

Abstract
Ras promotes the accumulation of the cyclin-dependent kinase inhibitor p21Waf1/Cip1 (p21). Previous studies reported that acute Raf/MEK/ERK activation elevates p21 protein levels by increased transcription. However, we have found that p21 induction in Ras-transformed murine fibroblasts occurs principally by a post-translational mechanism. Chronic activation of the Raf/MEK/ERK pathway blocked proteasome-mediated p21 degradation, resulting in accumulation of p21 protein with an elevated half-life. The stabilization of p21 by Ras was accompanied by high levels of p21-associated cyclin D1 and, similarly to Ras, cyclin D1 was sufficient to inhibit the proteasome-mediated p21 degradation. Knock-down of cyclin D1 by RNA interference confirmed that Ras-induced p21 stabilization was dependent upon cyclin D1 expression. We show that p21 directly binds to the C8α subunit of the 20S proteasome complex and that by competing for binding, cyclin D1 inhibits p21 degradation by purified 20S complexes in vitro. Therefore, we propose that Ras stabilizes p21 by promoting the formation of p21–cyclin D1 complexes that prevent p21 association with, and subsequent degradation by, the 20S proteasome.
Keywords: cyclin/degradation/p21Waf1/Cip1/proteasome/Ras
Introduction
Ras genes (H-Ras, K-Ras and N-Ras) encode small molecular weight membrane-localized proteins that are often mutated in a variety of tumours, most frequently in carcinomas of the pancreas, colon and thyroid (Bos, 1989). These mutations inhibit GTP hydrolysis, making Ras constitutively GTP bound and consequently active. Ras proteins signal through effector pathways including the Raf/MEK/ERK mitogen-activated protein kinase cascade (Raf/MAPK), RalGDS and phosphatidylinositol 3-kinase (PI3K), to promote cellular transformation (Vojtek and Der, 1998).
Ras contributes to oncogenic transformation by influencing the cell cycle machinery. Ras-regulated pathways play integral roles in the induction of cyclin D1, a key event required for G1 phase progression (Kerkhoff and Rapp, 1998). In addition, the assembly of newly synthesized cyclin D1 with cyclin-dependent kinase (cdk) partners cdk4 and cdk6 requires Ras activation of the Raf/MAPK pathway (Cheng et al., 1998). Efficient assembly of these complexes requires the cdk ‘inhibitors’ of the Cip/Kip family (Cheng et al., 1999). Whereas p21Waf1/Cip1 (p21) and p27Kip1 (p27) inhibit the activity of other cyclin–cdk complexes, at low concentration they act positively on cyclin D–cdk complexes to promote assembly, stability and nuclear localization (LaBaer et al., 1997; Cheng et al., 1999; Alt et al., 2002). Consistent with a positive effect on cell cycle progression, p21 expression is transiently elevated in an ERK-dependent manner when entering the cell cycle after growth factor stimulation (Liu et al., 1996; Bottazzi et al., 1999).
In some circumstances, Ras signalling through the Raf/MAPK pathway elevates p21 to levels that arrest cell cycle progression. Introduction of activated Ras into primary rodent and human cells induces high p21 levels that impose a proliferative block associated with G1 arrest (Lloyd et al., 1997; Serrano et al., 1997; Olson et al., 1998). Studies using conditionally active Raf kinase indicate that high intensity ERK signalling also leads to p21-mediated growth arrest in rodent fibroblasts (Sewing et al., 1997; Woods et al., 1997) and human tumour cells (Ravi et al., 1999).
Ras-induced elevation of p21 has been attributed to transcriptional activation, which occurs by p53-dependent (Lloyd et al., 1997; Serrano et al., 1997) or -independent mechanisms (Sewing et al., 1997; Woods et al., 1997) depending on cell type. Although p21 transcription has been well studied (Gartel and Tyner, 1999), regulation of p21 levels by post-transcriptional mechanisms are less well defined. p21 is a short-lived protein that is degraded by the large multisubunit proteasome complex (Blagosklonny et al., 1996; Cayrol and Ducommun, 1998; Sheaff et al., 2000; Touitou et al., 2001). The 26S proteasome is composed of a barrel-shaped 20S catalytic chamber, ‘capped’ at each end by 19S complexes (Hershko and Ciechanover, 1998). Most proteins degraded by the proteasome require ubiquitin conjugation for targeting via the association of ubiquitin with the 19S lid (Hershko and Ciechanover, 1998). However, p21 is a novel ubiquitylation-independent proteasome target (Sheaff et al., 2000) that by-passes the need for ubiquitylation by binding directly to the C8α subunit of the 20S proteasome catalytic complex (Touitou et al., 2001).
Given that protein degradation plays a critical role in cell cycle control (reviewed in Yew, 2001), we wished to determine whether Ras contributed to elevated p21 levels by modulating protein stability in addition to promoting p21 transcription. We show that Ras-transformed fibroblasts have elevated p21 due to the post-transcriptional inhibition of proteasome-mediated degradation. Ras-induced stabilization of p21 protein was dependent on signalling through the Raf/MAPK pathway and required cyclin D1 expression. We show that cyclin D1 associates with p21 and prevents its degradation by the 20S proteasome in vitro. These data define a novel post-translational mechanism by which Ras regulates a regulatory component of the cell cycle machinery.
Results
Elevated p21 levels in Ras-transformed fibroblasts are not affected by proteasome inhibition
Under conditions in which p21 is degraded rapidly, p21 protein levels rise after treatment with proteasome inhibitors such as the Streptomyces lactacystinaeus metabolite lactacystin (LC) (Blagosklonny et al., 1996; Cayrol and Ducommun, 1998; Sheaff et al., 2000; Touitou et al., 2001). We wished to determine whether proteasome inhibition would elevate p21 levels further in Ras-transformed cells. Wild-type (S3T3) and V12 H-Ras- transformed Swiss 3T3 (Ras-S3T3) mouse fibroblasts (Leevers and Marshall, 1992) were serum starved either with or without LC treatment to determine the effect of proteasome inhibition on p21 levels (Figure 1A). p21 increased 8-fold after LC treatment in parental S3T3 cells; however, p21 levels were as high in untreated Ras-S3T3 cells as in LC-treated parental cells and there was no further elevation after proteasome inhibition. The lack of effect on p21 was not due to inefficient proteasome blockade by LC in Ras-S3T3 cells since the accumulation and appearance of high molecular weight ubiquitylated forms of another proteasome target, β-catenin, were apparent (Figure 1A). Consistent with previous reports, p21 ubiquitylation was not evident under any of the test conditions (Sheaff et al., 2000) (Figure 1A; data not shown). The observation that LC did not influence p21 levels in Ras-S3T3 cells suggested that the rate of p21 degradation by the proteasome was significantly reduced, consistent with a role for Ras in promoting p21 protein stability.
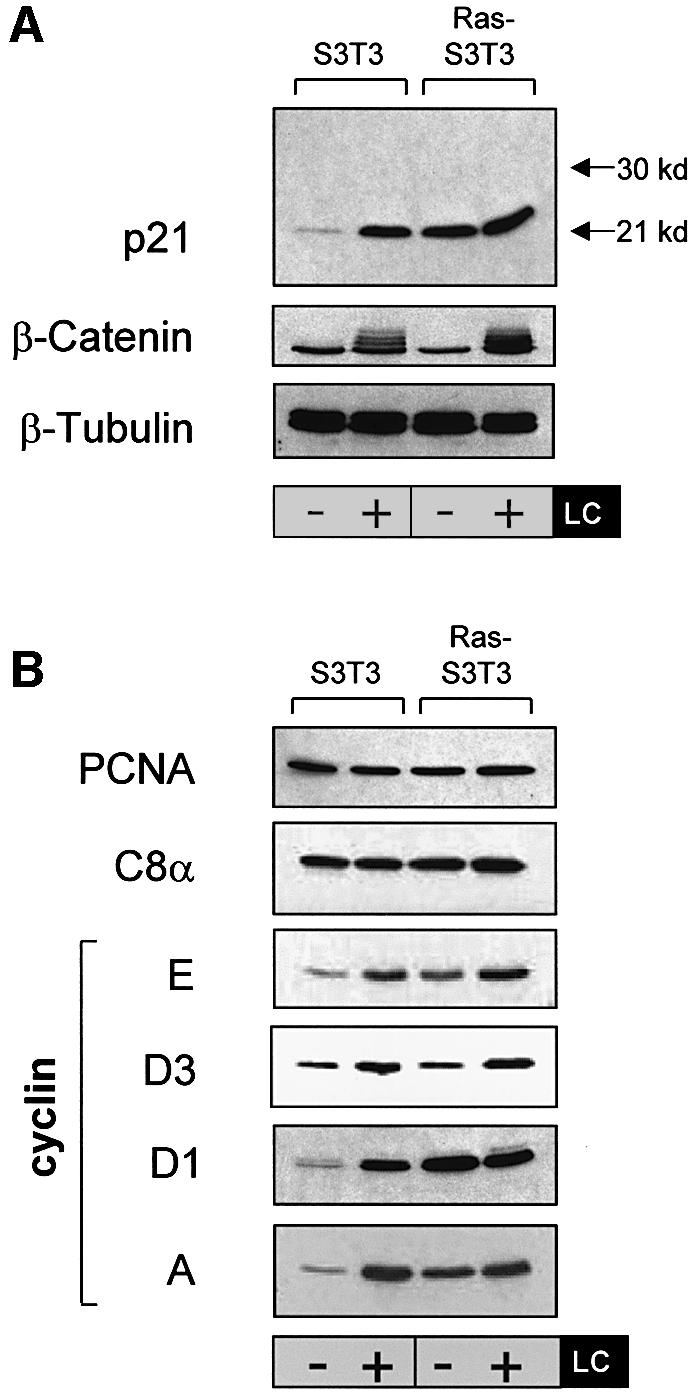

Fig. 1. Ras-induced p21 levels are uncoupled from the effects of proteasome inhibition and are elevated by a post-transcriptional mechanism. (A) Elevated p21 levels in Ras-transformed fibroblasts are not affected by proteasome inhibition. Parental (S3T3) and Ras-transformed S3T3 (Ras-S3T3) cells were serum starved for 16 h in the presence or absence of 10 µM lactacystin (LC) proteasome inhibitor. Extracts were western blotted as indicated. β-tubulin controlled for loading. (B) A subset of p21-binding partners are regulated by Ras in a manner similar to p21. Membranes shown in (A) were stripped and reprobed as indicated. (C) p21 mRNA is not elevated in Ras-S3T3 cells. Total RNA was isolated from serum-starved parental and Ras-S3T3 cells before northern blot analysis for p21 and cyclin D1. Expression levels were normalized to GAPDH levels and are presented as levels relative to the parental cell line.
Since protein–protein interactions may modulate p21 stability (Timchenko et al., 1997; Cayrol and Ducommun, 1998), we immunoblotted the samples from Figure 1A to determine how p21-binding protein levels were influenced by Ras and LC (Figure 1B). Although proliferating cell nuclear antigen (PCNA) has been reported to influence p21 protein stability (Cayrol and Ducommun, 1998; Touitou et al., 2001), we found no difference in levels between parental and Ras-S3T3 cells. Similarly, we detected little difference in cyclin D2, D3 and E levels or in cdk4, cdk6 or C8α proteasomal subunit levels (Figure 1B; data not shown). However, cyclins D1 and A levels paralleled p21, with significant elevation in Ras-S3T3 cells that were uncoupled from the effects of proteasome inhibition. These data are consistent with the possibility that Ras promotes p21 protein stability by increasing the pool of cyclins D1 and A available for p21 binding.
We analysed p21 mRNA by northern blotting to determine whether message accumulation contributed to elevated levels in Ras-S3T3 cells. Total RNA was isolated from serum-starved parental and Ras-S3T3 cells and blotted with p21, cyclin D1 or GAPDH probes as described in Materials and methods. Transcript levels were quantified, corrected for GAPDH loading, and are shown relative to untreated S3T3 cells (Figure 1C). In contrast to the large differences in protein levels shown in Figure 1A, steady-state p21 transcript levels were only 1.4-fold higher in Ras-S3T3 cells than in parental cells. In contrast, cyclin D1 mRNA was >6-fold higher in Ras-S3T3 cells (Figure 1C). Therefore, p21 levels are elevated in Ras-transformed cells primarily by inhibition of proteasome-mediated degradation rather than mRNA accumulation.
The Raf/MAPK pathway mediates Ras-induced post-transcriptional p21 regulation
Since p21 levels have been reported to be influenced by both the Raf/MAPK and PI3K pathways (Li et al., 2001; Rossig et al., 2002), we wished to determine whether these Ras effector pathways mediated the Ras-induced decrease in p21 degradation. The Raf/MAPK pathway was blocked with the MEK inhibitor UO126 in the presence or absence of LC for 16 h. Proteasome inhibition alone had no effect on p21 levels; however, UO126 treatment reduced p21 levels (Figure 2A, compare lanes 1 and 3) and re-established sensitivity to proteasome inhibition; LC treatment increased p21 levels in UO126-treated cells (compare lanes 3 and 4). UO126 also downregulated cyclins D1 and A and restored their responsiveness to LC (Figure 2A), but did not affect PCNA levels (data not shown). The drug vehicle dimethylsulfoxide (DMSO) did not affect any of the proteins examined. In contrast to MEK inhibition, PI3K inhibition with LY294002 did not alter p21, cyclin D1 or cyclin A levels in Ras-S3T3 cells (see Supplementary figure S1 available at The EMBO Journal Online).

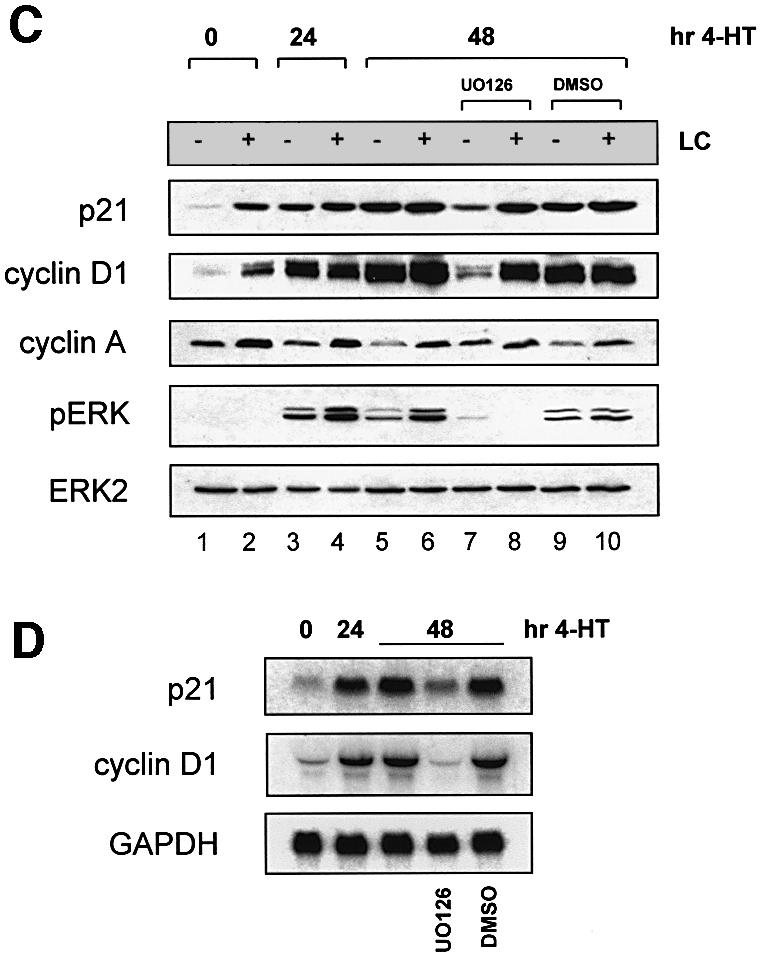
Fig. 2. The Raf/MAPK effector pathway mediates Ras-induced post-transcriptional p21 regulation. (A) Inhibition of MEK reduces Ras-induced p21 levels and restores sensitivity to LC. Ras-S3T3 cells were incubated with 10 µM MEK inhibitor UO126 or 0.1% DMSO vehicle control and LC for 16 h. Extracts were western blotted as indicated. ERK2 controlled for loading. (B) p21 mRNA levels in Ras-S3T3 cells are not affected by UO126. Total RNA was isolated from Ras-S3T3 cells treated with UO126 or DMSO vehicle control for northern blot analysis of p21 and cyclin D1. Expression levels were normalized to GAPDH and are presented as levels relative to the parental cell line. (C) Raf activation is sufficient to uncouple p21 expression from the effects of proteasome inhibition. Raf:ER S3T3 cells treated with 100 nM 4-HT for 24–48 h were incubated with UO126 and/or LC in the absence of serum for 16 h. Extracts were western blotted as indicated. (D) Acute Raf activation elevates p21 and cyclin D1 mRNA. Total RNA was isolated from Raf:ER S3T3 cells treated as indicated and northern blotted for p21, cyclin D1 and GAPDH.
Although U0126 reduced Ras-induction of p21 (Figure 2A), it did not affect p21 mRNA levels in Ras-S3T3 cells (Figure 2B), consistent with the northern blot data presented in Figure 1C. However, UO126 dramatically reduced cyclin D1 mRNA, consistent with previous reports linking Ras/Raf/MAPK signalling to the induction of cyclin D1 transcription (Kerkhoff and Rapp, 1998). These data confirm that Ras induction of p21 results largely from a post-transcriptional mechanism, and support the hypothesis that cyclins A and/or D1, but not PCNA, mediate Ras-induced p21 protein stabilization.
Raf activation is sufficient to uncouple p21 expression from the effects of proteasome inhibition
To determine whether Raf/MAPK signalling is sufficient to uncouple p21 from proteasome-mediated degradation, we used S3T3 cells expressing a conditionally active version of Raf-1 (Woods et al., 1997; Sahai et al., 2001). These cells (Raf:ER S3T3) express the Raf-1 kinase domain fused to a mutated oestrogen-binding domain of the human receptor, making the kinase responsive to the oestrogen analogue 4-hydroxytamoxifen (4-HT). Addition of 4-HT induced Raf activity, resulting in ERK phosphorylation and elevated p21 protein and mRNA (Figure 2C and D). Quantification of cells undergoing DNA synthesis with or without 4-HT indicated that Raf-induced p21 levels (Figure 2C, compare lanes 1 and 3) were associated with a proliferative block (Supplementary figure S2). Raf activation by 4-HT for 24 or 48 h was sufficient to uncouple p21 from the influence of LC (compare lanes 1 and 2 with lanes 3 and 4, and 5 and 6). Inhibition of MEK with UO126 significantly reduced p21 induction in cells stimulated with 4-HT for 48 h and re-sensitized p21 to LC (compare lanes 5 and 6 with lanes 7 and 8), similar to the ability of U0126 to re-establish LC responsiveness in Ras-S3T3 cells (Figure 2A). The drug vehicle DMSO had no effect on any protein examined. In addition, cyclin D1 and p21 were influenced by Raf activation and LC in parallel, similar to the matching effects on cyclin D1 and p21 of Ras and LC in S3T3 cells (Figure 1A and C). In contrast to cyclin D1, the levels of cyclin A dropped following Raf activation and were partially restored by UO126 treatment (compare lanes 5 and 7). Since cyclin A is induced upon S-phase entry and not directly by intracellular signalling pathways, the reduction in cyclin A probably results from the growth arrest induced by Raf:ER activation. In addition, the reciprocal patterns of cyclin A and p21 regulation by Raf activation indicate that cyclin A is not a major factor involved in mediating Ras/Raf-induced p21 stability.
Stabilization of p21 by the Raf/MAPK pathway and by cyclin D1
To determine whether the differential effects of proteasome inhibition by LC on p21 levels reflect quantitative differences in the rate of p21 turnover in parental versus Ras-S3T3 cells, we examined p21 decay in S3T3 and Ras-S3T3 cells by inhibiting protein synthesis with cycloheximide (CHX). Serum-starved cells were treated with 25 µg/ml CHX for the times indicated and p21 levels assessed by western blotting (Figure 3A). Ras-S3T3 cells expressed elevated p21 levels that did not decline significantly over the period of CHX treatment; 70% of the p21 remained 2 h after protein synthesis was blocked (Figure 3B). In contrast, parental S3T3 cells had lower p21 levels that rapidly decayed following CHX treatment. Densitometric analysis revealed that p21 half-life was extended from 1 h in S3T3 cells to >3 h in Ras-transformed S3T3 cells (Figure 3B).

Fig. 3. Analyses of p21 protein stability. (A) p21 turnover is reduced in Ras-S3T3 versus parental S3T3 cells. S3T3 cells were treated with 25 µg/ml CHX to inhibit new protein synthesis for the times indicated, and extracts probed for p21 and β-tubulin to control for loading. A longer p21 exposure is shown for parental S3T3 cells to allow comparison. (B) p21 expression quantified by densitometric analysis. Expression is represented as the percentage remaining relative to time zero. Half-life values were calculated using lines of best fit. (C) Co-expression of Ras or cyclin D1 is sufficient to stabilize p21. Pulse–chase analysis was undertaken in NIH-3T3 cells transfected with epitope-tagged p21, H-Ras D12 and/or cyclin D1. Cells were treated with 10 µM UO126 (UO), 20 µM LY294002 (LY), 10 µM lactacystin (LC) or vehicle controls where indicated. Shown is the percentage of 35S-labelled p21 remaining after 2 h of chase, relative to a control sample lysed at time zero.
In addition to the approach described above, we used a pulse–chase method to study the effect of co-expressed proteins on p21 stability in NIH-3T3 fibroblasts transfected with Flag epitope-tagged p21. NIH-3T3 cells were transfected, serum starved overnight and then labelled with [35S]methionine/cysteine (Met/Cys) and chased with an excess of cold Met/Cys as detailed in Materials and methods. The half-life of Flag-p21 was <2 h under these conditions (Supplementary figure S3). For comparison purposes, a 2 h time point post-chase was used subsequently (Figure 3C). Co-expression of activated Ras significantly increased the proportion of 35S-labelled p21 remaining 2 h post-chase (Figure 3C), consistent with the Ras stabilization data presented in Figure 3A. Similar to the data presented in Figure 2, inhibition of MEK activity with UO126 blocked Ras-induced p21 stabilization. In contrast, incubation with LY294002 only slightly impaired the ability of Ras to reduce p21 turnover.
As there was a coordinated pattern of p21 and cyclin D1 regulation by Ras and the Raf/MAPK pathway, and since p21-binding partners influence p21 degradation, we examined whether cyclin D1 would recapitulate the Ras effect on p21 stability. Figure 3C shows that ectopic cyclin D1 expression increased the amount of p21 remaining after 2 h to 77%, even more than did Ras (70%), and approached the maximal stabilization achieved by inhibition of proteasome activity with LC (90%). Therefore, like Ras, elevated expression of cyclin D1 stabilized p21 protein, consistent with the hypothesis that cyclin D1 may be the Ras target that blocks the proteasomal p21 degradation.
If Ras-induced p21 stabilization results from increased p21–cyclin D1 interactions, then the proportion of p21 bound to cyclin D1 should increase in cells with elevated Ras or Raf signalling. Immunoprecipitation of p21 from cells with active Ras or Raf followed by quantitative analysis of associated cyclin D1 revealed that Ras and Raf increase the ratio of p21 associated with cyclin D1 ∼5-fold (Figure 4A), consistent with Ras-induced cyclin D1 forming complexes that protect p21 from proteasome-mediated degradation.

Fig. 4. Ras-induced cyclin D1 binds and stabilizes p21. (A) Ras and Raf signalling increase the proportion of p21 associated with cyclin D1. p21 was precipitated from S3T3. RAS-S3T3 or Raf:ER cells treated with or without 4-HT for 24 hours. As a control, Ras-S3T3 lysates were incubated with the M2 anti-flag antibody (*). Immunoprecipitated proteins were quantified using the relevant primary antibodies and a Cy5-coupled secondary IgG as described in Materials and methods. Shown below are the amounts of co-purified p21 and cyclin D1 relative to the control cell line (average of three experiments). (B) Cyclin D1 knock-down in Ras-S3T3 cells results in decreased p21. Lysates from parental Ras-S3T3 cells, two independent pSuper control clones and two independent cyclin D1 knock-down clones (pSuper D1 RNAi) were western blotted for β-tubulin, cyclin D1 and p21. (C) Knock-down of cyclin D1 restores p21 sensitivity to LC. One empty vector and one cyclin D1 knock-down clone were serum starved with or without LC treatment for 16 h as indicated, and lysates were blotted for cyclin D1 or p21.
Cyclin D1 is required for Ras to uncouple p21 from proteasomal degradation
To determine whether Ras-induced cyclin D1 expression was required to maintain elevated p21 levels in Ras-S3T3 cells, we knocked-down cyclin D1 protein levels by RNA interference (RNAi) using the stable expression of short hairpin RNAs. Two targeting sequences in the cyclin D1-coding sequence were chosen to construct pSuper RNAi vectors (Brummelkamp et al., 2002). Both D1 pSuper knock-down vectors were transfected into Ras-S3T3 cells along with a puromycin resistance gene, and stable clones were selected. As a control, the empty pSuper vector was transfected along with the puromycin resistance gene into Ras-S3T3 cells. Figure 4B shows that the levels of cyclin D1 and p21 were comparable in parental Ras-S3T3 cells and two empty pSuper vector control clones. However, Ras-S3T3 clones expressing pSuper D1 RNAi had drastically decreased levels of cyclin D1 without accompanying reductions in active ERK (data not shown). Significantly, p21 levels were also reduced in the D1 knock-down clones (Figure 4B). Consistent with the proposal that cyclin D1 protects p21 from proteasomal degradation, knock-down of cyclin D1 restored proteasomal degradation of p21 in Ras-S3T3 cells since the reduced p21 levels were rendered responsive to LC treatment (Figure 4C), as we previously observed in untransformed S3T3 cells (Figure 1A). These data indicate that cyclin D1 is a critical regulator of p21 proteasomal degradation in Ras-S3T3 cells.
Cyclin D1 binds a C-terminal cyclin-binding site of p21
Ubiquitin-independent p21 degradation is mediated by the binding of a C-terminal degradation sequence to the C8α subunit of the 20S proteasome (Touitou et al., 2001). A previously characterized cyclin-binding site (Adams et al., 1996; Chen et al., 1996) is located within the minimal C8α association domain of p21 (residues 140–164) (Touitou et al., 2001). This cyclin-binding motif (amino acids 152–158) was named Cy2, as it followed the identification of the first N-terminal site (Chen et al., 1996). The Cy2 site binds cyclin A, D1 and E and their corresponding cdk complexes with lower affinity than the N-terminal site (Adams et al., 1996; Chen et al., 1996; Ball et al., 1997). One possibility, therefore, is that elevated cyclin D1 levels induced by Ras promote p21 stability by competing with the C8α subunit for binding to p21, resulting in decreased proteasome degradation. To determine whether cyclin D1 from Ras-S3T3 cells associates with the C-terminus of p21, we incubated GST fusion proteins of p27, p21, a p21 mutant lacking the N-terminal cyclin site (ΔNp21; amino acids 86–164) or a GST control with lysates from Ras-S3T3 cells, then captured protein complexes with glutathione–Sepharose beads and analysed them by western blotting (Figure 5A). Full-length p21 and p27 captured cyclins D1 and A, while ΔNp21 associated only with cyclin D1. Cdk4 (Figure 5A) and Cdk6 (data not shown) were also associated with p21 and p27, but significantly less with ΔNp21, suggesting that cyclin D1 binding to Cy2 may not require a cyclin–cdk complex under these conditions, in agreement with previous reports (Adams et al., 1996; Ball et al., 1997). Consistent with ΔNp21 containing a single Cy site and the sole PCNA-binding site, less cyclin D1 but equal PCNA were associated with the C-terminus compared with full-length p21. Selectivity was validated by the lack of GST binding to any proteins and the lack of p27 binding to PCNA. These observations indicate that cyclin D1, but not cyclin A, can associate with the p21 C-terminus.
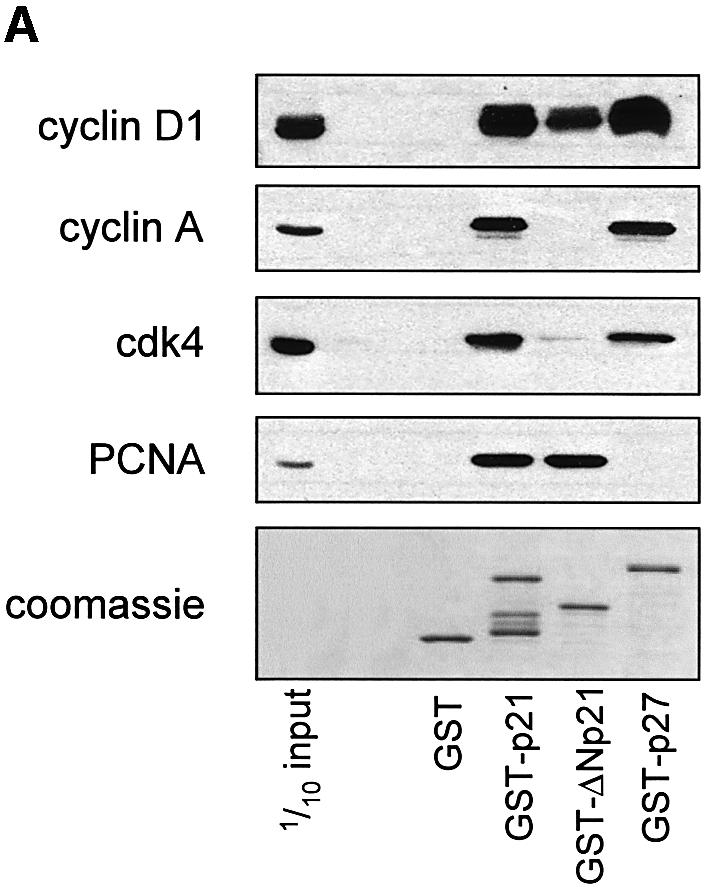

Fig. 5. Cyclin D1 binds to the p21 C-terminus and blocks the p21–C8α interaction. (A) Cyclin D1, but not cyclin A, binds to the Cy2 site of GST–p21. Ras-S3T3 extracts were incubated with the GST fusion proteins indicated, and captured complexes were western blotted for the p21-binding partners shown. A p21 mutant lacking the N-terminus was used (GST–ΔNp21) to determine which cyclins bind the C-terminal Cy2 site. (B) Association of p21 with C8α in vitro. IVTT-synthesized 35S-labelled p21 or luciferase control (L) were added to cyclin D1, C8α or Ral-His fusion proteins. Complexes were separated by SDS–PAGE, and His fusion-associated 35S-labelled proteins were detected by exposing membranes to autoradiography film. (C) Cyclin D1 competes with C8α for p21 binding. The assay described in (B) was repeated in the presence of 0, 2, 5 or 10 µg of cyclin D1, K-cyclin or GST–PCD fusions. Equal amounts of the different GST fusion proteins were added, as judged by western blot of one-tenth of the input.
Cyclin D1 competes with proteasome subunit C8α for p21 binding in vitro
The results above are consistent with the hypothesis that Ras-induced p21 stabilization is mediated by cyclin D1 binding to p21, thereby blocking interactions with the 20S proteasome C8α subunit. To test this, we initially confirmed that p21 produced by in vitro transcription/translation (IVTT) bound to recombinant His-tagged C8α. 35S-labelled p21 or luciferase controls (L) were incubated with His-tagged cyclin D1 or C8α bound to nickel– agarose beads before extensive washing and SDS–PAGE; then 35S-labelled proteins associated with His fusion proteins were detected by autoradiography. p21 bound cyclin D1 efficiently, and bound C8α less efficiently, but did not bind the luciferase control (Figure 5B). A control Ral His fusion protein bound neither p21 nor luciferase, confirming the specificity of these interactions (Figure 5B).
We next examined whether the p21–C8α association could be competitively inhibited with increasing concentrations of GST–cyclin D1 (Figure 5C). As a control, we used a GST fusion of K-cyclin, which is expressed by Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV8) and has significant homology to D-type cyclins (Cesarman et al., 1996). However, unlike D-type cyclin–cdk complexes, K-cyclin-associated kinase activity is resistant to inhibition by p21 due to significantly reduced affinity for cdk inhibitors (Swanton et al., 1997). An irrelevant fusion protein (GST–PCD; see Materials and methods) was also used to control for the addition of GST. While the addition of increasing amounts of K-cyclin or PCD to the binding reaction did not affect p21 capture by C8α, equivalent amounts of cyclin D1 competitively inhibited the C8α–p21 interaction (Figure 5C). Cyclin D1 blocking the C8α–p21 interaction did not result from association with C8α, however, as no GST fusion proteins were detected in the C8α complexes (data not shown). These observations indicate that binding of cyclin D1 to p21 blocks its association with the C8α proteasome subunit.
Cyclin D1 inhibits 20S proteasome-mediated p21 degradation in vitro
To determine whether cyclin D1 competing with C8α for p21 binding would block p21 degradation by the 20S proteasome, we assayed p21 degradation in vitro using IVTT p21 and purified 20S proteasome complexes (Figure 6). Initially, we added 35S-labelled p21 or p27 to 20S proteasomes at 30°C for the times indicated, before samples were separated by SDS–PAGE, transferred and exposed to autoradiography film. Degradation of p27 is a ubiquitin- and 26S proteasome-dependent process (Alessandrini et al., 1997), and p27 was not degraded in this assay by the 20S proteasome (Figure 6A). In contrast, 35S-labelled p21 was degraded over the 20 min period (Figure 6B), dependent upon the 20S proteasome (Figure 6A; compare lanes 4 and 5). Proteasome-mediated degradation of p21 was then assayed in the presence of cyclin D1 or K-cyclin. IVTT p21 was incubated with GST–cyclin D1 or GST–K-cyclin for 1 h at 30°C before the addition of 20S proteasome. Compared with control assays where only 25% of the p21 input remained after 20 min, cyclin D1 significantly retarded 20S proteasome-mediated proteolysis (Figure 6C). In contrast, addition of the same quantity of control K-cyclin had only a slight effect on p21 degradation, and was comparable with the effect of PCD (data not shown). To determine whether the inhibitory effect of cyclin D1 is specific for p21, we assayed β-casein degradation by the 20S proteasome in the presence or absence of GST–cyclin D1. Cyclin D1 had no effect on the rate of β-casein degradation (Supplementary figure S4), confirming that the inhibitory effect on proteasomal degradation is specific for p21. In summary, these results support the model that Ras-induced elevation of cyclin D1 stabilizes p21 by competing for binding with the 20S proteasome.

Fig. 6. Cyclin D1 inhibits 20S proteasome-mediated p21 degradation in vitro. (A) 20S proteasome activity degrades p21, but not p27, in vitro. IVTT p21 or p27 were incubated with 20S proteasome for the times indicated. Reactions were stopped with Laemmli buffer and analysed for 35S-labelled p21/p27 as described. (B) Proteasome-mediated degradation was quantified by PhosphorImager analysis of 35S-labelled proteins. Shown is the percentage of 35S-labelled protein remaining at each time point, relative to a sample at time zero. (C) Proteasome-mediated p21 degradation is blocked specifically by cyclin D1. The p21 degradation assay described in (A) was repeated in the presence of GST–cyclin D1 or GST–K-cyclin.
Discussion
Ras is a centrally positioned signal transduction switch that modulates the expression of many proteins. Following the discovery that ERK activation by Ras leads to the phosphorylation, nuclear translocation and activation of a variety of transcription factors, Ras regulation of gene transcription has been studied in great detail (Zuber et al., 2000, and references therein). However, an emerging concept is that Ras and its effector pathways also regulate protein levels by post-transcriptional mechanisms, in cluding influencing proteasome-mediated degradation. Examples include the inhibition of proteasome-mediated cyclin D1 degradation via the PI3K pathway (Diehl et al., 1998) and the Ras-induced stabilization of c-myc (Sears et al., 1999).
A common mechanism for regulating p21 degradation is through direct protein–protein interactions. We found that the regulation of cyclin D1 expression by Ras and the Raf/MAPK pathway paralleled that of p21 protein (Figures 1 and 2). Transient overexpression of cyclin D1 alone was sufficient to mimic Ras-induced stabilization of p21 (Figure 3) and knock-down of cyclin D1 in Ras-transformed cells both reduced p21 levels and re-sensitized p21 to the effects of proteasome inhibition (Figure 4B and C). The inhibitory effect of cyclin D1 on p21 degradation in vivo, together with the observations that cyclin D1 can associate with p21 at the Cy2 site and block p21 binding to the C8α proteasome subunit, led us to test whether cyclin D1 would inhibit p21 degradation by the 20S complex in vitro. We found that p21, but not the related cdk inhibitor p27, was degraded in a time- and proteasome-dependent manner and that cyclin D1 efficiently blocked p21 degradation (Figure 6).
These data indicate that cyclin D1 is a critical mediator of Ras-induced p21 stability in Ras-transformed cells (Figure 7). One possible mechanism is that cyclin D1–cdk complexes may protect p21 from proteasomal degradation. Alternatively, cdk-free cyclin D1 may be the stabilizing influence in vivo, consistent with the ability of cyclin D1 to prevent p21 degradation in vitro. Interestingly, cdk-independent roles for cyclin D1 in the regulation of transcription factors have been described (e.g. Zwijsen et al., 1997), and cyclin D1 free of cdk has been identified in transformed cells that lack functional pRb (Bates et al., 1994). Our attempts to determine whether a cdk-free pool of cyclin D1 exists in Ras-transformed cells by cdk4/6 immunodepletion were unsuccessful; therefore, it remains unclear whether cdk-free cyclin D1 is involved in Ras-induced p21 stabilization.

Fig. 7. Ras-induced stabilization of p21. (A) Low ERK activity. Under basal conditions, Ras-mediated cyclin D1 transcription and expression are low. The proportion of p21 bound to cyclin D1 is low and the C-terminus is free to bind the C8α subunit of the 20S proteasome. Constitutive proteasome-mediated degradation of p21 results in low levels of expression. (B) High ERK activity. Oncogenic mutation of Ras and constitutive activation of the Raf/ERK pathway drives high levels of cyclin D1 (or cyclin D1–cdk complexes) that may bind the C-terminal Cy2 site of p21, thereby masking the proteasome-binding motif. p21 association with C8α is blocked, thereby reducing 20S proteasome-mediated degradation.
Previous reports have shown that in growth factor-stimulated non-transformed cells, p21 primarily exists in a complex with cyclin D1–cdk and is unstable (Parry et al., 1999). This complex probably results from cyclin–cdk association with the high affinity binding sites at the N-terminus of p21. However, we show here that in Ras-transformed cells, cyclin D1 is required to protect p21 from proteasomal degradation. Consistent with its ability to competitively inhibit p21 binding to the C8α subunit, cyclin D1 (or cdk complex) may protect p21 from degradation via association with the C-terminal Cy2 site, which overlaps the C8α-binding site. We believe that association of cyclin D1 with the Cy1 and/or Cy2 site of p21 determines the impact of the complex on the stability of p21 in cells of differing transformation status. Detailed characterization of the p21 complexes formed in mitogen-stimulated non-transformed cells, versus those present in Ras-transformed cells, is the subject of future investigation.
Inhibition of p21 degradation via masking of the C8α-binding site may represent a general mechanism of p21 stabilization. For example, PCNA regulates p21 turnover in vivo (Cayrol and Ducommun, 1998; Touitou et al., 2001), and the PCNA-binding site is directly N-terminal to critical C8α-binding residues 156–161 (Touitou et al., 2001), suggesting that PCNA may stabilize p21 by interfering with C8α binding. Whether PCNA competitively inhibits the p21–C8α association in a manner similar to cyclin D1 has not been determined. As PCNA levels were not altered by Ras or UO126 (Figure 1; data not shown), PCNA probably did not play a significant part in Ras-induced p21 stabilization. In addition, elevated expression of the CCAAT/enhancer-binding protein α (C/EBPα) transcription factor mediates adipocyte and hepatocyte growth inhibition via increased p21 levels (Timchenko et al., 1997). Interestingly, p21 induction by C/EBPα is post-transcriptional and involves a direct C/EBPα–p21 interaction through the p21 C-terminus (Timchenko et al., 1997). Although the C/EBPα-binding site on p21 has not been mapped, we predict that C/EBPα prevents proteasome-mediated p21 degradation by blocking binding to the 20S proteasome. C/EBPα probably did not contribute to Ras-induced p21 stabilization as its expression is restricted to differentiated cells and was not detectable in S3T3 cells (data not shown).
Given that the Raf/MAPK pathway has also been shown to elevate p21 by transcriptional and post-transcriptional mechanisms such as enhanced mRNA stability (Ravi et al., 1999), it is the foremost signalling pathway regulating p21 expression at multiple levels. In Ras-transformed cells, enhanced protein stability was the principal contributing factor to the elevated p21 levels; mRNA levels were not increased relative to parental cells (Figure 1C). However, in agreement with previous reports (Sewing et al., 1997; Woods et al., 1997), p21 mRNA was increased in Raf:ER S3T3 cells following Raf activation for up to 48 h (Figure 2D). Elevated transcription combined with enhanced protein stability in the context of acute high-intensity Raf signalling led to high growth-inhibitory levels of p21 expression. The levels of p21 induced by Raf activation were higher than in Ras-S3T3 cells (Figure 4A; data not shown), which are not compromised for proliferation by p21 and where protein stabilization is the principal mechanism of p21 elevation. Therefore, part of the Ras transformation process appears to involve the repression of growth-inhibitory p21 transcription by the Raf/MAPK pathway.
What is the basis for the lack of p21 mRNA induction in cells stably transformed by Ras compared with cells in which the Raf/MAPK pathway is acutely activated? Fibroblasts transformed by oncogenic Ras or tumour cell lines harbouring oncogenic Ras alleles overcome the growth inhibitory action of the Raf/MAPK pathway via the small GTPase Rho (Olson et al., 1998; Sahai et al., 2001). Sahai et al. (2001) showed recently that sustained activation of Raf selects for cells with high levels of Rho activity. Rho represses p21 induction (Adnane et al., 1998; Olson et al., 1998; Sahai et al., 2001), in part through inhibition of its transcription, although other post-transcriptional mechanisms may also contribute (our unpublished observations). Therefore, sustained Raf/MAPK signalling imposes a p21-mediated cell cycle arrest by transcriptional and post-translational mechanisms. Cells with high Rho activity repress the transcriptional component sufficiently such that cell cycle progression is permitted and these selected cells expand out into a transformed population. Consistent with this model, inhibition of Rho activity in Ras-transformed S3T3 cells results in elevated p21 mRNA and protein levels and consequent growth inhibition (our unpublished observations; see also Sahai et al., 2001).
Elevated p21 levels have been associated with transformation and poor patient prognosis with a variety of tumours, including prostate, ovarian, cervical and breast, where p21 provocatively has been said to act as an oncogene (Roninson, 2002). We propose that the elevated p21 expression in some tumour cells probably reflects the increased expression of C-terminal p21-binding partners such as cyclin D1 that modify its degradation by the proteasome. Interestingly, 60% of breast cancers overexpress p21, and this overexpression does not correlate with p53 expression (Rey et al., 1998). However, p21 does correlate with cyclin D1 overexpression; 93% of breast tumours that overexpressed D-type cyclins also overexpressed p21 (Russell et al., 1999), suggesting that a relationship exists between p21 and cyclin D1 levels in some human malignancies. In addition, activation of Raf signalling in MCF10A human breast epithelial cells elevated cyclin D1 mRNA and protein levels and increased p21 protein expression without affecting p21 mRNA levels (Schulze et al., 2001). Also, the stability of p21 in the MCF7 breast cancer cell line was enhanced relative to other tumour lines, and correlated with the overexpression of cyclin D1 in these cells (Russell et al., 1999).
What might be the functional role of elevated p21 levels in human tumour cells? Increased p21 expression may maintain the transformed state through positive effects on cell proliferation and protection from apoptosis (Roninson, 2002). Cells with elevated cyclin D1 levels probably require additional p21 for assembly and nuclear localization of cyclin D1 complexes with cdk4 or cdk6. We observed that the Ras/MAPK pathway uncouples cyclin D1 from the effects of proteasome inhibition (Figures 1B and 2C). Interestingly, cyclin D1 has been reported to be less stable in cells lacking p21 and p27, suggesting that the Cip/Kip proteins are required to maintain cyclin D1 protein levels (Cheng et al., 1999). p21-induced stabilization probably works by inhibiting cyclin D1 nuclear export, a requisite step for its ubiquitylation and degradation (Alt et al., 2002). In addition, through its reported ability to inhibit pro-apoptotic proteins such as caspases and apoptosis signal-regulating kinase 1 (ASK1) (Roninson, 2002), elevated p21 may protect from apoptosis induced by a variety of stimuli.
In conclusion, we have provided evidence for a novel mechanism by which Ras regulates protein levels through modulation of proteasome-mediated degradation. These data highlight the importance of further studies into the role of Ras signalling in the post-translational regulation of protein expression and its contribution to the transformed phenotype.
Materials and methods
Plasmids
pGEX K-cyclin, pcDNA3 p27 and pcDNA3 Flag-p21 were from S.Mittnacht, Institute of Cancer Research, London, UK. pGEM-2 p21 and pGEX-2T C8α were from M.Allday, Ludwig Institute of Cancer Research, London, UK. pBABE-puro Raf-1 [DD]:ER was from M.McMahon, UCSF Cancer Centre. pGEX p21 and p27 were from X.Lu, Ludwig Institute for Cancer Research, London, UK. pSuper was from R.Agami, Netherlands Cancer Institute. Murine cyclin D1 knock-down pSuper vectors targeted coding sequence bases 334–356 (D1A) and 646–668 (D1B). Details of cloning procedures for pGEX-2T ΔNp21 (amino acids 86–164), pRK5 cyclin D1-Myc, pGEX-6P1 cyclin D1, pET32a-cyclin D1, pET32a-C8α, pSuperD1A and pSuperD1B are available on request.
Cell culture and treatment
S3T3 fibroblasts were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) at 37°C and 10% CO2. V12-H-Ras-transformed S3T3 cells have been described previously (Leevers and Marshall, 1992). S3T3 cells stably expressing Raf-1[DD]:ER were from E.Sahai, Institute of Cancer Research, London, UK. Fibroblasts were serum starved to avoid Rho-dependent repression of p21 (Olson et al., 1998; Sahai et al., 2001; data not shown). NIH-3T3 fibroblasts were cultured and transfected as described previously (Coleman et al., 2001). Ras-transformed Swiss 3T3 cells were transfected with pSuperD1A and pSuperD1B, or empty pSuper, along with pBABE puro and selected with 2.5 µg/ml puromycin. LC was purchased from Professor E.J.Corey, Harvard University, Boston, MA. UO126 was from Promega and LY294002 was from Biomol. All other reagents were from Sigma unless stated otherwise.
Protein extraction, immunoprecipitation and western blotting
Cells were washed in phosphate-buffered saline (PBS) before lysis as described previously (Coleman et al., 2001). For GST pull-down assays, cells were lysed in Triton lysis buffer (TLB; 250 mM NaCl, 50 mM HEPES pH 7, 5 mM EDTA, 10 mM β-glycerol phosphate, 10 mM Naf, 1 mM sodium vanadate, 0.5 mM DTT, 0.2% Triton X100 plus protease inhibitors). Protein concentrations of whole-cell lysates (WCLs) were determined using BCA protein assay reagent (Pierce). Samples were probed with goat anti-p21 (SC-397G), cyclin D1 (SC-450), cyclin D1 (SC-718), PCNA (SC-56), cyclin D3 (SC-6283), cyclin E (SC-481), cyclin A (SC-596), Cdk4 (SC-601) (all from Santa Cruz), ERK2 (Ab122; C.J.M.), phospho-ERK (M8159: Sigma), cyclin D2 (Research Diagnostics), C8α (PW8110; Affiniti), β-catenin (C19220; Trans duction Labs) or anti-β-tubulin (N357; Amersham Pharmacia). Western blots were developed with the enhanced chemiluminescence (ECL) kit (Amersham Pharmacia). For quantitative immunoprecipitation, p21 was purified from TLB-derived lysates using monoclonal mouse p21 antibody Ab-4 (OP76, Oncogene Research) and immunoblotted for goat anti-p21 and rabbit anti-cyclin D1. Membranes were incubated with rabbit anti-goat IgG bridge prior to probing with Cy5-conjugated anti-rabbit IgG (Jackson Immunoresearch) and quantification with a STORM 850 scanner (Molecular Dynamics).
Pulse–chase analysis
Transfected NIH-3T3 cells were serum-starved overnight before incubating in DMEM minus Met/Cys for 30 min and then labelling with 150 µC/ml EXPRE35S35S labelling mix (NEN) for 1 h at 37°C. 35S-labelled cells were then chased in excess cold Met/Cys (DMEM plus 10× l-Met and l-Cys; Gibco-BRL) for 2 h. Where indicated, cells were incubated with UO126, LY or vehicle controls, and treatments maintained during the pulse–chase. Flag-tagged p21 was immunoprecipitated from 100 µg of WCL with 2 µg of anti-Flag antibody (M2; Kodak) for 2 h at 4°C. Following SDS–PAGE, transfer and exposure to a PhosphorImager screen, 35S-labelled p21 was quantified using a STORM 850 scanner and Imagequant software.
Northern blotting
Total RNA was isolated with the Qiagen RNeasy kit according to the manufacturer’s instructions. RNA (10 µg) was separated by denaturing agarose gel electrophoresis and transferred to BrightStar-Plus nylon membranes (Ambion) before being probed using the contents and instructions of the NorthernMax Kit (Ambion). p21 probe was made by PCR from mouse cDNA using the primers sense, 5′-CAATCCTGGTGATGTCCGACCTG-3′; and antisense, 5′-GCAGAAGACCAATC TGCGCTTGG-3′ (40 cycles, annealing 52°C). Mouse cyclin D1 probe was made with the primers sense, 5′-CTGACACCAATCTCCTCAACGA-3′; and antisense, 5′-TAGCAGGAGAGGAAGTTGTTGG-3′ (40 cycles, annealing 55°C). Purified PCR products were labelled with the Megaprime DNA labelling system (Amersham Pharmacia). The GAPDH probe was from G.Mavria, Institute of Cancer Research, London, UK.
Recombinant protein preparations
His-Ral and GST were from S.Hooper, Institute of Cancer Research, London, UK. Cultures of BL21(DE3) Escherichia coli (Stratagene) were induced with 30 µM isopropyl-β-d-thiogalactopyranoside (IPTG) for 16 h at 25°C. Cells were lysed in 5 mM MgCl2, 1 mM dithiothreitol (DTT) and protease inhibitors in Tris-buffered saline (TBS). Lysate supernatants were incubated with glutathione–Sepharose beads (Amersham Pharmacia) for 2 h at 4°C. Beads were washed extensively with lysis buffer before eluting GST with 10 mM glutathione and dialysis against TBS at 4°C. For His fusions, cells were induced with 0.1 mM IPTG at 37°C for 2 h, before lysis in 50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole, 20 mM β-mercaptoethanol and protease inhibitors (pH 8). Lysates were sonicated and clarified before incubating supernatants with Ni-NTA–agarose beads (Qiagen) for 1 h at 4°C. Beads were vortexed in lysis buffer before eluting His with 250 mM imidazole and dialysis against 50 mM NaH2PO4/300 mM NaCl, for 16 h at 4°C. Protein quality and concentration were determined by Coomassie staining SDS–PAGE gels with bovine serum albumin (BSA) as standard.
In vitro translation, association assays and pull-downs
For IVTT, pGEM-2 p21, pCDNA3 p27 or luciferase (TnT T7 control) plasmid DNA were added to the TnT-coupled reticulocyte lysate system (Promega; L4610) according to the manufacturer’s instructions and 35S-labelled with l-[35S]methionine (Amersham; AG1094). His fusion protein (2 µg) was bound to Ni-NTA beads for 2 h at 4°C before blocking in 1 mg/ml BSA for 20 min at 25°C. Beads were resuspended in HisBind buffer (250 mM NaCl, 10 mM imidazole, 10 mM β-mercaptoethanol, 0.5% Triton X-100, 10 mM β-glycerol phosphate, 10 mM NaF, 10 mM sodium vanadate and protease inhibitors) before adding IVTT p21 (5 µl) or luciferase for 1 h at 4°C. Competition between cyclins and C8α for p21 was investigated by adding increasing quantities of GST–cyclin D1, GST–K-cyclin or GST–PCD control to samples at the same time as p21. GST–PCD encodes the PAK1 CRIB domain fused to GST (E.Sahai). Beads were washed in HisBind before associated proteins were eluted with Laemmli buffer. GST pull-down experiments were undertaken using 2 µg of GST fusion per 1 mg of protein derived from cells lysed in ELB and incubated for 16 h at 4°C.
20S proteasomal assay of p21 degradation
20S proteasome purified from human erythrocytes was purchased from Affiniti (PW8720). IVTT p21 (1 µl) or p27 (0.75 µl) and 2 µg of either GST–cyclin D1 or GST–K-cyclin were made up to 20 µl with PBS. Binding was permitted for 60 min at 30°C before the addition of 1 µg of 20S proteasome and incubation at 30°C for the times indicated. Reactions were stopped with Laemmli buffer.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank M.Allday, X.Lu, M.McMahon, R.Agami and S.Mittnacht for plasmids, S.Hooper and E.Sahai for reagents and technical advice, members of the Olson and Marshall laboratories for many helpful discussions, and E.Sahai, D.Croft and P.Frankel for critical reading of the manuscript. Research was supported by Cancer Research UK. M.L.C. was supported by The Institute of Cancer Research. M.F.O. was a Mr and Mrs John Jaffe Donation University Research Fellow of The Royal Society.
References
- Adams P.D., Sellers,W.R., Sharma,S.K., Wu,A.D., Nalin,C.M. and Kaelin,W.G.,Jr (1996) Identification of a cyclin–cdk2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Mol. Cell. Biol., 16, 6623–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adnane J., Bizouarn,F.A., Qian,Y., Hamilton,A.D. and Sebti,S.M. (1998) p21WAF1/CIP1 is upregulated by the geranylgeranyltransferase I inhibitor GGTI-298 through a transforming growth factor β- and Sp1-responsive element: involvement of the small GTPase rhoA. Mol. Cell. Biol., 18, 6962–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandrini A., Chiaur,D.S. and Pagano,M. (1997) Regulation of the cyclin-dependent kinase inhibitor p27 by degradation and phosphorylation. Leukemia, 11, 342–345. [DOI] [PubMed] [Google Scholar]
- Alt J.R., Gladden,A.B. and Diehl,J.A. (2002) p21Cip1 promotes cyclin D1 nuclear accumulation via direct inhibition of nuclear export. J. Biol. Chem., 277, 8517–8523. [DOI] [PubMed] [Google Scholar]
- Ball K.L., Lain,S., Fahraeus,R., Smythe,C. and Lane,D.P. (1997) Cell-cycle arrest and inhibition of Cdk4 activity by small peptides based on the carboxy-terminal domain of p21WAF1. Curr. Biol., 7, 71–80. [DOI] [PubMed] [Google Scholar]
- Bates S., Parry,D., Bonetta,L., Vousden,K., Dickson,C. and Peters,G. (1994) Absence of cyclin D1/cdk complexes in cells lacking functional retinoblastoma protein. Oncogene, 9, 1633–1640. [PubMed] [Google Scholar]
- Blagosklonny M.V., Wu,G.S., Omura,S. and el-Deiry,W.S. (1996) Proteasome-dependent regulation of p21WAF1/CIP1 expression. Biochem. Biophys. Res. Commun., 227, 564–569. [DOI] [PubMed] [Google Scholar]
- Bos J.L. (1989) ras oncogenes in human cancer: a review. Cancer Res., 49, 4682–4689. [PubMed] [Google Scholar]
- Bottazzi M.E., Zhu,X., Bohmer,R.M. and Assoian,R.K. (1999) Regulation of p21cip1 expression by growth factors and the extracellular matrix reveals a role for transient ERK activity in G1 phase. J. Cell Biol., 146, 1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp T.R., Bernards,R. and Agami,R. (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science, 296, 550–553 [DOI] [PubMed] [Google Scholar]
- Cayrol C. and Ducommun,B. (1998) Interaction with cyclin-dependent kinases and PCNA modulates proteasome-dependent degradation of p21. Oncogene, 17, 2437–2444. [DOI] [PubMed] [Google Scholar]
- Cesarman E., Nador,R.G., Bai,F., Bohenzky,R.A., Russo,J.J., Moore,P.S., Chang,Y. and Knowles,D.M. (1996) Kaposi’s sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi’s sarcoma and malignant lymphoma. J. Virol., 70, 8218–8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Saha,P., Kornbluth,S., Dynlacht,B.D. and Dutta,A. (1996) Cyclin-binding motifs are essential for the function of p21CIP1. Mol. Cell. Biol., 16, 4673–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M., Sexl,V., Sherr,C.J. and Roussel,M.F. (1998) Assembly of cyclin D-dependent kinase and titration of p27Kip1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc. Natl Acad. Sci. USA, 95, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M., Olivier,P., Diehl,J.A., Fero,M., Roussel,M.F., Roberts,J.M. and Sherr,C.J. (1999) The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J., 18, 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman M.L., Sahai,E.A., Yeo,M., Bosch,M., Dewar,A. and Olson,M.F. (2001) Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol., 3, 339–345. [DOI] [PubMed] [Google Scholar]
- Diehl J.A., Cheng,M., Roussel,M.F. and Sherr,C.J. (1998) Glycogen synthase kinase-3β regulates cyclin D1 proteolysis and subcellular localization. Genes Dev., 12, 3499–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartel A.L. and Tyner,A.L. (1999) Transcriptional regulation of the p21WAF1/CIP1 gene. Exp. Cell Res., 246, 280–289. [DOI] [PubMed] [Google Scholar]
- Hershko A. and Ciechanover,A. (1998) The ubiquitin system. Annu. Rev. Biochem., 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Kerkhoff E. and Rapp,U.R. (1998) Cell cycle targets of Ras/Raf signalling. Oncogene, 17, 1457–1462. [DOI] [PubMed] [Google Scholar]
- LaBaer J., Garrett,M.D., Stevenson,L.F., Slingerland,J.M., Sandhu,C., Chou,H.S., Fattaey,A. and Harlow,E. (1997) New functional activities for the p21 family of CDK inhibitors. Genes Dev., 11, 847–862. [DOI] [PubMed] [Google Scholar]
- Leevers S.J. and Marshall C.J. (1992) Activation of extracellular signal-regulated kinase, ERK2, by p21ras oncoprotein. EMBO J., 11, 569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Dowbenko,D. and Lasky,L.A. (2001) AKT/PKB phosphorylation of p21Cip1/WAF1 enhances protein stability of p21Cip1/WAF1 and promotes cell survival. J. Biol. Chem., 276, 52. [DOI] [PubMed] [Google Scholar]
- Liu Y., Martindale,J.L., Gorospe,M. and Holbrook,N.J. (1996) Regulation of p21WAF1/CIP1 expression through mitogen-activated protein kinase signaling pathway. Cancer Res., 56, 31–35. [PubMed] [Google Scholar]
- Lloyd A.C., Obermuller,F., Staddon,S., Barth,C.F., McMahon,M. and Land,H. (1997) Cooperating oncogenes converge to regulate cyclin/cdk complexes. Genes Dev., 11, 663–677. [DOI] [PubMed] [Google Scholar]
- Olson M.F., Paterson,H.F. and Marshall,C.J. (1998) Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature, 394, 295–299. [DOI] [PubMed] [Google Scholar]
- Parry D., Mahony,D., Wills,K. and Lees,E. (1999) Cyclin D–CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol. Cell. Biol., 19, 1775–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi R.K., McMahon,M., Yangang,Z., Williams,J.R., Dillehay,L.E., Nelkin,B.D. and Mabry,M. (1999) Raf-1-induced cell cycle arrest in LNCaP human prostate cancer cells. J. Cell. Biochem., 72, 458–469. [DOI] [PubMed] [Google Scholar]
- Rey M.J. et al. (1998) p21WAF1/Cip1 is associated with cyclin D1CCND1 expression and tubular differentiation but is independent of p53 overexpression in human breast carcinoma. J. Pathol., 184, 265–271. [DOI] [PubMed] [Google Scholar]
- Roninson I.B. (2002) Oncogenic functions of tumour suppressor p21Waf1/Cip1/Sdi1: association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer Lett., 179, 1–14. [DOI] [PubMed] [Google Scholar]
- Rossig L., Badorff,C., Holzmann,Y., Zeiher,A.M. and Dimmeler,S. (2002) Glycogen synthase kinase-3 couples AKT-dependent signaling to the regulation of p21Cip1 degradation. J. Biol. Chem., 277, 9684–9689. [DOI] [PubMed] [Google Scholar]
- Russell A., Hendley,J. and Germain,D. (1999) Inhibitory effect of p21 in MCF-7 cells is overcome by its coordinated stabilization with D-type cyclins. Oncogene, 18, 6454–6459. [DOI] [PubMed] [Google Scholar]
- Sahai E., Olson,M.F. and Marshall,C.J. (2001) Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J., 20, 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze A., Lehmann,K., Jefferies,H.B.J, McMahon,M. and Downward,J. (2001) Analysis of the transcriptional program induced by Raf in epithelial cells. Genes Dev., 15, 981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears R., Leone,G., DeGregori,J. and Nevins,J.R. (1999) Ras enhances Myc protein stability. Mol. Cell, 3, 169–179. [DOI] [PubMed] [Google Scholar]
- Serrano M., Lin,A.W., McCurrach,M.E., Beach,D. and Lowe,S.W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell, 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Sewing A., Wiseman,B., Lloyd,A.C. and Land,H. (1997) High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol. Cell. Biol., 17, 5588–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheaff R.J., Singer,J.D., Swanger,J., Smitherman,M., Roberts,J.M. and Clurman,B.E. (2000) Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol. Cell, 5, 403–410. [DOI] [PubMed] [Google Scholar]
- Swanton C., Mann,D.J., Fleckenstein,B., Neipel,F., Peters,G. and Jones,N. (1997) Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature, 390, 184–187. [DOI] [PubMed] [Google Scholar]
- Timchenko N.A., Harris,T.E., Wilde,M., Bilyeu,T.A., Burgess-Beusse,B.L., Finegold,M.J. and Darlington,G.J. (1997) CCAAT/enhancer binding protein α regulates p21 protein and hepatocyte proliferation in newborn mice. Mol. Cell. Biol., 17, 7353–7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touitou R., Richardson,J., Bose,S., Nakanishi,M., Rivett,J. and Allday,M.J. (2001) A degradation signal located in the C-terminus of p21WAF1/CIP1 is a binding site for the C8 α-subunit of the 20S proteasome. EMBO J., 20, 2367–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojtek A.B. and Der,C.J. (1998) Increasing complexity of the Ras signaling pathway. J. Biol. Chem., 273, 19925–19928. [DOI] [PubMed] [Google Scholar]
- Woods D., Parry,D., Cherwinski,H., Bosch,E., Lees,E. and McMahon,M. (1997) Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol. Cell. Biol., 17, 5598–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yew P.R. (2001) Ubiquitin-mediated proteolysis of vertebrate G1- and S-phase regulators. J. Cell Physiol., 187, 1–10. [DOI] [PubMed] [Google Scholar]
- Zuber J., Tchernitsa,O.I., Hinzmann,B., Schmitz,A.C., Grips,M., Hellriegel,M., Sers,C., Rosenthal,A. and Schafer,R. (2000) A genome-wide survey of RAS transformation targets. Nat. Genet., 24, 144–152. [DOI] [PubMed] [Google Scholar]
- Zwijsen R.M., Wientjens,E., Klompmaker,R., van der Sman,J., Bernards,R. and Michalides,R.J. (1997) CDK-independent activation of estrogen receptor by cyclin D1. Cell, 88, 405–415. [DOI] [PubMed] [Google Scholar]