions, the addition of (52–96)Vpr stimulated homologous integration which reached 85% of the overall yield even though the increase factor was only 1.5 (lane 6). In conclusion, the stimulatory effect of the (52–96)Vpr peptide does not depend upon the order in which the components are added. Finally, IN was incubated with the mini-viral DNA whilst (52–96) was incubated with the plasmid DNA. Both protein–DNA complexes were mixed and the reaction was started immediately (lane 7). In these conditions, the proportion of homologous integration products was comparable with that obtained in the absence of (52–96)Vpr. Thus, no stimulation was observed when Vpr was pre-bound to the plasmid DNA. This result rules out the possibility that the stimulatory effect was due to the displacement of IN from the plasmid DNA. Altogether, these data suggest that Vpr stimulates the formation of a competent complex on the mini-viral DNA.
Figure 6.

Effect of the pre-incubation of the partners on the (52–96)Vpr stimulation of the long DNA strand transfer. Reactions were performed with 2 pmol IN, 10 ng of U3U5 substrate and 40 ng of pSP70 target in the presence (lanes 2, 4, 6 and 7) or absence (lanes 1, 3 and 5) of (52–96)Vpr. Either all the partners are pre-incubated for 10 min on ice before 1 h of incubation at 37°C (lanes 1 and 2), U3U5 DNA donor is omitted from the pre-incubation (lanes 3 and 4) or the pSP70 target is omitted from the pre-incubation (lanes 5 and 6). Finally, IN and U3U5 on one side and (52–96)Vpr with pSP70 on the other side were pre-incubated for 10 min on ice before being mixed together (lane 7). The nature of the integration product is reported on the right: 32P-labeled U3U5 substrate (a), homologous integration products (b) and heterologous integration products (c).
(52–96) and IN bind synergistically to DNA
To obtain new insights into the possibility that the binding of DNA stimulates the IN on the DNA, we performed an electrophoretic mobility gel shift assay of the mini-viral DNA in the presence of the non-specific target DNA. As previously shown, the interaction of long DNA with either IN or (52–96)Vpr led to the formation of large complexes that eventually formed aggregates which could be recovered at the top of an agarose gel (30,34). In the presence of increasing concentrations of IN, no shift of the mini-viral DNA was observed, indicating that no stable IN–DNA complex could be recovered for concentrations up to 0.6 µM (Fig. 7, lanes 1–5). Conversely, increasing concentrations of (52–96)Vpr led to the formation of a complex that could be recovered at the top of a gel (Fig. 7, lanes 6–10). No discrete shifted bands were detected, indicating the presence of large insoluble complexes (30).
Figure 7.

Electrophoretic mobility gel shift assay with IN and (52–96) peptide. Incubations were performed for 30 min on ice with 10 ng of radiolabeled U3U5 substrate and 40 ng of pSP70 target in a buffer containing 20 mM HEPES pH 7.0, 2 mM DTT and 10 mM MnCl2. Gel shift assay with increasing concentrations of IN: 0.1, 0.2, 0.4 and 0.6 µM (lanes 2–5). Lane 1, no IN (left panel). Gel shift assay with increasing concentrations of (52–96)Vpr: 0.2, 0.4, 0.8 and 1.6 µM (lanes 7–10). Lane 6, no (52–96)Vpr (center panel). Gel shift assay with increasing concentrations of (52–96)Vpr in the presence of IN at the concentration of 0.2 µM (right panel). The concentrations of (52–96)Vpr were identical to those in the center panel. Asterisks indicate discrete, stable complexes which were observed only in the concomitant presence of IN and (52–96)Vpr.
The experiment was carried out in the presence of 0.2 µM IN and increasing concentrations of (52–96)Vpr. DNA was retained even at the lowest concentration of (52–96)Vpr (Fig. 7, lane 12). For higher concentrations of the peptide, the amount of complex was much higher than previously observed in the absence of IN at equivalent concentrations (compare lanes 3, 8 and 13). The quantity of complex recovered at the top of the gel was dose dependent (lanes 12–14) and, at the highest concentration, all the DNA molecules were trapped within protein–DNA complexes. This result indicated a cooperative binding of IN and (52–96)Vpr to the mini-viral DNA. Moreover, discrete shifted bands were observed, suggesting that stable complexes were formed between IN, (52–96)Vpr and the mini-viral DNA.
DISCUSSION
In this study, we attempted to characterize the effect of Vpr on the integration process in vitro. When a unique 21mer oligonucleotide was used as a DNA donor and acceptor, micromolar quantities of both Vpr and its C-terminal DNA-binding domain, (52–96)Vpr, inhibited the reaction. Steady-state fluorescence anisotropy showed that both full-length and (52–96)Vpr bind to short ODNs with comparable affinities, thereby confirming previous observations with long nucleic acids (29,30). Binding of both Vpr and (52–96)Vpr to the short substrate was positively cooperative and independent of the sequence. It is therefore likely that the peptide–ODN complexes encompass the entire oligonucleotide sequence. Moreover, the concentrations of the two proteins required to inhibit IN activities were consistent with their affinities for the short ODN containing the IN cognate binding site. Altogether, these results suggest that Vpr and (52–96)Vpr impair the interaction of the enzyme with its short DNA substrate. The use of a 492 bp mini-viral DNA substrate confirmed that Vpr can inhibit the reaction. However, the activity of the DNA-binding domain (52–96)Vpr differed notably from when the short U5 substrate was present, as low concentrations of this peptide stimulated the integration reaction. The finding that the short and longer substrates had opposite effects is not unprecedented as the NCp7 protein can stimulate IN activity on long but not on short substrates in vitro (10).
(52–96)Vpr shares common features with DNA-binding proteins that stimulate IN-mediated strand transfer. First, no direct interaction of either Vpr or its subdomains with IN has been reported to date. Secondly, the stimulation effect was maximal for a precise ratio of the reaction components, and increasing the protein concentration led to inhibition. However, unlike other DNA-binding proteins such as NCp7 or HMGY(I), which stimulate homologous and heterologous strand transfer integration indiscriminately, the (52–96) domain specifically stimulated homologous integration resulting from the joining of two donor DNA substrates. This effect has never been reported, although it has been shown that HMG-2 may stimulate the avian sarcoma virus IN-mediated heterologous integration more specifically (5).
Strand transfer experiments performed with IN purified from viral particles have shown that the relative amounts of heterologous and homologous strand transfer products can be affected by the pre-incubation conditions (7). For example, homologous strand transfer stimulation was observed when IN was pre-incubated with the acceptor DNA, thus showing that an early commitment with the donor DNA was responsible for the stimulation of homologous integration. This result suggested that (52–96)Vpr acts similarly by stimulating the specific binding of IN to the mini-viral DNA. An alternative model is that IN is displaced from the plasmid target by the non-specific binding of (52–96)Vpr, thus increasing the percentage of strand transfer events taking place within the mini-viral DNA. However, three lines of evidence support the first hypothesis. (i) Homologous integration was always stimulated and was not dependent upon the order in which the components were added. Furthermore, the pre-formation of a complex between (52–96)Vpr and the plasmid target did not reinforce the stimulation, thus suggesting that the stimulatory effect did not arise from concealing the plasmid DNA from IN. Finally, the homologous integration yield was optimized when (52–96)Vpr was pre-incubated together with IN and the mini-viral DNA. This result indicates that the effect of (52–96) is probably due to the formation of a complex involving these three partners. (ii) The specific displacement of IN from the plasmid target but not from the mini-viral DNA would require Vpr to display a differential affinity between the two DNA molecules. This was not the case as stimulation was not affected by the nature of the target DNA. Moreover, it has been shown previously that Vpr binds to DNA in a non-specific way, thus rendering this hypothesis implausible (29,30). (iii) The DNA gel shift assay demonstrated that the apparent affinity of IN for the mini-viral DNA was enhanced when both proteins were present simultaneously. Moreover, the presence of (52–96)Vpr gave rise to shifted bands within the gel, suggesting the presence of discrete stable complexes. Thus, we propose that (52–96)Vpr stabilizes the complexes formed following the binding of IN to its cognate specific site at the extremities of the mini-viral DNA. This enrichment of IN–viral DNA complexes could lead to an increase of homologous integration activity as previously observed with IN purified from viral particles (7).
The full-length Vpr did not display this activity, suggesting that a functional motif within the (52–96) peptide is unmasked in the absence of the N-terminal moiety of the protein. (52–96)Vpr and Vpr did not behave similarly at low concentrations, as attested by the unique ability of (52–96)Vpr to promote DNA localization to the nuclear compartment of transfected cells (31). This difference may be due to steric hindrance of the C-terminal part of Vpr by its (1–51) sequence (35). Indeed, an internal folding of the protein has been evidenced by NMR spectroscopy. This could inhibit partly the interactions of Vpr with other protein targets, which occur mostly by its N-terminal domain (21,36–38). This folded structure is obviously absent in (52–96)Vpr, accounting for its better efficiency. Recent data suggest that the phosphorylation of the C-terminal part of Vpr is a prerequisite for its viral DNA translocation activity (39). This post-translational modification may be responsible for this local change.
Finally, it is noteworthy that Vpr is a probable component of the PIC. As homologous integration results from the connection of two DNA substrates containing the viral extremities, it is tempting to imagine that Vpr could contribute to the arrangement of the PICs by closing the two DNA extremities of the viral DNA. However, Vpr-defective virions are able to replicate in established cell lines and primary dividing cells, suggesting that Vpr is not necessarily required for proviral DNA integration. Moreover, no specific effect of either Vpr or (52–96)Vpr on integration was reported in vivo. Incidentally, the stimulation of homologous integration, although presumably thwarted by cellular factors such as barrier-to-autointgeration factor (40), is most probably not a relevant physiological activity as this effect would be detrimental for viral replication. Vpr is assumed to play a determinant role along with IN and the MAp24 proteins during the course of the nuclear importation of the PIC in non-dividing cells (41–43). One mechanism stipulates that Vpr increases the affinity for the karyopherin pathway of the nuclear localization signal-containing components of the PIC such as IN (28). Thus, the in vitro stimulation of homologous integration may rather reflect an interaction required for the importation of PIC into the nucleus. As this interaction is mediated by the C-terminal domain, the N-terminal part of Vpr would remain free to interact with the components of the karyopherin pathway. We currently are investigating this type of interaction.
Acknowledgments
ACKNOWLEDGEMENTS
We acknowledge skillful technical support from Françoise Simon. This work was supported by funding from the Agence Nationale de Recherche sur le Sida (ANRS), the Centre National de la Recherche Scientifique (CNRS) and Ensemble Contre le Sida (ECS). J.B. is the recipient of a fellowship from the ANRS.
REFERENCES
- 1.Brown P.O., Bowerman,B., Varmus,H.E. and Bishop,J.M. (1989) Retroviral integration: structure of the initial covalent product and its precursor and a role for the viral IN protein. Proc. Natl Acad. Sci. USA, 86, 2525–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fujiwara T. and Mizuuchi,K. (1988) Retroviral DNA integration: structure of an integration intermediate. Cell, 54, 497–504. [DOI] [PubMed] [Google Scholar]
- 3.Carteau S., Gorelick,R.J. and Bushman,F.D. (1999) Coupled integration of human immunodeficiency virus type 1 cDNA ends by purified integrase in vitro: stimulation by the viral nucleocapsid protein. J. Virol., 73, 6670–6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cherepanov P., Surratt,D., Toelen,J., Pluymers,W., Griffith,J., De Clercq,E. and Debyser,Z. (1999) Activity of recombinant HIV-1 integrase on mini-HIV DNA. Nucleic Acids Res., 27, 2202–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hindmarsh P., Ridky,T., Reeves,R., Andrake,M., Skalka,A.M. and Leis,J. (1999) HMG protein family members stimulate human immunodeficiency virus type 1 and avian sarcoma virus concerted DNA integration in vitro. J. Virol., 73, 2994–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinha S., Pursley,M.H. and Grandgenett,D.P. (2002) Efficient concerted integration by recombinant human immunodeficiency virus type 1 integrase without cellular or viral cofactors. J. Virol., 76, 3105–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodarzi G., Im,G.J., Brackmann,K. and Grandgenett,D. (1995) Concerted integration of retrovirus-like DNA by human immunodeficiency virus type 1 integrase. J. Virol., 69, 6090–6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellison V., Abrams,H., Roe,T., Lifson,J. and Brown,P. (1990) Human immunodeficiency virus integration in a cell-free system. J. Virol., 64, 2711–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansen M.S. and Bushman,F.D. (1997) Human immunodeficiency virus type 2 preintegration complexes: activities in vitro and response to inhibitors. J. Virol., 71, 3351–3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carteau S., Batson,S.C., Poljak,L., Mouscadet,J.F., De,R.H., Darlix,J.L., Roques,B.P., Kas,E. and Auclair,C. (1997) Human immunodeficiency virus type 1 nucleocapsid protein specifically stimulates Mg2+-dependent DNA integration in vitro. J. Virol., 71, 6225–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farnet C.M. and Bushman,F.D. (1997) HIV-1 cDNA integration: requirement of HMG I(Y) protein for function of preintegration complexes in vitro. Cell, 88, 483–492. [DOI] [PubMed] [Google Scholar]
- 12.Hindmarsh P. and Leis,J. (1999) Reconstitution of concerted DNA integration with purified components. Adv. Virus Res., 52, 397–410. [DOI] [PubMed] [Google Scholar]
- 13.Kondo E., Mammano,F., Cohen,E.A. and Gottlinger,H.G. (1995) The p6gag domain of human immunodeficiency virus type 1 is sufficient for the incorporation of Vpr into heterologous viral particles. J. Virol., 69, 2759–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen E.A., Dehni,G., Sodroski,J.G. and Haseltine,W.A. (1990) Human immunodeficiency virus vpr product is a virion-associated regulatory protein. J. Virol., 64, 3097–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heinzinger N.K., Bukinsky,M.I., Haggerty,S.A., Ragland,A.M., Kewalramani,V., Lee,M.A., Gendelman,H.E., Ratner,L., Stevenson,M. and Emerman,M. (1994) The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc. Natl Acad. Sci. USA, 91, 7311–7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Rocquigny H., Petitjean,P., Tanchou,V., Decimo,D., Drouot,L., Delaunay,T., Darlix,J.L. and Roques,B.P. (1997) The zinc fingers of HIV nucleocapsid protein NCp7 direct interactions with the viral regulatory protein Vpr. J. Biol. Chem., 272, 30753–30759. [DOI] [PubMed] [Google Scholar]
- 17.Tung H.Y., De,R.H., Zhao,L.J., Cayla,X., Roques,B.P. and Ozon,R. (1997) Direct activation of protein phosphatase-2A0 by HIV-1 encoded protein complex NCp7:vpr. FEBS Lett., 401, 197–201. [DOI] [PubMed] [Google Scholar]
- 18.Gallay P., Swingler,S., Song,J., Bushman,F. and Trono,D. (1995) HIV nuclear import is governed by the phosphotyrosine-mediated binding of matrix to the core domain of integrase. Cell, 83, 569–576. [DOI] [PubMed] [Google Scholar]
- 19.Cohen E.A., Terwilliger,E.F., Jalinoos,Y., Proulx,J., Sodroski,J.G. and Haseltine,W.A. (1990) Identification of HIV-1 vpr product and function. J. AIDS, 3, 11–18. [PubMed] [Google Scholar]
- 20.Wang L., Mukherjee,S., Jia,F., Narayan,O. and Zhao,L.J. (1995) Interaction of virion protein Vpr of human immunodeficiency virus type 1 with cellular transcription factor Sp1 and trans-activation of viral long terminal repeat. J. Biol. Chem., 270, 25564–25569. [DOI] [PubMed] [Google Scholar]
- 21.Agostini I., Navarro,J.M., Rey,F., BouHamdan,M., Spire,B., Vigne,R. and Sire,J. (1996) The human immunodeficiency virus type 1 Vpr transactivator: cooperation with promoter-bound activator domains and binding to TFIIB. J. Mol. Biol., 261, 599–606. [DOI] [PubMed] [Google Scholar]
- 22.He J., Choe,S., Walker,R., Di Marzio,P., Morgan,D.O. and Landau,N.R. (1995) Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol., 69, 6705–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jowett J.B., Planelles,V., Poon,B., Shah,N.P., Chen,M.L. and Chen,I.S. (1995) The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J. Virol., 69, 6304–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emerman M. (1996) HIV-1, Vpr and the cell cycle. Curr. Biol., 6, 1096–1103. [DOI] [PubMed] [Google Scholar]
- 25.Jacotot E., Ravagnan,L., Loeffler,M., Ferri,K.F., Vieira,H.L., Zamzami,N., Costantini,P., Druillennec,S., Hoebeke,J., Briand,J.P. et al. (2000) The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med., 191, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart S.A., Poon,B., Jowett,J.B. and Chen,I.S. (1997) Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol., 71, 5579–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Depienne C., Roques,P., Creminon,C., Fritsch,L., Casseron,R., Dormont,D., Dargemont,C. and Benichou,S. (2000) Cellular distribution and karyophilic properties of matrix, integrase and Vpr proteins from the human and simian immunodeficiency viruses. Exp. Cell Res., 260, 387–395. [DOI] [PubMed] [Google Scholar]
- 28.Bukrinsky M. and Adzhubei,A. (1999) Viral protein R of HIV-1. Rev. Med. Virol., 9, 39–49. [DOI] [PubMed] [Google Scholar]
- 29.Zhang S., Pointer,D., Singer,G., Feng,Y., Park,K. and Zhao,L.J. (1998) Direct binding to nucleic acids by Vpr of human immunodeficiency virus type 1. Gene, 212, 157–166. [DOI] [PubMed] [Google Scholar]
- 30.De Rocquigny H., Caneparo,A., Delaunay,T., Bischerour,J., Mouscadet,J.F. and Roques,B.P. (2000) Interactions of the C-terminus of viral protein R with nucleic acids are modulated by its N-terminus. Eur. J. Biochem., 267, 3654–3660. [DOI] [PubMed] [Google Scholar]
- 31.Kichler A., Pages,J.C., Leborgne,C., Druillennec,S., Lenoir,C., Coulaud,D., Delain,E., Le Cam,E., Roques,B.P. and Danos,O. (2000) Efficient DNA transfection mediated by the C-terminal domain of human immunodeficiency virus type 1 viral protein R. J. Virol., 74, 5424–5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Cam E., Coulaud,D., Delain,E., Petitjean,P., Roques,B.P., Gerard,D., Stoylova,E., Vuilleumier,C., Stoylov,S.P. and Mely,Y. (1998) Properties and growth mechanism of the ordered aggregation of a model RNA by the HIV-1 nucleocapsid protein: an electron microscopy investigation. Biopolymers, 45, 217–229. [DOI] [PubMed] [Google Scholar]
- 33.Leh H., Brodin,P., Bischerour,J., Deprez,E., Tauc,P., Brochon,J.C., LeCam,E., Coulaud,D., Auclair,C. and Mouscadet,J.F. (2000) Determinants of Mg2+-dependent activities of recombinant human immunodeficiency virus type 1 integrase. Biochemistry, 39, 9285–9294. [DOI] [PubMed] [Google Scholar]
- 34.Brodin P., Pinskaya,M., Buckle,M., Parsch,U., Romanova,E., Engels,J., Gottikh,M. and Mouscadet,J.F. (2002) Disruption of HIV-1 integrase–DNA complexes by short 6-oxocytosine-containing oligonucleotides. Biochemistry, 41, 1529–1538. [DOI] [PubMed] [Google Scholar]
- 35.Wecker K., Morellet,N., Bouaziz,S. and Roques,B.P. (2002) NMR structure of the HIV-1 regulatory protein Vpr in H2O/trifluoroethanol. Comparison with the Vpr N-terminal (1–51) and C-terminal (52–96) domains. Eur. J. Biochem., 269, 3779–3788. [DOI] [PubMed] [Google Scholar]
- 36.Stark L.A. and Hay,R.T. (1998) Human immunodeficiency virus type 1 (HIV-1) viral protein R (Vpr) interacts with Lys-tRNA synthetase: implications for priming of HIV-1 reverse transcription. J. Virol., 72, 3037–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.BouHamdan M., Benichou,S., Rey,F., Navarro,J.M., Agostini,I., Spire,B., Camonis,J., Slupphaug,G., Vigne,R., Benarous,R. et al. (1996) Human immunodeficiency virus type 1 Vpr protein binds to the uracil DNA glycosylase DNA repair enzyme. J. Virol., 70, 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sato A., Yoshimoto,J., Isaka,Y., Miki,S., Suyama,A., Adachi,A., Hayami,M., Fujiwara,T. and Yoshie,O. (1996) Evidence for direct association of Vpr and matrix protein p17 within the HIV-1 virion. Virology, 220, 208–212. [DOI] [PubMed] [Google Scholar]
- 39.Agostini I., Popov,S., Hao,T., Li,J.H., Dubrovsky,L., Chaika,O., Chaika,N., Lewis,R. and Bukrinsky,M. (2002) Phosphorylation of Vpr regulates HIV type 1 nuclear import and macrophage infection. AIDS Res. Hum. Retroviruses, 18, 283–288. [DOI] [PubMed] [Google Scholar]
- 40.Popov S., Rexach,M., Ratner,L., Blobel,G. and Bukrinsky,M. (1998) Viral protein R regulates docking of the HIV-1 preintegration complex to the nuclear pore complex. J. Biol. Chem., 273, 13347–13352. [DOI] [PubMed] [Google Scholar]
- 41.Chen H. and Engelman,A. (1998) The barrier-to-autointegration protein is a host factor for HIV type 1 integration. Proc. Natl Acad. Sci. USA, 95, 15270–15274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Popov S., Rexach,M., Zybarth,G., Reiling,N., Lee,M.A., Ratner,L., Lane,C.M., Moore,M.S., Blobel,G. and Bukrinsky,M. (1998) Viral protein R regulates nuclear import of the HIV-1 pre-integration complex. EMBO J., 17, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vodicka M.A., Koepp,D.M., Silver,P.A. and Emerman,M. (1998) HIV-1 Vpr interacts with the nuclear transport pathway to promote macrophage infection. Genes Dev., 12, 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]