

Abstract
Studies have shown that expression of cystic fibrosis transmembrane conductance regulator (CFTR) is associated with enhanced glutathione (GSH) efflux from airway epithelial cells, implicating a role for CFTR in the control of oxidative stress in the airways. To define the mechanism underlying CFTR-associated GSH flux, we studied wild-type and mutant CFTR proteins expressed in Sf9 membranes, as well as purified and reconstituted CFTR. We show that CFTR-expressing membrane vesicles mediate nucleotide-activated GSH flux, which is disrupted in the R347D pore mutant, and in the Walker A K464A and K1250A mutants. Further, we reveal that purified CFTR protein alone directly mediates nucleotide-dependent GSH flux. Interestingly, although ATP supports GSH flux through CFTR, this activity is enhanced in the presence of the non-hydrolyzable ATP analog AMP-PNP. These findings corroborate previous suggestions that CFTR pore properties can vary with the nature of the nucleotide interaction. In conclusion, our data demonstrate that GSH flux is an intrinsic function of CFTR and prompt future examination of the role of this function in airway biology in health and disease.
Keywords: CFTR/glutathione/purified protein/R347D pore mutant/Walker A mutants
Introduction
Cystic fibrosis (CF) is a lethal autosomal recessive disease caused by mutations within the cystic fibrosis transmembrane conductance regulator (CFTR) gene (Boat et al., 1989). Most CF patients typically die of respiratory failure, which results from chronic inflammation and infection of the lung (Khan et al., 1995). Although mutations throughout the CFTR gene are known to cause disease, there is considerable debate regarding the primary defect(s) responsible for the pathogenesis of the CF respiratory phenotype.
CFTR is a member of the ATP-binding cassette (ABC) superfamily, which includes other clinically important members, such as P-glycoprotein (Pgp) and the multidrug resistance protein, MRP1 (Riordan et al., 1985, 1989; Cole et al., 1992). All members of this family share a common domain organization, containing a minimum of two transmembrane domains (TMDs) and two nucleotide-binding domains (NBDs; Higgins, 1995). In contrast to Pgp and MRP1, which act as efflux pumps, CFTR protein is a phosphorylation- and nucleotide-regulated chloride channel (Anderson et al., 1991b; Tabcharani et al., 1991; Bear et al., 1992). Chloride channel activity by CFTR participates in the elaboration of salt and water transport at multiple sites in the respiratory epithelium. There is a general consensus that chloride conduction through CFTR has an essential role in driving fluid secretion by the submucosal glands underlying the surface epithelium of the airways (Boat et al., 1989; Frizzell, 1999). Conversely, on the apical membrane of the surface epithelium, CFTR is thought to provide the path for chloride absorption to accompany sodium absorption, probably through the ENaC channel (Boucher, 1999). The balance between these two transport functions is thought to lead to an optimal airway surface liquid (ASL) volume to promote ciliary clearance of mucus and bacteria. In CF, loss of functional CFTR expression is thought to disturb this balance between fluid secretion and absorption, leading to net volume depletion, increased viscosity and ineffective bacterial clearance (Boucher, 1999).
Several reports have postulated that glutathione (GSH) deficiency also plays an essential role in the onset and progression of CF. High GSH concentrations, in the range of 400 µM, are found normally in ASL covering the respiratory epithelium (Cantin et al., 1987). However, decreased GSH levels have been detected in the ASL of patients with CF as well as in patients with other pulmonary diseases (Roum et al., 1993; Behr et al., 1995; Rahman and MacNee, 2000). Similarly, Gao et al. (1999) showed that GSH efflux from cells expressing mutant CFTR protein was reduced relative to that measured from cells transfected with wild-type CFTR. Moreover, in comparative in vivo studies of wild-type and CFTR knock-out mice, it was determined that GSH concentrations in the ASL obtained from mutant mice were significantly lower than those obtained from the normal mice (Velsor et al., 2001). Collectively, these data support the hypothesis that CFTR has an important role in the regulation of GSH flux from epithelial cells into the ASL.
GSH is considered to be one of the body’s most important intra- and extracellular antioxidants, providing protection against exposure to high levels of reactive oxygen species, to tissues such as the lung (Kelly, 1999). The redox status of GSH has also been implicated in the regulation of inflammation and the immune response, with GSH deficiency exacerbating inflammation and damage of the airways (Droge et al., 1991; Rahman and MacNee, 2000). In addition to GSH acting as an antioxidant, it also contributes to maintaining normal airway physiology by modifying mucus viscosity through the reduction of disulfide bonds in secreted mucins (Hudson, 2001). Moreover, recent results suggest that CFTR-dependent GSH flux could contribute to the differential sensitivity of epithelial cells to oxidative stress-induced apoptosis (Jungas et al., 2002). Therefore, in summary, there has been considerable speculation that mutations in CFTR may contribute, at least partially, to the low GSH levels observed in the ASL of CF patients and, subsequently, to the pathogenesis of CF respiratory disease.
Linsdell and Hanrahan (1998b) were the first to demonstrate that CFTR expression was associated with GSH conductance. These authors showed that ‘macro’ membrane patches excised from cells transfected with CFTR could mediate GSH currents. Similar to the chloride channel function of CFTR, the GSH currents were activated by the addition of both cAMP-dependent kinase and MgATP (agents required for CFTR chloride channel function) and were blocked by chloride channel blockers, such as DNDS (dinitrostilbene-2,2′-disulfonate) and glibenclamide. However, the authors found that the GSH conductance was asymmetric, as inward GSH currents were minimal relative to outward currents. They had observed this phenomenon with a number of other large organic anions in their previous work (Linsdell and Hanrahan, 1998a). Further, as GSH blocked chloride currents through CFTR from the intracellular solution, the authors hypothesized that GSH directly accesses the CFTR channel pore to compete with chloride ion permeation. However, these studies did not establish that CFTR itself could mediate GSH flux directly. It is well known that other chloride channel activities can be regulated by phosphorylation and MgATP (e.g. ORCC and pseudo-CFTR channels; Schwiebert et al., 1994; Jovov et al., 1995; Marvao et al., 1998; Tsumura et al., 1998) and inhibited by DNDS (e.g. ORCC and Ca-activated channels; Venglarik et al., 1994; Fuller et al., 2001; Paradiso et al., 2001) or glibenclamide (Yamazaki and Hume, 1997; Gyomorey et al., 2000). Furthermore, significant data have been amassed over the years which suggest that CFTR can interact with and modify the activity of associated chloride transport proteins such as the HCO3–/Cl– antiporter (Lee et al., 1999; Ahn et al., 2001) and ClC-3 (Ogura et al., 2002). These observations raise the possibility that the GSH flux measured in CFTR-transfected cells may be mediated by a CFTR-regulated transporter.
The primary goal of the present studies was to determine whether CFTR could mediate GSH flux directly. We tested this hypothesis by measuring nucleotide-regulated GSH flux into ‘inside-out’ plasma membrane vesicles prepared from Sf9 cells, expressing either wild-type or mutant CFTR proteins. Furthermore, to assess directly the intrinsic ability of CFTR to mediate GSH flux, we studied nucleotide-dependent GSH flux into proteoliposomes containing purified and functionally reconstituted CFTR.
Results
Expression of CFTR in Sf9 membranes confers a pathway that mediates GSH flux
To assess the role of CFTR in GSH permeation, we compared GSH uptake by ‘inside-out’ membrane vesicles prepared from Sf9 cells expressing CFTR with GSH uptake by ‘inside-out’ membrane vesicles prepared from non-transfected cells. Uptake by ‘inside-out’ membrane vesicles can be equated to efflux from the cytosol to the extracellular solution. In both cases, the vesicles were pre-phosphorylated using the catalytic subunit of protein kinase A (PKA) in the presence of 1 mM ATP, to reproduce the conditions under which GSH conductance was detected originally in patch clamp studies (Linsdell and Hanrahan, 1998b). Uptake was initiated by the addition of [35S]GSH plus either MgATP (10 mM) or MgAMP-PNP (10 mM) in the presence of 1 mM cold GSH, a concentration previously shown not to have non-specific effects on CFTR function (Kogan et al., 2001). As shown in Figure 1A, no significant GSH flux was conferred by CFTR expression in the presence of MgATP. On the other hand, in the presence of MgAMP-PNP, there was an unequivocal increase in GSH flux mediated by CFTR-expressing Sf9 membranes, relative to Sf9 membranes not expressing CFTR (P = 0.0004). Therefore, the subsequent kinetic studies of CFTR-dependent GSH flux were conducted in the presence of MgAMP-PNP.

Fig. 1. Sf9 membrane vesicles expressing phosphorylated CFTR mediate nucleotide-dependent GSH flux. (A) PKA-phosphorylated membrane vesicles either expressing or not expressing wild-type CFTR protein were incubated with [35S]GSH and 1 mM cold GSH in the presence of either MgATP or MgAMP-PNP (n = 4–6). GSH flux in membrane vesicles containing equal amounts of CFTR was expressed as a percentage of GSH flux measured in membrane vesicles with no CFTR. The asterisk represents a statistically significant difference in CFTR-mediated GSH flux in vesicles treated with MgATP versus vesicles treated with MgAMP-PNP (Student’s t-test; *P = 0.0004). (B) GSH uptake into vesicles expressing (filled squares) or not expressing CFTR (filled circles) was measured in the presence of MgAMP-PNP. GSH flux was normalized according to the amount of total protein in each treatment group. Curve fitting was performed by non-linear regression analysis, using the Michaelis–Menten equation. The data points represent the mean of duplicate values. (C) Kinetic parameters of CFTR-dependent GSH flux in the presence of MgAMP-PNP. A graph representing GSH flux by wild-type CFTR in the presence of MgAMP-PNP was obtained by subtracting GSH uptake values of vesicles with no CFTR from vesicles expressing CFTR (B). GSH flux values were expressed relative to the amount of CFTR in different preparations. Curve fitting was performed by non-linear regression analysis, using the Michaelis–Menten equation, to yield the following kinetic parameters: Km = 0.47 mM, Vmax = 612 pmol GSH/µg CFTR/h, r2 = 0.92. For all panels, CFTR quantitation is as described in Materials and methods.
As shown in Figure 1B, membrane vesicles expressing CFTR exhibited concentration-dependent GSH flux, which saturated at increasing GSH concentrations. GSH uptake by membrane vesicles prepared from non-transfected cells was significantly less than that measured for vesicles expressing CFTR. In this figure, GSH flux in preparations with or without CFTR was normalized according to the amount of total protein in each treatment group. Subtraction of the uptake data measured in non-transfected vesicles from those measured in CFTR-transfected vesicles yielded GSH flux values that are mediated specifically by CFTR protein. These flux values, therefore, are expressed relative to the amount of CFTR protein, determined as described in Materials and methods. GSH flux by CFTR in the presence of MgAMP-PNP could be described by a Michaelis– Menten curve with a Km of 0.47 mM and a Vmax of 612 pmol GSH/µg CFTR/h (Figure 1C).
The time dependence of GSH accumulation in Sf9 membrane vesicles expressing phosphorylated CFTR was determined in the presence of 10 mM MgAMP-PNP. Figure 2A shows an appreciable increase of GSH uptake by 5 min, reaching 0.7 pmol/µg total protein at 60 min. In contrast, membrane vesicles prepared from non-transfected Sf9 cells showed no appreciable [35S]GSH uptake over this time period, supporting the idea that this function is conferred specifically by CFTR.

Fig. 2. Characterization of CFTR-dependent GSH flux in the presence of MgAMP-PNP. (A) Time dependence of [35S]GSH uptake by phosphorylated membrane vesicles either expressing wild-type CFTR protein (filled circles) or not expressing CFTR (open circles). All uptake reactions contained 65 µg of total protein and were carried out for 1, 5, 10, 30 or 60 min. GSH flux was normalized according to the amount of total protein in each treatment group. Flux values are expressed as the mean ± SEM (n = 2–4). (B) The osmotic sensitivity of [35S]GSH uptake in the presence of MgAMP-PNP was determined by incubating CFTR-expressing vesicles in transport buffer containing various sucrose concentrations, ranging from zero to 750 mM. Values are expressed as the mean ± SEM (n = 2–4). Uptake values were normalized according to the amount of CFTR protein.
To confirm that GSH uptake by CFTR-expressing vesicles indeed represents uptake into the internal compartment of the vesicles, rather than surface binding, we examined the effect of increasing osmotic sensitivity on GSH uptake. Increasing the osmolarity of the transport buffer will shrink the vesicles’ lumen, thus causing decreased accumulation of ions (Loe et al., 1996). As shown in Figure 2B, increasing the sucrose concentrations in the transport buffer from 0 to 750 mM caused a significant decrease in GSH accumulation in CFTR-containing vesicles, from 558 to 313 pmol/µg CFTR/h, respectively. Therefore, the observed increase in GSH accumulation in CFTR-expressing vesicles represents uptake into the vesicles’ lumen.
Mutant CFTR proteins expressed in Sf9 membranes exhibit defective GSH flux
Arginine at position 347 is thought to contribute to the chloride channel pore of CFTR either by providing a binding site for chloride (Tabcharani et al., 1993) or by stabilizing the overall pore architecture (Cotten and Welsh, 1999). We postulated that if GSH can permeate CFTR directly, GSH uptake would be impaired in vesicles expressing this variant. As seen in Figure 3A (insert), the R347D variant was expressed well in Sf9 membranes. We compared [35S]GSH uptake by vesicles containing either phosphorylated wild-type or R347D CFTR proteins in the presence of 1 mM GSH, as this concentration is close to the Km(GSH) and is known not to have any non-specific effects on non-pore regions of CFTR. Specifically, our previous studies showed that the ATPase activity of purified CFTR is not affected by the addition of 1 mM GSH (Kogan et al., 2001). In the current study, we found that [35S]GSH uptake was significantly decreased in membrane vesicles expressing the R347D mutant protein, relative to those expressing wild-type CFTR, with rates of GSH flux of 207 and 498 pmol/µg CFTR/h, respectively (Figure 3A; P < 0.0001). This observation suggests that the AMP-PNP-dependent GSH flux in Sf9 membranes expressing CFTR is due predominantly to CFTR function. Furthermore, as shown in Figure 3B, a significant decrease in GSH uptake by vesicles expressing the mutant protein was also observed throughout a wide range of GSH concentrations. Kinetic analyses revealed that vesicles with the R347D mutation exhibited an ∼75% decrease in the Vmax of GSH uptake compared with vesicles with wild-type CFTR, corresponding to 176 and 612 pmol GSH/µg CFTR/h, respectively (Table I). These analyses also suggest that GSH interaction with the pore of the R347D mutant is ∼4 times stronger than that with the wild-type pore, with Km(GSH) values of 0.11 and 0.47 mM, respectively. Modified affinity of GSH for the mutant channel is consistent with previous reports, suggesting altered interaction of GSH with the R347D pore (Kogan et al., 2001).
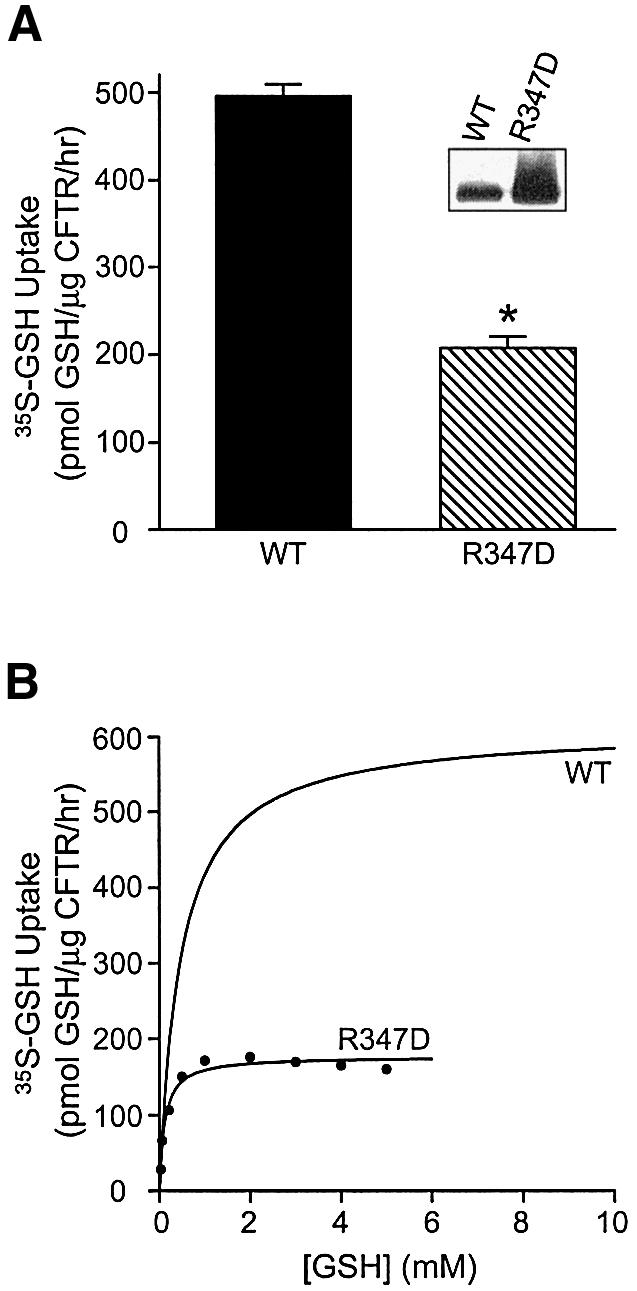
Fig. 3. Comparison of GSH flux by wild-type CFTR versus CFTR R347D protein. (A) Membrane vesicles expressing phosphorylated wild-type or R347D CFTR proteins were incubated with 20 nM [35S]GSH and 1 mM cold GSH, in the presence of MgAMP-PNP. The asterisk represents statistically significant differences in [35S]GSH flux, relative to vesicles expressing wild-type CFTR (Student’s t-test; *P < 0.0001). Values are expressed as the mean ± SEM (n = 5 for wild-type CFTR, n = 2 for R347D). Inset: expression of CFTR in membranes from Sf9 cells transfected with wild-type or R347D CFTR constructs. Membranes were solubilized in 2% SDS and Laemmli solubilizer and subjected to 4–12% SDS–PAGE. Immunoblotting was performed using the monoclonal anti-CFTR antibody M3A7 diluted 1:1000 (Chemicon International, Tamecula, CA). (B) Effect of increasing substrate concentration on [35S]GSH uptake by vesicles expressing phosphorylated wild-type CFTR or the R347D variant, in the presence of MgAMP-PNP. A graph representing GSH flux by wild-type CFTR is described in Figure 1C. [35S]GSH uptake by vesicles expressing the R347D protein was also obtained by subtracting GSH uptake values of vesicles with no CFTR from those of vesicles expressing the R347D variant. Curve fitting was performed by non-linear regression analysis, using the Michaelis–Menten equation, to yield the following kinetic parameters for the R347D mutant: Km = 0.11 mM, Vmax = 176 pmol GSH/µg CFTR/h, r2 = 0.97. GSH flux values were expressed relative to the amount of CFTR in different preparations.
Table I. Kinetic parameters of GSH uptake by Sf9 membrane vesicles containing PKA-phosphorylated wild-type or R347D CFTR proteins, in the presence of MgAMP-PNP.
| Variables | Wild type | R347D |
|---|---|---|
| Vmax (pmol/µg CFTR/h) | 612 | 176 |
| Km (mM) | 0.47 | 0.11 |
To assess the nucleotide dependence of GSH permeation through CFTR, we determined the consequences of lysine mutations in the conserved Walker A consensus motifs for ATP binding in NBD1 and NBD2: K464A and K1250A, respectively. The function of these CFTR mutant proteins has been studied extensively, and it is well established that both mutations exert a disruptive effect on nucleotide interaction with CFTR (Ramjeesingh et al., 1999b; Aleksandrov et al., 2002). In Figure 4, we show that both the K464A and K1250A mutants exhibit similar significant reductions in GSH flux. We observed that GSH uptake in both the K464A and K1250A membrane vesicles was 3- to 4-fold lower than in vesicles expressing wild-type CFTR protein, yielding permeability values of 132 and 120 pmol/µg CFTR/h, respectively (P < 0.001). These findings support the notion that CFTR mediates nucleotide-regulated GSH flux.
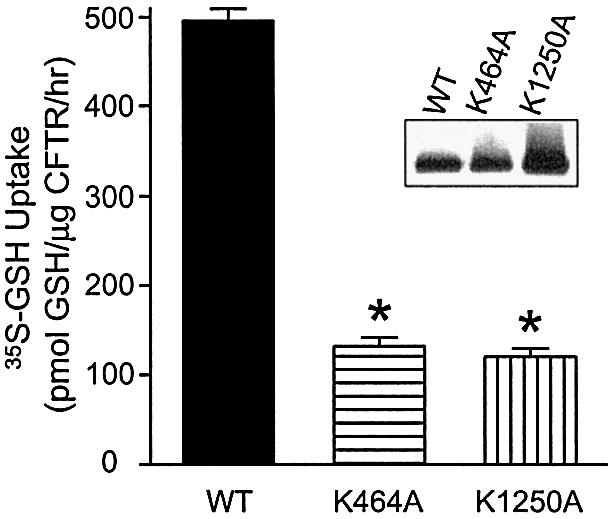
Fig. 4. Relative contribution of the two NBDs to GSH flux by CFTR protein. Membrane vesicles expressing phosphorylated wild-type, K464A or K1250A CFTR were incubated with 20 nM [35S]GSH and 1 mM cold GSH in CFTR transport buffer, in the presence of MgAMP-PNP. [35S]GSH uptake by different preparations was analyzed by one-way ANOVA, followed by Bonferroni’s multiple comparison test. The asterisks represent statistically significant differences in [35S]GSH uptake relative to vesicles expressing wild-type CFTR (*P < 0.001). Values shown represent the mean activity (± SEM; for K464A and K1250A, n = 4; for wild-type CFTR, n = 5). Inset: expression of CFTR in membranes from Sf9 cells transfected with wild-type, K464A or K1250A CFTR constructs. Western blot procedures are as described in Figure 3A.
Purified and reconstituted wild-type CFTR protein supports GSH permeation into proteoliposomes
To confirm that GSH permeability is indeed intrinsic to CFTR and to characterize further the nucleotide dependence of this property, GSH accumulation was measured in proteoliposomes containing purified and reconstituted wild-type CFTR protein. CFTR protein containing a polyhistidine tag was expressed in Sf9 cells, purified and reconstituted into pure phospholipid liposomes, as previously described (Ramjeesingh et al., 1999a; Kogan et al., 2002). Examination of CFTR-containing proteoliposomes by electron microscopy and negative staining revealed that most were spherical with a diameter of 30–60 nm, and a few with a larger diameter of ∼100 nm (Figure 5A). Radioactive GSH uptake experiments with purified and reconstituted CFTR protein were performed with PKA-phosphorylated protein to ensure that CFTR channels could gate to the open configuration (Ramjeesingh et al., 1999a). After the phosphorylation reaction, both PKA and MgATP were removed by dialysis.
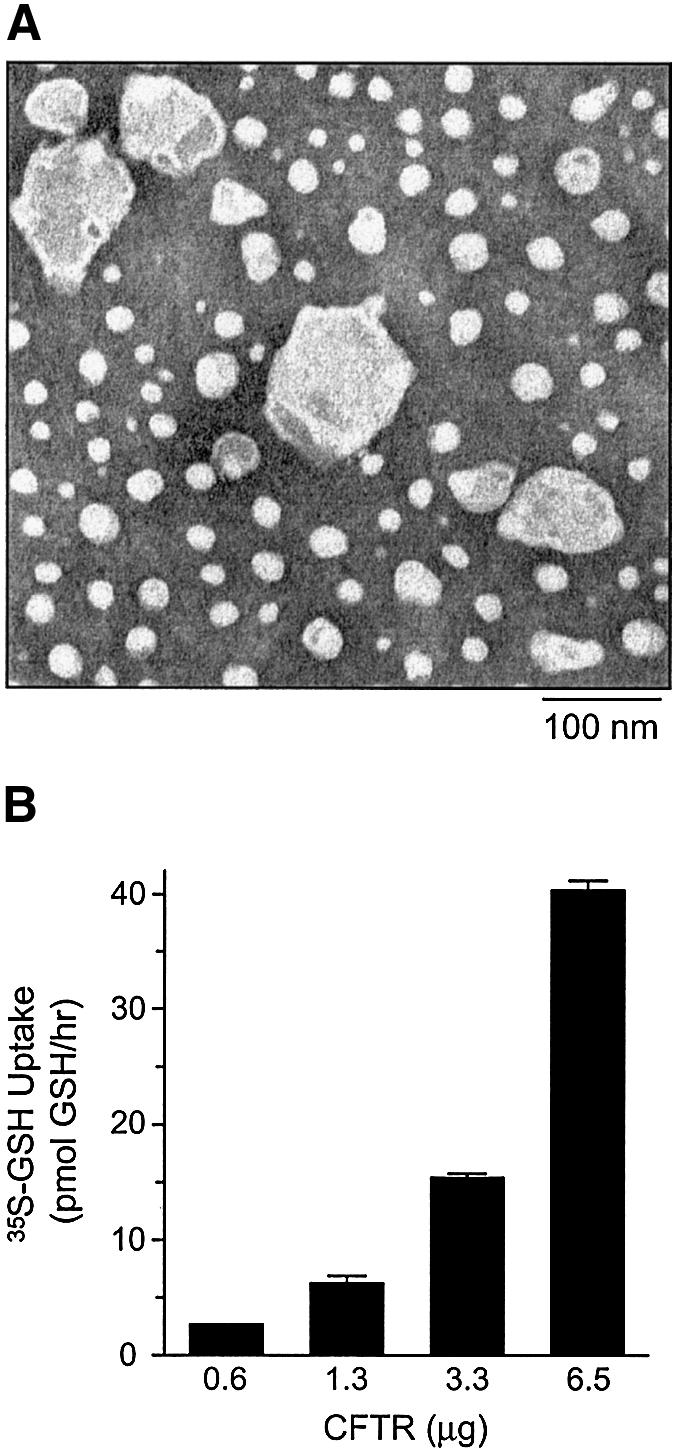
Fig. 5. GSH flux by purified and reconstituted CFTR protein. (A) Electron microscopy of purified and reconstituted wild-type CFTR. Proteoliposomes on carbon formvar-coated grids were stained with 2% uranyl acetate and visualized by negative staining. Magnification is ×150 000. Bar = 100 nm. (B) Increasing concentrations of reconstituted and phosphorylated wild-type CFTR protein (0.6–6.5 µg) were incubated with 20 nM [35S]GSH and 1 mM cold GSH in CFTR transport buffer, in the presence of MgAMP-PNP. Each value represents the mean of duplicate determinations.
As shown in Figure 5B, the degree of MgAMP-PNP-stimulated [35S]GSH flux mediated by proteoliposomes was dependent on the quantity of reconstituted CFTR protein. These data, therefore, indicate that CFTR can mediate GSH flux directly. We then compared the nucleotide-dependent regulation of [35S]GSH flux and 36Cl– flux in parallel studies of purified, reconstituted CFTR protein (Figure 6). In both assays, CFTR protein was pre-phosphorylated, as previously described, and the uptake initiated by addition of either [35S]GSH or 36Cl–. Uptake by proteoliposomes was assessed under three conditions: no added nucleotide, 1 mM MgATP or 6 mM MgAMP-PNP. For both anions, uptake was enhanced by the addition of nucleotide, confirming that both [35S]GSH flux and 36Cl– flux activities are nucleotide dependent. As predicted on the basis of substantial electrophysiological data (Vergani et al., 2003), 36Cl– flux was markedly stimulated by the addition of MgATP (Figure 6B; P < 0.001). On the other hand, no significant 36Cl– flux was mediated by proteoliposomes treated with MgAMP-PNP. On the contrary, the results obtained in our studies of [35S]GSH flux showed different nucleotide specificity. While MgATP stimulated [35S]GSH flux considerably, MgAMP-PNP caused a significantly greater response (Figure 6A; P < 0.05 and P < 0.001, respectively). The mean values for GSH permeation in the presence of MgATP or MgAMP-PNP are 3.5 and 9.8 pmol/µg CFTR/h, respectively. Collectively, our data support the concept that there may be differential permeation properties of CFTR that are mediated by interaction with MgATP or MgAMP-PNP. The 50-fold decrease in GSH flux observed in vesicles expressing purified and reconstituted wild-type CFTR, as compared with Sf9 membrane vesicles expressing CFTR, can be accounted for by the fact that only ∼50% of the reconstituted proteins display an ‘inside-out’ orientation. Further, it is estimated that only ∼10% of reconstituted CFTR in the proteoliposomes is truly active (Bear et al., 1992). Taking these considerations into account, the overall GSH flux in CFTR-expressing membrane vesicles is only 2- to 3-fold greater, as compared with that measured in CFTR proteoliposomes.

Fig. 6. Nucleotide-regulated GSH versus chloride flux by purified and reconstituted CFTR protein. (A) Proteoliposomes containing purified, reconstituted and phosphorylated CFTR were incubated with 20 nM [35S]GSH and 1 mM cold GSH at 33°C in CFTR transport buffer, in the presence of MgATP, MgAMP-PNP or no nucleotides for 60 min. GSH uptake values were normalized according to the amount of CFTR in each preparation. Values shown represent the mean activity (± SEM; n = 2–6). [35S]GSH uptake by different preparations was analyzed by one-way ANOVA, followed by Bonferroni’s multiple comparison test. The asterisks represent statistically significant differences in [35S]GSH flux relative to vesicles treated with no nucleotides (*P < 0.05; **P < 0.001). (B) 36Cl– flux in purified, reconstituted and phosphorylated CFTR was measured at 33°C in the presence of MgATP, MgAMP-PNP or no nucleotides, as previously described (Li, 1996). Flux values were normalized according to the amount of CFTR in each preparation. Values shown represent the mean activity (± SEM; n = 3). Chloride flux by different preparations was analyzed by one-way ANOVA, followed by Bonferroni’s multiple comparison test. The asterisks represent statistically significant differences in flux relative to vesicles treated with no nucleotides (*P < 0.001).
Discussion
Since the discovery of CFTR in 1989, there has been considerable debate regarding the spectrum of its biological function(s). Initially, the primary issue lay in establishing whether CFTR was a chloride channel or a channel regulator. Now, a large body of evidence is available indicating that CFTR is both an anion channel and a regulator of ion transporters and channels (Anderson et al., 1991a; Bear et al., 1992; Egan et al., 1992; Stutts et al., 1995; Lee et al., 1999; Schwiebert et al., 1999). Therefore, when it was shown recently that expression of CFTR led to the appearance of GSH currents (Linsdell and Hanrahan, 1998b), it became imperative to determine if CFTR mediated these currents directly. Resolving this issue is important because CFTR-mediated flux of this major antioxidant could provide a clear link between CFTR expression and the airway inflammation characteristic of CF. In the present studies of Sf9 membrane vesicles expressing CFTR, as well as purified and reconstituted CFTR protein, we have shown that CFTR alone is capable of directly mediating nucleotide-regulated flux of GSH. Hence, the absence or dysfunction of CFTR may account, at least partially, for the low GSH levels observed in the ASL of CF patients and, subsequently, to the pathogenesis of CF respiratory disease (Hudson, 2001).
Comparison of GSH efflux rates measured in Sf9 membrane vesicles expressing CFTR (present study) with efflux rates determined for GSH across the apical membrane of airway epithelial monolayers (Gao et al., 1999) permits a crude estimate of the relative contribution of CFTR to this function in physiological systems. Based on the analysis of the data in Figure 1, we determined that the maximal GSH efflux associated with CFTR expression across Sf9 membranes (i.e. in the presence of MgAMP-PNP) is ∼0.73 pmol/µg total protein/h (Vmax). Estimates of the rate of GSH efflux associated with CFTR expression in human bronchiolar epithelial cells reported by Gao et al. (1999) are in the range of 1.4 pmol/µg total protein/h. Hence, the GSH efflux rates in the two systems are similar. However, such extrapolations are problematic, given our uncertainty about the proportion of functional CFTR protein in the membranes of both expression systems. Interestingly, the efflux rate for GSH conferred by CFTR expression in Sf9 membranes, in the present work, is approximately three orders of magnitude greater than that determined for GSH efflux, mediated by vesicles prepared from MRP-transfected cells (∼1 pmol/mg protein/h; Loe et al., 2000). The current data, therefore, further support the claim that GSH flux is an important function of CFTR.
Our comparative studies of the GSH and chloride fluxes mediated by purified, phosphorylated CFTR revealed differences in the nucleotide dependence of these two functions. While both GSH and chloride fluxes were activated by the addition of MgATP, MgAMP-PNP was ineffective in activating chloride flux, but caused a greater activation of GSH flux compared with MgATP. The nucleotide dependence of CFTR-mediated chloride flux observed in the current study is consistent with data from several other laboratories (Anderson et al., 1991b; Gunderson and Kopito, 1995; Vergani et al., 2003), and suggests that MgATP binding plus secondary conformational changes (possibly induced by hydrolysis) are required for channel opening and chloride conduction. However, it is noteworthy that considerable controversy exists in the field with regards to the regulation of CFTR chloride channel activity (Schultz et al., 1996; Aleksandrov et al., 2000; Ikuma and Welsh, 2000).
Our finding that GSH flux is activated by MgAMP-PNP is consistent, in part, with the results from patch–clamp studies by Linsdell and Hanrahan (1998b). As previously mentioned, these authors reported that GSH conductance conferred by phosphorylated CFTR was increased in both the inward and outward direction by addition of MgAMP-PNP or pyrophosphate (PPi), relative to the conductance activated by MgATP (Linsdell and Hanrahan, 1998a). Furthermore, in a separate report, they showed that the permeability of the large organic anion gluconate was increased relative to chloride, upon addition of MgAMP-PNP. Based on these data, the authors suggested that the properties of the CFTR pore changed upon inhibition of its ATPase activity (Linsdell and Hanrahan, 1998a). Therefore, it is reasonable that GSH flux through CFTR may be favored by nucleotide binding rather than hydrolysis, reflecting different pore properties associated with differential nucleotide interactions.
The concept that differential nucleotide interactions with the NBDs of ABC proteins can induce specific conformations within their membrane domains is supported in models based on recent structures solved for the prokaryotic ABC proteins (Chang and Roth, 2001; Locher et al., 2002). For example, the BtuCD structure provides a mechanistic scheme for ATP-powered vitamin B12 import in which ATP binding and/or hydrolysis by the NBDs alters an existing interface, evoking a conformational change in the membrane-spanning domains and opening of an inner vestibule of this transporter. Further, lower resolution structures of Pgp, obtained by analysis of two-dimensional crystals, reveal that AMP-PNP interaction at the NBDs induces a unique conformation in the membrane-spanning domains, which is distinct from that observed in the presence of ADP/vanadate, which should mimic the post-hydrolytic, transition state (Rosenberg et al., 2001). Hence, it is reasonable to speculate that the translocation path through the TMD of CFTR would also undergo distinct conformational changes in response to changes in nucleotide interaction.
Based on our current findings, we propose that under certain conditions, modifications of CFTR that reduce its intrinsic ATP hydrolysis could inhibit CFTR-mediated chloride flux and promote CFTR-mediated GSH flux. For example, physiological concentrations of GSH (5–10 mM) are known to inhibit CFTR ATPase activity (Kogan et al., 2001). Consequently, it is possible that GSH itself may promote its translocation through the pore by decreasing CFTR’s intrinsic ATPase activity. We therefore believe that there may be certain physiological conditions that favor distinct transport functions of CFTR. This idea is consistent with that of Reddy and Quinton (2001), who recently suggested that distinct agonists and/or modifications of CFTR can selectively activate CFTR-mediated chloride or bicarbonate currents.
Although our findings suggest that CFTR is capable of mediating GSH flux directly, we cannot rule out the possibility that total GSH flux across the apical membrane of native epithelia may also involve other membrane proteins, in addition to CFTR. However, according to the literature, other ABC family members thought to mediate GSH flux, e.g. MRP1 and MRP2/cMOAT, are either localized on the basolateral membrane of respiratory epithelia or expressed at very low levels in this tissue, respectively (Wright et al., 1998; Cherrington et al., 2002). Moreover, to the best of our knowledge, GSH transport by non-ABC proteins, such as the organic anion-transporting peptides (OATPs), has not been reported in the lung. Hence, we believe that therapeutic strategies aimed at enhancing GSH flux through mutant CFTR may be useful in the treatment of CF. For example, certain CFTR openers appear to exert similar effects on channel gating to those of AMP-PNP and PPi, e.g. NS004, NS1619 (Al-Nakkash et al., 2001) and genistein (Hwang and Sheppard, 1999). Hence, we predict that these agents may act to promote GSH flux through wild-type and certain CFTR mutant proteins. We plan to test the ability of these agents to correct defective GSH flux by CFTR mutants in future experiments.
Materials and methods
Membrane vesicle preparation
‘Inside-out’ membrane vesicles were prepared according to methods described previously (Loe et al., 2000), with a few modifications. An Sf9 cell pellet (0.5 l) expressing either recombinant CFTR-His proteins (wild type or mutant: R347D, K464A, K1250A) or no CFTR was solubilized in 30 ml of homogenization buffer containing 250 mM sucrose, 50 mM Tris–HCl, 0.25 mM CaCl2 pH 7.5 and protease inhibitors (Roche Diagnostics GmbH, Mannheim, Germany). Solubilized cell pellets were disrupted by French Press (French® Pressure Cell Press; SLM-AMINCO spectronic instruments) at 1000 p.s.i. EDTA was added to yield a final concentration of 1 mM, and cells were centrifuged for 10 min at 500 g at 4°C. The supernatant was layered over 35% sucrose in 50 mM Tris–HCl (w/w), and centrifuged for 1 h at 100 000 g at 4°C. The interface was resuspended in 25 mM sucrose, 50 mM Tris–HCl pH 7.5 and centrifuged for 30 min at 100 000 g at 4°C. The membrane pellet was washed in CFTR transport buffer (TSB) containing 125 mM NaCl, 50 mM Tris–HCl pH 7.5, and centrifuged for 20 min at 125 000 g at 4°C. The resulting membrane pellet was resuspended in CFTR TSB and passed 20 times through a 27 gauge needle for ‘inside-out’ vesicle formation.
Uptake of glutathione in membrane vesicles
CFTR in membrane vesicles was activated by phosphorylation using the catalytic subunit of PKA (Promega, Madison, WI) for 10 min at room temperature, as previously described (Kogan et al., 2002). After CFTR phosphorylation, reactions containing acivicin (Sigma) at a final concentration of 0.5 mM were incubated for 10 min at 37°C, to decrease GSH catabolism by γ-glutamyl transpeptidase during the uptake assay (Smith et al., 1995). GSH uptake was carried out at 33°C for 60 min in a final volume of 110 µl, and contained the following final concentrations: 10 mM ATP, 6 mM MgCl2, 1 mM GSH, [35S]GSH (25–30 nM, 3–6 µCi; Perkin Elmer Lifescience Can) and an ATP-regenerating system containing 10 mM creatine phosphate and 100 µg/ml creatine kinase in CFTR TSB (Loe et al., 2000). In some uptake reactions, 10 mM AMP-PNP was substituted for ATP. For the time course experiments, reactions were incubated for 1, 5, 10, 30 and 60 min. At the end of the incubation period, vesicles were centrifuged through Sephadex G-50 columns equilibrated with CFTR TSB to separate external [35S]GSH from radioactive GSH that accumulated inside the vesicles (Ramjeesingh et al., 1999b). Intra vesicular radioactivity was counted and normalized according to the concentration of either CFTR or total protein in each preparation. For measuring GSH flux under increasing substrate concentrations, CFTR-expressing membrane vesicles were incubated with [35S]GSH and increasing concentrations of cold GSH to yield GSH concentrations ranging from 0.1 nM to 10 mM (13 different concentrations).
Quantitation of CFTR in membrane vesicles
CFTR standards were prepared from Sf9 cells expressing wild-type CFTR protein. The protein was purified according to the methods described previously (Ramjeesingh et al., 1999a; Kogan et al., 2002), and quantitated using amino acid analysis (Alberta Peptide Institute, Alberta, Canada). Membranes expressing CFTR were solubilized in 2% SDS and Laemmli solubilizer, and subjected to 4–12% SDS–PAGE, along with purified CFTR standards of known concentration. Immunoblotting was performed using the monoclonal anti-CFTR antibody M3A7 diluted 1:1000 (Chemicon International, Tamecula, CA). Densitometric analyses (NIH image) were used for quantitation.
Purification and reconstitution of CFTR-His proteins
Detailed protocols regarding the generation of CFTR-His proteins are described elsewhere (Ramjeesingh et al., 1999a). Frozen Sf9 cell pellet from 1 l, expressing recombinant CFTR-His proteins, was thawed and solubilized in a buffer containing 250 mM sucrose, 50 mM Tris–HCl, 0.25 mM CaCl2 pH 7.5 and protease inhibitors (Loe et al., 2000). Cells were then disrupted using a French Press set at 1000 p.s.i. and centrifuged for 20 min at 500 g at 4°C. Supernatant containing crude membranes was centrifuged for 2 h at 100 000 g at 4°C to yield a plasma membrane-enriched fraction. Membranes were solubilized overnight in 8% pentadecafluorooctanoic acid (PFO), 25 mM phosphate pH 8.0. Procedures for purification, reconstitution and phosphorylation of purified CFTR-His were as described elsewhere (Ramjeesingh et al., 1999a; Kogan et al., 2002). Phosphorylated samples were dialyzed overnight to remove PKA and/or ATP, as reported previously (Ramjeesingh et al., 1999a; Kogan et al., 2002).
Uptake of glutathione by proteoliposomes
GSH uptake reactions were carried out at 33°C for 60 min in a final volume of 170 µl, and contained the following final concentrations: 1 mM ATP or 6 mM AMP-PNP, 2 mM MgCl2, 1 mM GSH and [35S]GSH (25–30 nM, 3–6 µCi; Perkin Elmer Lifescience Can) in 150 mM KCl. At the end of the incubation period, 130 µl from the uptake reaction were centrifuged through Sephadex G-50 columns equilibrated with 150 mM KCl (Ramjeesingh et al., 1999a). Intravesicular radioactivity was counted and normalized according to the CFTR concentration in each preparation.
Uptake of chloride by proteoliposomes
A concentrative tracer uptake assay, described previously (Li et al., 1996), was used to measure 36Cl– flux into proteoliposomes containing purified and reconstituted CFTR protein. Intravesicular 36Cl– was assayed after incubation of proteoliposomes at 33°C for 60 min, in the presence of MgATP, MgAMP-PNP or no nucleotides.
Statistical analysis
Results are expressed as the mean ± SEM. Statistical significance was assessed using two-tailed Student’s t-test or one-way analysis of variance (ANOVA), with Bonferroni’s multiple comparison test for comparing uptake in pairs of different treatments. Curve generation was carried out using Prism Software (San Diego, CA).
Acknowledgments
Acknowledgements
We are grateful to Yu Ming Heng and Cameron Ackerley for the electron microscopy images and to Dr M.Corey for assistance with the statistical analyses. The authors are also grateful to Prof. Frances Sharom (University of Guelph) for her suggestions and advice with respect to experimental design. Dr G.Lukacs (University of Toronto) also provided valuable comments regarding the experimental work and the manuscript. This work was supported by an operating grant awarded to C.E.B. by the Canadian Cystic Fibrosis Foundation (CCFF) and a CIHR grant to S.P.C.C. (10519). I.K. was supported by a CCFF Studentship Award, J.F.K. by a CCFF Fellowship Award, and E.M.L. by a CIHR Doctoral Award.
References
- Ahn W., Kim,K.H., Lee,J.A., Kim,J.Y., Choi,J.Y., Moe,O.W., Milgram,S.L., Muallem,S. and Lee,M.G. (2001) Regulatory interaction between the cystic fibrosis transmembrane conductance regulator and HCO3– salvage mechanisms in model systems and the mouse pancreatic duct. J. Biol. Chem., 276, 17236–17243. [DOI] [PubMed] [Google Scholar]
- Al-Nakkash L., Hu,S., Li,M. and Hwang,T.C. (2001) A common mechanism for cystic fibrosis transmembrane conductance regulator protein activation by genistein and benzimidazolone analogs. J. Pharmacol. Exp. Ther., 296, 464–472. [PubMed] [Google Scholar]
- Aleksandrov A.A., Chang,X., Aleksandrov,L. and Riordan,J.R. (2000) The non-hydrolytic pathway of cystic fibrosis transmembrane conductance regulator ion channel gating. J. Physiol., 528, 259–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrov L., Aleksandrov,A.A., Chang,X.B. and Riordan,J.R. (2002) The first nucleotide binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover. J. Biol. Chem., 277, 15419–15425. [DOI] [PubMed] [Google Scholar]
- Anderson M., Gregory,R., Thompson,S., Souza,D., Paul,S., Mulligan,R., Smith,A. and Welsh,M. (1991a) Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science, 253, 202–205. [DOI] [PubMed] [Google Scholar]
- Anderson M.P., Berger,H.A., Rich,D.P., Gregory,R.J., Smith,A.E. and Welsh,M.J. (1991b) Nucleoside triphosphates are required to open the CFTR chloride channel. Cell, 67, 775–784. [DOI] [PubMed] [Google Scholar]
- Bear C.E., Li,C., Kartner,N., Bridges,R., Jensen,T., Ramjeesingh,M. and Riordan,J. (1992) Purification and functional reconstitution of the cystic fibrosis transmembrane conductance regulator (CFTR). Cell, 68, 809–818. [DOI] [PubMed] [Google Scholar]
- Behr J., Degenkolb,B., Maier,K., Braun,B., Beinert,T., Krombach,F., Vogelmeier,C. and Fruhmann,G. (1995) Increased oxidation of extracellular glutathione by bronchoalveolar inflammatory cells in diffuse fibrosing alveolitis. Eur. Respir. J., 8, 1286–1292. [DOI] [PubMed] [Google Scholar]
- Boat T.F., Welsh,M.J. and Beaudet,A.L. (1989) Cystic fibrosis. In Scriver,C.R., Beaudet,A.L., Sly,W.S. and Valle,D. (eds), The Metabolic Basis of Inherited Disease. McGraw-Hill, New York, NY, pp. 2649–2680.
- Boucher R.C. (1999) Molecular insights into the physiology of the ‘thin film’ of airway surface liquid. J. Physiol., 516, 631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin A.M., North,S.L., Hubbard,R.C. and Crystal,R.G. (1987) Normal alveolar epithelial lining fluid contains high levels of glutathione. J. Appl. Physiol., 63, 152–157. [DOI] [PubMed] [Google Scholar]
- Chang G. and Roth,C.B. (2001) Structure of MsbA from E.coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science, 293, 1793–1800. [DOI] [PubMed] [Google Scholar]
- Cherrington N.J., Hartley,D.P., Li,N., Johnson,D.R. and Klaassen,C.D. (2002) Organ distribution of multidrug resistance proteins 1, 2 and 3 (Mrp1, 2 and 3) mRNA and hepatic induction of Mrp3 by constitutive androstane receptor activators in rats. J. Pharmacol. Exp. Ther., 300, 97–104. [DOI] [PubMed] [Google Scholar]
- Cole S.P. et al. (1992) Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science, 258, 1650–1654. [DOI] [PubMed] [Google Scholar]
- Cotten J.F. and Welsh,M.J. (1999) Cystic fibrosis-associated mutations at arginine 347 alter the pore architecture of CFTR. Evidence for disruption of a salt bridge. J. Biol. Chem., 274, 5429–5435. [DOI] [PubMed] [Google Scholar]
- Droge W., Eck,H.P., Gmunder,H. and Mihm,S. (1991) Modulation of lymphocyte functions and immune responses by cysteine and cysteine derivatives. Am. J. Med., 91, 140S–144S. [DOI] [PubMed] [Google Scholar]
- Egan M., Flotte,T., Afione,S., Solow,R., Zeitlin,P.L., Carter,B.J. and Guggino,W.B. (1992) Defective regulation of outwardly rectifying Cl– channels by protein kinase A corrected by insertion of CFTR. Nature, 358, 581–584. [DOI] [PubMed] [Google Scholar]
- Frizzell R.A. (1999) Ten years with CFTR. Physiol. Rev., 79, S1–S2. [DOI] [PubMed] [Google Scholar]
- Fuller C.M., Ji,H.L., Tousson,A., Elble,R.C., Pauli,B.U. and Benos,D.J. (2001) Ca2+-activated Cl– channels: a newly emerging anion transport family. Pflugers Arch., 443, S107–110. [DOI] [PubMed] [Google Scholar]
- Gao L., Kim,K.J., Yankaskas,J.R. and Forman,H.J. (1999) Abnormal glutathione transport in cystic fibrosis airway epithelia. Am. J. Physiol., 277, L113–L118. [DOI] [PubMed] [Google Scholar]
- Gunderson K.L. and Kopito,R.R. (1995) Conformational states of CFTR associated with channel gating: the role of ATP binding and hydrolysis. Cell, 82, 231–239. [DOI] [PubMed] [Google Scholar]
- Gyomorey K., Rozmahel,R. and Bear,C.E. (2000) Amelioration of intestinal disease severity in cystic fibrosis mice is associated with improved chloride secretory capacity. Pediatr. Res., 48, 731–734. [DOI] [PubMed] [Google Scholar]
- Higgins C. (1995) The ABC of channel regulation. Cell, 82, 693–696. [DOI] [PubMed] [Google Scholar]
- Hudson V.M. (2001) Rethinking cystic fibrosis pathology: the critical role of abnormal reduced glutathione (GSH) transport caused by CFTR mutation. Free Radic. Biol. Med., 30, 1440–1461. [DOI] [PubMed] [Google Scholar]
- Hwang T.C. and Sheppard,D.N. (1999) Molecular pharmacology of the CFTR Cl– channel. Trends Pharmacol. Sci., 20, 448–453. [DOI] [PubMed] [Google Scholar]
- Ikuma M. and Welsh,M.J. (2000) Regulation of CFTR Cl– channel gating by ATP binding and hydrolysis. Proc. Natl Acad. Sci. USA, 97, 8675–8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovov B., Ismailov,I.I., Berdiev,B.K., Fuller,C.M., Sorscher,E.J., Dedman,J.R., Kaetzel,M.A. and Benos,D.J. (1995) Interaction between cystic fibrosis transmembrane conductance regulator and outwardly rectified chloride channels. J. Biol. Chem., 270, 29194–29200. [DOI] [PubMed] [Google Scholar]
- Jungas T., Motta,I., Duffieux,F., Fanen,P., Stoven,V. and Ojcius,D.M. (2002) Glutathione levels and BAX activation during apoptosis due to oxidative stress in cells expressing wild-type and mutant CFTR. J. Biol. Chem., 277, 27912–27918. [DOI] [PubMed] [Google Scholar]
- Kelly F.J. (1999) Gluthathione: in defence of the lung. Food Chem. Toxicol., 37, 963–966. [DOI] [PubMed] [Google Scholar]
- Khan T.Z., Wagener,J.S., Bost,T., Martinez,J., Accurso,F.J. and Riches,D.W. (1995) Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med., 151, 1075–1082. [DOI] [PubMed] [Google Scholar]
- Kogan I., Ramjeesingh,M., Huan,L.J., Wang,Y. and Bear,C.E. (2001) Perturbation of the pore of the cystic fibrosis transmembrane conductance regulator (CFTR) inhibits its ATPase activity. J. Biol. Chem., 276, 11575–11581. [DOI] [PubMed] [Google Scholar]
- Kogan I., Ramjeesingh,M., Li,C. and Bear,C.E. (2002) Studies of the molecular basis for cystic fibrosis using purified reconstituted CFTR protein. Methods Mol. Med., 70, 143–157. [DOI] [PubMed] [Google Scholar]
- Lee M.G., Wigley,W.C., Zeng,W., Noel,L.E., Marino,C.R., Thomas,P.J. and Muallem,S. (1999) Regulation of Cl–/HCO3– exchange by cystic fibrosis transmembrane conductance regulator expressed in NIH 3T3 and HEK 293 cells. J. Biol. Chem., 274, 3414–3421. [DOI] [PubMed] [Google Scholar]
- Li C., Ramjeesingh,M. and Bear,C.B. (1996) Purified cystic fibrosis transmembrane conductance regulator (CFTR) does not function as an ATP channel. J. Biol. Chem., 271, 11623–11626. [DOI] [PubMed] [Google Scholar]
- Linsdell P. and Hanrahan,J. (1998a) Adenosine triphosphate-dependent asymmetry of anion permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. J. Gen. Physiol., 111, 601–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsdell P. and Hanrahan,J. (1998b) Glutathione permeability of CFTR. Am. J. Physiol., 275, C323–C326. [DOI] [PubMed] [Google Scholar]
- Locher K.P., Lee,A.T. and Rees,D.C. (2002) The E.coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science, 296, 1091–1098. [DOI] [PubMed] [Google Scholar]
- Loe D.W., Almquist,K.C., Deeley,R.G. and Cole,S.P. (1996) Multidrug resistance protein (MRP)-mediated transport of leukotriene C4 and chemotherapeutic agents in membrane vesicles. Demonstration of glutathione-dependent vincristine transport. J. Biol. Chem., 271, 9675–9682. [DOI] [PubMed] [Google Scholar]
- Loe D.W., Oleschuk,C.J., Deeley,R.G. and Cole,S.P. (2000) Structure–activity studies of verapamil analogs that modulate transport of leukotriene C(4) and reduced glutathione by multidrug resistance protein MRP1. Biochem. Biophys. Res. Commun., 275, 795–803. [DOI] [PubMed] [Google Scholar]
- Marvao P., De Jesus Ferreira,M.C., Bailly,C., Paulais,M., Bens,M., Guinamard,R., Moreau,R., Vandewalle,A. and Teulon,J. (1998) Cl– absorption across the thick ascending limb is not altered in cystic fibrosis mice. A role for a pseudo-CFTR Cl– channel. J. Clin. Invest., 102, 1986–1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura T., Furukawa,T., Toyozaki,T., Yamada,K., Zheng,Y.J., Katayama,Y., Nakaya,H. and Inagaki,N. (2002) ClC-3B, a novel ClC-3 splicing variant that interacts with EBP50 and facilitates expression of CFTR-regulated ORCC. FASEB J., 16, 863–865. [DOI] [PubMed] [Google Scholar]
- Paradiso A.M., Ribeiro,C.M. and Boucher,R.C. (2001) Polarized signaling via purinoceptors in normal and cystic fibrosis airway epithelia. J. Gen. Physiol., 117, 53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I. and MacNee,W. (2000) Oxidative stress and regulation of glutathione in lung inflammation. Eur. Respir. J., 16, 534–554. [DOI] [PubMed] [Google Scholar]
- Ramjeesingh M., Garami,E., Galley,K., Li,C., Wang,Y. and Bear,C.E. (1999a) Purification and reconstitution of epithelial chloride channel cystic fibrosis transmembrane conductance regulator. Methods Enzymol., 294, 227–246. [DOI] [PubMed] [Google Scholar]
- Ramjeesingh M., Li,C., Garami,E., Huan,L.J., Galley,K., Wang,Y. and Bear,C.E. (1999b) Walker mutations reveal loose relationship between catalytic and channel-gating activities of purified CFTR (cystic fibrosis transmembrane conductance regulator). Biochemistry, 38, 1463–1468. [DOI] [PubMed] [Google Scholar]
- Reddy M.M. and Quinton,P.M. (2001) Selective activation of cystic fibrosis transmembrane conductance regulator Cl– and HCO3– conductances. JOP, 2, 212–218. [PubMed] [Google Scholar]
- Riordan J.R., Deuchars,K., Kartner,N., Alon,N., Trent,J. and Ling,V. (1985) Amplification of P-glycoprotein genes in multidrug-resistant mammalian cell lines. Nature, 316, 817–819. [DOI] [PubMed] [Google Scholar]
- Riordan J. et al. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science, 245, 1066–1073. [DOI] [PubMed] [Google Scholar]
- Rosenberg M.F. et al. (2001) Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J., 20, 5615–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roum J.H., Buhl,R., McElvaney,N.G., Borok,Z. and Crystal,R.G. (1993) Systemic deficiency of glutathione in cystic fibrosis. J. Appl. Physiol., 75, 2419–2424. [DOI] [PubMed] [Google Scholar]
- Schultz B.D., Bridges,R.J. and Frizzell,R.A. (1996) Lack of conventional ATPase properties in CFTR chloride channel gating. J. Membr. Biol., 151, 63–75. [DOI] [PubMed] [Google Scholar]
- Schwiebert E.M., Flotte,T., Cutting,G.R. and Guggino,W.B. (1994) Both CFTR and outwardly rectifying chloride channels contribute to cAMP-stimulated whole cell chloride currents. Am. J. Physiol., 266, C1464–C1477. [DOI] [PubMed] [Google Scholar]
- Schwiebert E.M., Benos,D.J., Egan,M.E., Stutts,M.J. and Guggino,W.B. (1999) CFTR is a conductance regulator as well as a chloride channel. Physiol. Rev., 79, S145–S166. [DOI] [PubMed] [Google Scholar]
- Smith T.K., Ikeda,Y., Fujii,J., Taniguchi,N. and Meister,A. (1995) Different sites of acivicin binding and inactivation of γ-glutamyl transpeptidases. Proc. Natl Acad. Sci. USA, 92, 2360–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutts M.J., Caneesa,C., Olsen,J., Hamrick,M., Cohn,J., Rossier,B., Rossier,R. and Boucher,B. (1995) CFTR as a cAMP-dependent regulator of sodium channels. Science, 269, 847–850. [DOI] [PubMed] [Google Scholar]
- Tabcharani J.A., Chang,X.B., Riordan,J.R. and Hanrahan,J.W. (1991) Phosphorylation-regulated Cl– channel in CHO cells stably expressing the cystic fibrosis gene. Nature, 352, 628–631. [DOI] [PubMed] [Google Scholar]
- Tabcharani J., Rommens,J., Hou,Y., Chang,X., Tsui,L., Riordan,J. and Hanrahan,J. (1993) Multi-ion pore behaviour in the CFTR chloride channel. Nature, 366, 79–82. [DOI] [PubMed] [Google Scholar]
- Tsumura T., Hazama,A., Miyoshi,T., Ueda,S. and Okada,Y. (1998) Activation of cAMP-dependent C1– currents in guinea-pig paneth cells without relevant evidence for CFTR expression. J. Physiol., 512, 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velsor L.W., van Heeckeren,A. and Day,B.J. (2001) Antioxidant imbalance in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am. J. Physiol. Lung Cell Mol. Physiol., 281, L31–L38. [DOI] [PubMed] [Google Scholar]
- Venglarik C.J., Singh,A.K. and Bridges,R.J. (1994) Comparison of -nitro versus -amino 4,4′-substituents of disulfonic stilbenes as chloride channel blockers. Mol. Cell. Biochem., 140, 137–146. [DOI] [PubMed] [Google Scholar]
- Vergani P., Nairn,A.C. and Gadsby,D.C. (2003) On the mechanism of MgATP-dependent gating of CFTR Cl– channels. J. Gen. Physiol., 121, 17–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S.R., Boag,A.H., Valdimarsson,G., Hipfner,D.R., Campling, B.G., Cole,S.P. and Deeley,R.G. (1998) Immunohistochemical detection of multidrug resistance protein in human lung cancer and normal lung. Clin. Cancer Res., 4, 2279–2289. [PubMed] [Google Scholar]
- Yamazaki J. and Hume,J.R. (1997) Inhibitory effects of glibenclamide on cystic fibrosis transmembrane regulator, swelling-activated and Ca2+-activated Cl– channels in mammalian cardiac myocytes. Circ. Res., 81, 101–109. [DOI] [PubMed] [Google Scholar]