

Abstract
Vertebrate cells are highly susceptible to infection by obligate intracellular parasites such as Toxoplasma gondii, yet the mechanism by which these microbes breach the confines of their target cell is poorly understood. While it is thought that Toxoplasma actively invades by secreting adhesive proteins from internal organelles called micronemes, no genetic evidence is available to support this contention. Here, we report successful disruption of M2AP, a microneme protein tightly associated with an adhesive protein called MIC2. M2AP knockout parasites were >80% impaired in host cell entry. This invasion defect was likely due to defective expression of MIC2, which partially accumulated in the parasite endoplasmic reticulum and Golgi. M2AP knockout parasites were also unable to rapidly secrete MIC2, an event that normally accompanies parasite attachment to a target cell. These findings indicate a critical role for the MIC2–M2AP protein complex in parasite invasion.
Keywords: adhesion/Apicomplexa/Golgi/knockout/parasite
Introduction
Toxoplasma gondii is a pathogenic protozoan capable of infecting a wide range of vertebrate hosts including humans and hence is one of the most successful parasites known. In immunocompromised individuals, T.gondii causes life-threatening encephalitis or pneumonia as an opportunistic infection frequently associated with AIDS (Luft and Remington, 1992). Congenitally infected individuals can also suffer severe cognitive and visual impairment (Martin, 2001). In addition to its impact as a human pathogen, this genetically tractable organism has recently gained attention as an important model for intracellular parasitism in general (Sinai and Joiner, 1997). Moreover, discoveries from T.gondii often serve as paradigms for other key pathogens within the phylum Apicomplexa, such as the malaria parasites, Plasmodium spp. However, despite its development as a model parasite, the molecular mechanisms that T.gondii uses to enter host cells remain poorly understood.
Unlike pathogenic bacteria, T.gondii is not known to produce cytopathic compounds or toxins that allow it to establish infection and exacerbate disease. However, T.gondii is highly adept at infecting and replicating in a variety of host cell types, which die from necrotic lysis when progeny tachyzoites (a rapidly dividing form responsible for acute disease) exit the cell after replicating for 24–48 h (Black and Boothroyd, 2000). Accordingly, lytic destruction of host cells is thought to be an important pathogenic mechanism in toxoplasmosis. Therefore, it is reasonable to expect that a detailed understanding of how T.gondii invades host cells will lead to new strategies for blocking infection at the level of host cell entry.
Toxoplasma gondii invasion is a remarkably rapid event (<30 s) that features three virtually contiguous steps: use of gliding motility to approach a host cell, tight binding of host receptors during attachment and active penetration into the host cell. Tachyzoite invasion is highly directional with attachment occurring at the parasite’s anterior or apical end. Immediately following apical attachment, the tachyzoite uses its actin–myosin-based motility machinery, a motor complex aptly positioned just under the parasite plasma membrane, to actively penetrate into the host cell (recently reviewed in Opitz and Soldati, 2002). As the parasite enters, it drags in the host plasma membrane (Suss-Toby et al., 1996; Mordue et al., 1999) to create a parasitophorous vacuole in which it replicates for the duration of its intracellular cycle. Video microscopy studies have shown that there is little or no delay between attachment and the onset of penetration (Morisaki et al., 1995; Hakansson et al., 1999; Black and Boothroyd, 2000). This seamless and rapid transition from attachment to penetration implies that the parasite expresses surface adhesive molecules that participate in both steps by concertedly interacting with host receptors and the actin–myosin machinery.
An emerging theme is that T.gondii and related parasites withhold adhesive proteins in secretory organelles, micronemes (MIC), until they are discharged onto the surface during parasite apical attachment to a host cell (Carruthers and Sibley, 1997; Garcia-Réguet et al., 2000; Rabenau et al., 2001). Micronemes release their contents at the parasite’s apical tip in response to elevated parasite intracellular calcium (Carruthers and Sibley, 1999; Carruthers et al., 1999a; Lovett et al., 2000), presumably through a signaling pathway that is activated upon initial contact with a target cell. Compounds that neutralize intracellular calcium or block other signaling components, such as kinases, strongly inhibit microneme secretion and markedly impair invasion, mainly by blocking attachment (Carruthers et al., 1999a). Furthermore, several microneme proteins exhibit a remarkable likeness to vertebrate adhesive proteins since they possess integrin-like A/I domains (MIC2), thrombospondin-like domains (MIC1 and MIC2), epidermal growth factor-like domains (MIC3 and MIC6–9) and lectin-like domains (MIC3 and MIC8) (Fourmaux et al., 1996; Wan et al., 1997; Donahue et al., 2000; Garcia-Réguet et al., 2000; Hehl et al., 2000; Reiss et al., 2001; Meissner et al., 2002a). In some cases, these MIC proteins have been shown to bind receptors on host cells (Carruthers et al., 1999a; Garcia-Réguet et al., 2000; Brecht et al., 2001). Additionally, several MIC proteins are membrane anchored and have a short cytoplasmic domain (C domain), making them candidates for connecting the actin–myosin machinery with a substrate for gliding or host receptors for penetration. Although these features seemingly suggest that microneme contents are important for invasion, antibodies to microneme proteins have failed to block T.gondii entry into host cells; the only exception being antibodies to apical membrane antigen 1 (AMA1), which reduce invasion by ∼40% (Hehl et al., 2000). Moreover, despite the availability of knockout strains impaired in expression of MIC1, MIC3, MIC4 or MIC6, there remains no genetic evidence that microneme proteins are necessary for T.gondii invasion (Reiss et al., 2001; Meissner et al., 2002a).
We now report that genetic disruption of the microneme protein M2AP, which is partnered with the adhesive protein MIC2, reduces expression and rapid secretion of MIC2 and significantly compromises tachyzoite invasion of human cells.
Results
Targeted disruption of M2AP
Repeated attempts to directly disrupt the gene encoding the adhesive protein MIC2 were unsuccessful (M.Lingnau and L.D.Sibley, unpublished data), suggesting that MIC2 is an essential gene. We recently discovered that MIC2 is permanently associated with another protein called MIC2-associated protein, M2AP (Figure 1A) (Rabenau et al., 2001). The MIC2–M2AP complex is formed <15 min after the proteins are synthesized in the rough endoplasmic reticulum (ER). The complex traffics through the parasite Golgi where a propeptide is removed from the N-terminus of M2AP before transport to the micronemes. When the parasite encounters a host cell, the MIC2–M2AP complex is mobilized onto the parasite’s apical surface where it presumably engages host receptors. During penetration, the complex redistributes backward towards the parasite’s posterior end where a parasite-derived protease called MPP1 is thought to release the complex in a soluble form by cleaving MIC2 near the base of its ectodomain (Carruthers et al., 2000b). This step reduces the affinity of MIC2 for host receptors (Carruthers et al., 1999a) and is presumably necessary to disengage receptors to allow the parasite to complete the invasion event (Soldati et al., 2001; Brossier et al., 2003).

Fig. 1. Targeted disruption and genetic complementation of M2AP. (A) The MIC2–M2AP complex. MIC2 is a type I membrane protein that anchors the complex to the microneme/plasma membrane. MIC2 consists of a C-terminal cytoplasmic (C) domain, a motif (M) domain comprising six tandem segments similar to the type I repeat of thrombospondin and an integrin-like I-domain (I). M2AP has an N-terminal propeptide, which is removed (small arrow) in the parasite Golgi, a central domain consisting of β-sheets (β) and a C-terminal domain comprised of random coil sequence (coil). An anonymous parasite proteinase, MPP1, cleaves (large arrow) MIC2 near or within the transmembrane anchor, releasing the complex from the parasite surface. (B) Schematic depiction of the M2AP knockout and complementation strategy. The knockout plasmid, pm2apKO, contains an HXGPRT expression cassette for drug selection and targeting sequences designed to mediate double-crossover homologous recombination at the M2AP locus of the parental strain, ΔHX, thereby replacing M2AP with HXGPRT in the knockout clone, m2apKO. For complementation, m2apKO was co-transfected with a 1:10 ratio of pCAT and pM2AP and, after selection, parasites expressing CAT alone (2G11) or CAT and M2AP (1C4) were cloned and analyzed. (C) Southern blot analysis of m2apKO and associated clones. Genomic DNA was digested with NcoI and NaeI and hybridized with probes for HXGPRT, M2AP and CAT, respectively. The HXGPRT probe (left panel) hybridized to the endogenous HXGPRT locus disrupted by an insertional mutation in ΔHX (Donald and Roos, 1998). An additional band corresponding to incorporated HXGPRT at the M2AP locus is seen in m2apKO, 2G11 and 1C4. The M2AP probe (middle panel) hybridized to two bands of the expected size from the M2AP locus in ΔHX, which was deleted in m2apKO and 2G11 and restored by incorporation of pM2AP (expressing the M2AP cDNA) in 1C4. The faint bands in the upper part of the middle panel are due to incomplete stripping of the HXGPRT probe. Hybridization with a CAT probe (right panel) revealed incorporation of pCAT in 2G11 and 1C4. (D) Western blot probed with anti-actin and anti-M2AP antibodies. M2AP expression was undetectable in m2apKO and 2G11 but restored in 1C4. (E) Phase contrast and indirect immunofluorescence images of intracellular tachyzoites showing M2AP in the micronemes of ΔHX, loss of expression in m2apKO and 2G11, and re-expression of micronemal M2AP in 1C4. Scale bar, 5 µm. (F) Growth rate of m2apKO was similar to ΔHX, 2G11 and 1C4, based on the number of parasites in vacuoles determined 26 h after inoculation in HFF. Data represent three independent experiments, each from counting six fields/clone at 600× total magnification.
Although M2AP itself does not have recognizable adhesive sequences, its close association with MIC2 suggests that it plays an accessory role in invasion. To test this hypothesis, we created a knockout plasmid, pm2apKO, which contains the selectable marker hypoxanthine phosphoribosyl transferase (HXGPRT) flanked on either side by ∼3 kb of targeting sequence derived from the M2AP locus (Figure 1B). pm2apKO was designed to precisely replace M2AP with HXGPRT in the parasite’s haploid genome by double-crossover homologous recombination. To allow selection for HXGPRT, pm2apKO was electroporated into a parasite strain (ΔHX) in which the HXGPRT locus was disrupted by partial gene deletion (Donald and Roos, 1998). After isolating a knockout clone, m2apKO, we genetically complemented this clone by re-introducing M2AP on a plasmid (pM2AP) containing the native M2AP promoter, which is important for proper expression (Soldati et al., 2001). A complementation clone (1C4) was isolated by co-transfecting pM2AP with a selectable plasmid, pCAT. To control for expression of the selectable marker chloramphenicol acetyl transferase (CAT), a clone (2G11) expressing CAT, but not M2AP, was also isolated.
Southern hybridizations revealed that pm2apKO correctly disrupted M2AP in m2apKO and that pM2AP integrated randomly into the genome of the complementation clone, 1C4 (Figure 1C). (M2AP probe hybridization to three bands in 1C4 indicates that this clone probably acquired two tandemly linked copies of pM2AP.) Western blotting (Figure 1D) showed loss of M2AP expression in m2apKO and 2G11 without affecting expression of parasite actin, a loading control. Re-expression of M2AP in 1C4 appeared to be at appropriate levels. Immuno fluorescence staining of intracellular tachyzoites confirmed the absence of M2AP expression in m2apKO and 2G11 and re-expression in the micronemes of 1C4 (Figure 1E). The loss of M2AP had no detectable effect on the rate of parasite replication (Figure 1F). Collectively, these results demonstrate successful modulated M2AP expression and that M2AP is not essential for in vitro propagation of T.gondii.
Disruption of M2AP causes partial retention of MIC2 in the ER/Golgi
Reiss et al. (2001) recently reported that disruption of MIC1 caused retention of a partner protein, MIC6, in the Golgi, suggesting that protein interactions are important for correct targeting to the micronemes of T.gondii. To test whether MIC2 transport to the micronemes was similarly dependent on M2AP, we stained the m2apKO and associated clones with antibodies to MIC2 and various other secretory proteins. As expected, ΔHX parasites exhibited apical co-localization of MIC2 with another known microneme protein, AMA1 (Figure 2A) (Donahue et al., 2000; Hehl et al., 2000). However, ∼48% of the m2apKO parasites showed mislocalization of MIC2 to a discrete site anterior to the nucleus, consistent with the location of the Golgi apparatus. A minority of m2apKO parasites (∼14%) showed perinuclear staining in the region of the ER or staining of both the Golgi and ER. The remaining parasites (∼38%) showed diffuse staining that was not exclusively confined to the Golgi, ER or micronemes and therefore likely represents accumulation within multiple compartments. The ER/Golgi retention phenotype appeared to be specific for MIC2 since, in addition to AMA1 (Figure 2A), MIC1, MIC4, MIC5, MIC6, MIC8 and MIC10 all showed normal localization to the micronemes (data not shown). While expression of CAT in 2G11 had no effect, re-expression of M2AP in 1C4 parasites reversed the retention phenotype and resulted in correct targeting of MIC2 to the micronemes. These results demonstrate that the retention phenotype was due directly to the absence of M2AP and not to a random mutation associated with the m2apKO clone or expression of CAT in 1C4. This retention phenotype was not restricted to replicating parasites since extracellular tachyzoites of m2apKO and 2G11 also showed MIC2 accumulation in the ER/Golgi (data not shown). Quantitative immunolabeling of ultrathin cryosections confirmed that MIC2 was significantly enriched (P = 0.0007, Mann–Whitney test) in the Golgi and depleted (P = 0.0005, Mann–Whitney test) in the micronemes of m2apKO parasites compared with ΔHX or 1C4 parasites (Figure 2C–E). Together, these results show that efficient trafficking of MIC2 to the micronemes is dependent on expression of M2AP.

Fig. 2. Disruption of M2AP causes MIC2 retention in the ER/Golgi. (A) Photomicrographs of intracellular tachyzoites showing co-localization of MIC2 and AMA1 in ΔHX micronemes, but retention of MIC2 in the Golgi (closed arrowhead) or ER (open arrowhead) of m2apKO and 2G11. M2AP re-expression in 1C4 restored MIC2 localization to the micronemes (bottom row). Scale bar, 5 µm. (B) Illustration of a tachyzoite indicating the approximate positions of sections shown in (C) including a longitudinal section of Golgi (box with broken line) and a cross-section through the apical region (broken line). (C) Colloidal gold immunolabeling of ultrathin sections revealed normal localization of MIC2 in the apical micronemes of ΔHX, but an abnormal abundance of MIC2 in the Golgi of m2apKO with correspondingly less MIC2 in the micronemes. Re-expression of M2AP in 1C4 restored normal localization of MIC2 to the micronemes. Scale bar, 0.2 µm. (D) Quantification of immunogold labeling confirmed significantly enhanced (P = 0.0005, Mann–Whitney test) labeling of MIC2 in the Golgi of m2apKO compared with ΔHX and 1C4. Twelve randomly chosen sections were quantified. (E) Quantification of 10 randomly chosen apical sections revealed significantly less (P = 0.0007, Mann–Whitney test) MIC2 in the micronemes of m2apKO compared with ΔHX and 1C4. Bars represent mean values in (D) and (E).
M2AP is necessary for proper expression and rapid secretion of MIC2
Few studies have examined the regulation of gene expression in T.gondii, and very little is known about gene regulatory networks in this parasite or about how the absence of one protein affects the expression levels of other interacting proteins. To test whether expression of MIC2 or other secretory proteins is altered in m2apKO, we probed western blots with antibodies to actin (loading control), GRA1 (a dense granule protein), AMA1, MIC4 or MIC2. While expression of other microneme and dense granule proteins was normal, m2apKO parasites appeared to express less MIC2 than ΔHX (Figure 3A). Re-expression of M2AP in 1C4 restored MIC2 expression to nearly the same level as ΔHX. Although 2G11 parasites appeared to express slightly less MIC2 than m2apKO parasites, it is not clear whether this is due directly to expression of CAT or if it is a natural consequence of clonal variation. Semi-quantification by western blotting revealed that m2apKO parasites express 25–50% less MIC2 compared with ΔHX (Figure 3B, left panel). Interestingly, a longer exposure revealed that, in addition to the 115 kDa major MIC2 species, m2apKO exclusively showed an additional ∼95 kDa minor band (Figure 3B, right panel). If this band is a degradation product, it could explain the lower abundance of MIC2 in m2apKO and is consistent with the possibility that MIC2 is misfolded in the absence of M2AP. Interestingly, domain-specific antibodies demonstrated that the 95 kDa product resulted from removal of the cytosolic C-terminal domain. This intriguingly mirrors MPP1 processing of MIC2 on the parasite surface (Carruthers et al., 2000b), which normally releases MIC2 into the surrounding medium as a 95–100 kDa soluble product. Thus, an alternative explanation for the presence of this processed MIC2 species in m2apKO cells is that a sub-population of MIC2 might abnormally encounter MPP1 within the parasite, presumably in a sorting compartment or a non-micronemal secretory vesicle. In support of the latter possibility, the 95 kDa MIC2 species is constitutively secreted by m2apKO parasites even when microneme secretion is completely blocked by chelating intracellular calcium (data not shown). Also, MIC2 was occasionally detected by immunoelectron microscopy in the dense granules of m2apKO, but not in ΔHX or 1C4 (data not shown). This implies that a small proportion of MIC2 in m2apKO is constitutively secreted through the dense granules, which is the default secretory pathway in T.gondii.

Fig. 3. MIC2 requires M2AP for proper expression and rapid secretion. (A) Western blot showing equal expression of actin, GRA1, AMA1 and MIC4 in all clones, but reduced expression of MIC2 in m2apKO and 2G11 compared with ΔHX and 1C4. (B) Short exposure (left panel) of a western blot loaded with 1.0, 0.75 and 0.5 relative amounts of ΔHX tachyzoite lysate compared with a 1.0 equivalent (normalized to actin) of m2apKO lysate. A longer exposure (right panel) of the same blot shows an additional ∼95 kDa MIC2 band (arrow) exclusively in m2apKO. Relative molecular masses are indicated in kDa. (C) Western blots of parasite lysates probed with an I-domain-specific mAb 6E9 (left panel) or a C-domain-specific antibody MαC-dom. (right panel, intentionally overexposed). Absence of reactivity with MαC-dom. suggests that the ∼95 kDa species is a degradation product resulting from proteolytic removal of the C domain. (D) Western blots of culture supernatants from constitutively secreting (Const./Basal Sec’n) tachyzoites or from tachyzoites induced for microneme secretion by ethanol (EtOH-Ind. Sec’n) or A23187 (A23187-Ind. Sec’n). While constitutive/basal secretion (top left panel) of MIC2 was not substantially affected by disruption of M2AP, induced secretion was severely impaired (top center and top right panels). Lower panels show the same blots probed with antibodies to GRA1 as a loading control.
Since less MIC2 reaches the micronemes of m2apKO, we predicted that rapid apical secretion of MIC2 is compromised in this clone. To test this, we immunoblotted culture supernatants from purified extracellular tachyzoites incubated at 37°C for 30 min in the absence of stimulation (constitutive secretion). In parallel, we briefly (2.5 min) induced secretion of micronemes with the calcium agonist ethanol (200 mM) (Carruthers et al., 1999b) or A23187 (200 nM) (Carruthers and Sibley, 1999). The relative amounts of non-stimulated release of MIC2 (Figure 3D, top left panel) were proportional to expression levels in the corresponding parasites (Figure 3A, top panel). Under these non-stimulatory conditions, MIC2 secretion from m2apKO and 2G11 parasites is presumably the sum of its constitutive release from the dense granules and basal secretion from the micronemes. In contrast, rapid micronemal secretion of MIC2 induced by either ethanol or A23187 was drastically reduced in m2apKO and 2G11 compared with ΔHX and 1C4 (Figure 3D). This effect was not due to sampling or loading errors since GRA1 secretion was equivalent in all clones. Therefore, m2apKO parasites are incapable of normally deploying MIC2 from the micronemes. This defect offered an opportunity to test the importance of the MIC2–M2AP complex in T.gondii invasion.
m2apKO parasites are defective in invasion
To test the ability of m2apKO parasites to invade host cells, we used a red/green invasion assay where external, attached parasites were stained red while internal, penetrated parasites were stained green (Figure 4A). Host cells were enumerated by staining nuclei blue with 4′,6 diamidino-2-phenylindole (DAPI). This assay can readily distinguish between an effect on attachment versus penetration. An effect on attachment is manifested by a reduction in both invaded (green) and attached (red) parasites, as exemplified by treatment with the calcium agonist BAPTA-AM, which blocks attachment by disabling microneme secretion (Figure 4B) (Carruthers et al., 1999a). A penetration defect is indicated by a reduction in invaded parasites and a large increase in attached parasites. This result is seen when parasites are treated with cytochalasin D, which cripples the actin-based machinery the parasite uses for motility and cell penetration (Dobrowolski and Sibley, 1996). Strikingly, disruption of M2AP caused an 80% reduction in invaded parasites (green) and a 50% reduction in attached parasites (red), making m2apKO one of the most severely invasion-defective T.gondii mutants isolated to date. This decline in invaded and attached parasites mirrors the effects of BAPTA-AM treatment, suggesting that m2apKO parasites are defective in attachment. Notably, 2G11 appeared to be even less invasive than m2apKO and this correlates directly with its lower expression of MIC2 relative to m2apKO. Re-expression of M2AP in 1C4 largely rescued the defect, but did not completely restore it to the same level as seen in ΔHX. This inability to fully regain invasion competence might be due, in part, to 1C4’s slightly less than normal expression of total MIC2 (Figure 3A) and micronemal MIC2 (Figure 2E).
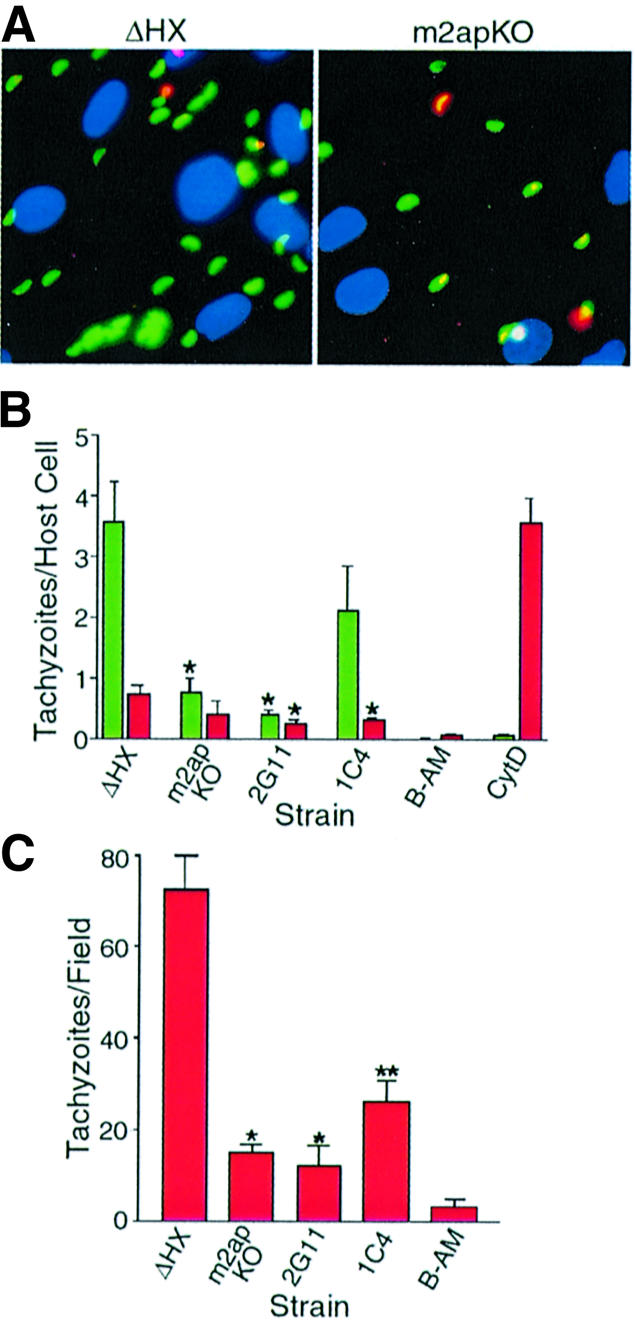
Fig. 4. Disruption of M2AP sharply reduces T.gondii attachment to host cells. (A) Representative fluorescence images of fibroblast monolayers infected with ΔHX and m2apKO parasites using a red/green invasion assay (see Materials and methods). External tachyzoites are stained red, internal tachyzoites are stained green and host nuclei are stained blue. (B) Quantification of invasion using the red/green assay. Red bars represent external, attached parasites while green bars represent internal, penetrated parasites. Asterisks indicate that attachment or penetration was significantly lower (P < 0.05, two-tailed Student’s t-test) than ΔHX. Tachyzoites treated with BAPTA-AM (B-AM) or cytochalasin D (CytD) were included as positive controls for defects in attachment or penetration, respectively. Data are mean values ± SEM of four independent experiments, counting six randomly selected fields for each sample. (C) Tachyzoite attachment to glutaraldehyde-fixed fibroblasts. A single asterisk indicates that attachment was significantly lower (P < 0.05, two-tailed Student’s t-test) than ΔHX and a double asterisk indicates that attachment was significantly higher than m2apKO (P < 0.05, two-tailed Student’s t-test). Data are mean values ± SEM of four independent experiments, counting six randomly selected fields for each sample.
To verify that disruption of M2AP impaired parasite attachment, we tested the ability of parasites to attach to host cells fixed with glutaraldehyde, which blocks parasite penetration by rigidifying the target cells (Grimwood et al., 1996). As expected, m2apKO and 2G11 parasites were unable to attach efficiently to fixed host cells, and 1C4 parasites significantly (P < 0.05) recovered attachment activity (Figure 4C). Collectively, these results demonstrate that the MIC2–M2AP complex participates in parasite attachment and is necessary for efficient invasion of host cells.
Discussion
Intracellular pathogens have evolved a variety of strategies for entering target cells. In most cases, the invading organism exploits the host’s endocytic or phagocytic pathways for entry. However, parasitic apicomplexans including T.gondii have devised a unique strategy in which active penetration is central to host cell entry. Active penetration is highly polarized and is initiated exclusively through the anterior or apical pole of the parasite. Recent studies have suggested that Toxoplasma accomplishes this polarized invasion by secreting adhesive protein complexes from micronemes, which are discharged from the apical tip of the parasite (Carruthers and Sibley, 1997, 1999; Garcia-Réguet et al., 2000). Micronemal adhesive protein complexes are thought to engage host receptors to form the moving junction during apical attachment (Soldati et al., 2001; Carruthers, 2002). Consistent with this notion, we found that disruption of the MIC2–M2AP adhesive complex markedly impaired parasite attachment to human fibroblasts. These findings offer important genetic support for the model that microneme proteins are key mediators of Toxoplasma attachment and entry into host cells.
Because disruption of M2AP markedly affected several properties of MIC2, it is unclear whether the attachment defect exhibited by m2apKO parasites was due directly to disruption of M2AP or indirectly to effects on MIC2. However, there are several indicators that MIC2 plays a more pivotal role in invasion than M2AP. For example, MIC2 is a member of the thrombospondin-related adhesive protein (TRAP) family, which is one of only a few secretory protein families that are phylogenetically conserved across the Apicomplexa. This conservation strongly suggests that MIC2 and other TRAP family proteins play a fundamental role in cell invasion by this group of parasites. Moreover, malaria parasites bearing a null mutation in TRAP are profoundly deficient in motility, invasion and virulence (Sultan et al., 1997; Kappe et al., 1999). Since MIC2 appears to be refractory to genetic disruption, it is presumably as important to Toxoplasma as TRAP is to malaria parasites. However, in contrast to TRAP, which is only expressed in the sporozoite stage of malaria, MIC2 is produced by all the invasive stages (tachyzoites, bradyzoites and sporozoites) that infect intermediate hosts such as humans. This broad expression pattern precludes knocking out the gene in a developmental stage in which the protein is not expressed, as was done for TRAP (Sultan et al., 1997; Kappe et al., 1999). Instead, directly testing the role of MIC2 in invasion will require using a conditional knockout strategy, such as the recently reported tetracycline repressible transactivator system (Meissner et al., 2002b). We are currently implementing this system to define directly the function of MIC2 in invasion.
Unlike MIC2, M2AP does not appear to have a counterpart in malaria parasites, based on searches of the nearly complete Plasmodium genome. M2AP is, however, conserved among parasites that are more closely related to Toxoplasma, arguing that it has a more specialized function than MIC2 (Rabenau et al., 2001). Although the precise role that M2AP plays in invasion remains elusive, our studies establish that M2AP is crucial for proper expression, localization and secretion of MIC2. These data indicate that the two proteins are inextricably dependent on one another for proper functioning in parasite adhesion.
m2apKO parasites were able to invade cells sufficiently well for recovery of the null mutant, indicating that M2AP is not an essential protein. The residual invasion activity of m2apKO parasites could be due to the presence of other micronemal adhesive protein complexes such as MIC1/4/6 and MIC3/8 (Reiss et al., 2001; Meissner et al., 2002a). MIC1 is a lactose-specific lectin that could mediate residual parasite invasion by binding to lactose-containing glycoproteins on the host cell surface (Lourenco et al., 2001). MIC3 possesses a lectin-like domain that is capable of binding unidentified receptors on host cells (Garcia-Réguet et al., 2000; Cerede et al., 2002) and therefore it could also contribute to host cell invasion in the absence of a functional MIC2–M2AP complex. Residual invasion could also be supported by the glycolipid-anchored surface antigens, such as SAG1, SAG2 and SAG3, which appear to participate in attachment (Grimwood and Smith, 1992, 1996; Dzierszinski et al., 2000). It is also conceivable that the modicum of MIC2 that reaches the surface of m2apKO parasites is capable of sustaining invasion at a low level. In this case, a lower density of MIC2 on the parasite surface would presumably result in less efficient binding, making this a rate-limiting step in invasion. Indeed, m2apKO parasites appear to eventually (over the course of several hours) invade host cells to nearly the same levels as parental parasites and thus they appear to have a delayed invasion phenotype. It will be interesting to determine whether this phenomenon is unique to m2apKO or is shared by other mutants that are partially defective in invasion, such as SAG3 knockout clones (Dzierszinski et al., 2000).
There are several possible explanations of why disruption of the MIC2–M2AP complex substantially impaired invasion whereas disruption of MIC1/4/6 or MIC3/8 failed to affect cell entry (Reiss et al., 2001; Meissner et al., 2002a). The MIC2–M2AP complex might bind to a key receptor on host cells, while the other adhesive protein complexes might recognize supplemental receptors. Indeed, our recent findings (J.M.Harper, E.F.Hoff and V.B.Carruthers, manuscript in preparation) suggest that the I domain of MIC2 binds glycosaminoglycans on heparan sulfate proteoglycans (HSPGs), which are known receptors for T.gondii invasion (Ortega-Barria and Boothroyd, 1999; Carruthers et al., 2000a). Alterna tively, the MIC1/4/6 and MIC3/8 complexes could be necessary for invading specific cell types other than those that have been tested thus far. This possibility is consistent with the idea that T.gondii has evolved multiple ligands to ensure that it is capable of invading many different cell types, each possessing at least one receptor recognized by a ligand in its repertoire. Finally, the MIC1/4/6 and MIC3/8 adhesive complexes might be functionally redundant, particularly if they recognize the same receptor. This possibility warrants further investigation since MIC1 is a lactose-binding lectin and MIC3 and MIC8 both possess lectin-like domains (Fourmaux et al., 1996; Garcia-Réguet et al., 2000; Meissner et al., 2002a). In this case, disruption of one or the other complex would not affect invasion whereas a double knockout mutant might be defective in invasion.
MIC2 retention in early secretory compartments (ER and Golgi) of m2apKO parasites implies that Toxoplasma possesses quality control (QC) systems for identifying incorrectly assembled protein complexes in the secretory system. Since individual proteins in multi-protein complexes are often dependent on each other for proper folding, it is also possible that MIC2 is misfolded in the absence of M2AP. The presence of QC systems in the secretory pathway of Toxoplasma is further supported by an earlier study that demonstrated an accumulation of MIC6 in the ER and Golgi of parasites lacking MIC1 (Reiss et al., 2001). In most eukaryotic organisms, the ER is the primary site for QC in the secretory system where recognition of hydrophobic patches on partially folded or misfolded proteins is mediated by the molecular chaperone BiP/GRP78 (Blond-Elguindi et al., 1993). Consistent with this notion, a recent study showed that T.gondii BiP possesses the ER retention motif, HDEL, at its C-terminus. Thus, it is possible that MIC2 is withheld in the ER by interaction with BiP, which could recognize misfolded MIC2 or hydrophobic patches on MIC2 that might normally form the dimer interface with M2AP. Alternatively, like yeast, Toxoplasma could rely heavily on a Golgi QC system involving Rer1p, which recognizes polar amino acid residues in the transmembrane domain(s) of improperly folded membrane proteins (Arvan et al., 2002). Our finding that the majority of m2apKO parasites showed accumulation of MIC2 in the Golgi apparatus supports this contention.
In conclusion, the present report provides the first strong genetic evidence that at least one microneme protein complex plays a crucial role in attachment and invasion of host cells by T.gondii. The design and implementation of novel strategies of interfering with MIC2–M2AP- mediated adhesion may create important new treatment options for controlling toxoplasmosis based on impairing parasite invasion of host cells.
Materials and methods
Plasmid constructs
pm2apKO was constructed by inserting 5′ and 3′ sequences that flank M2AP into pminiHXGPRT (Donald and Roos, 1998). The M2AP 5′ flank (3009 bp) was PCR amplified from cosmid cB6-23 (provided by J.Ajioka, Cambridge University, UK) using primers M2AP.-3009.ApaI.F (5′-GATCGGGCCCAACAGAGCTCTCCTAGC-3′) and M2AP-1.ApaI.R (5′-GATCGGGCCCGTTGGGAGAGTGACGTGAG-3′). (Note: the number in each primer name corresponds to the position of the distal nucleotide in the primer relative to the adenosine in the start codon of M2AP, which is designated 1.) The M2AP 3′ flank (2860 bp) was PCR amplified using primers M2AP.1834.BamHI.F (5′-GATCGGATCCAGATTCGCGACGCACCTG-3′) and M2AP.4688.NotI.R (5′-GATCGCGGCCGCAAGCACAACTCTGGGCGT-3′). PCR products were cloned into pminiHXGPRT using homologous restriction sites and standard cloning procedures.
pM2AP was created by replacing the HXGPRT expression cassette in pm2apKO with the M2AP ORF, which was PCR amplified from an RH strain cDNA library (V.B.Carruthers, unpublished data) using primers M2AP.+3.NsiI.F (5′-GATCATGCATAAACTCGCTGCCGTGTCC-3′) and M2AP.933.Pac1.R (5′-GATCTTAATTAAGCCTCATCGTCACT-3′). The amplified M2AP ORF translates into the same product as wild-type M2AP except for a lysine to histidine substitution at amino acid position 2 introduced as part of the NsiI restriction site. pCAT is equivalent to pTUB5CATSag1, kindly provided by Dominique Soldati (Imperial College, London, UK). All plasmids were verified by restriction digests and sequencing. Preparative scale purifications were performed using a Qiagen Mega plasmid kit according to the manufacturer’s instructions.
Parasites, transfection and selection
All parasites were cultured by passage in primary human foreskin fibroblasts (HFF). ΔHX (equivalent to RHhxgprt–; Donald and Roos, 1998) parasites were purified by membrane filtration, washed and resuspended in cytomix buffer (2 mM EDTA, 120 mM KCl, 0.15 mM CaCl2, 10 mM K2HPO4/KH2PO4, 25 mM HEPES, 5 mM MgCl2·6H2O; pH 7.6). Parasites (3.5 × 107) were electroporated with 25 µg NotI-linearized pm2apKO using a Bio-Rad GenePulser II (settings 1.5 kV, 25 µF, no resistance). After overnight growth, transformants were selected with 25 µg/ml mycophenolic acid and 50 µg/ml xanthine for one passage before cloning by limiting dilution under drug selection. Ninety-six clones were picked and expanded in a 96-well plate of HFF. When parasites emerged, 40% of each culture was transferred to a new 96-well plate of HFF and the remaining parasites were washed three times with Dulbecco’s modified Eagle’s medium (DMEM) + 10 mM HEPES and lysed in 100 µl of 100°C SDS–PAGE running buffer. Lysates were immobilized on nitrocellulose membranes in duplicate using a Bio-Rad Dot Blot apparatus. Dot blots were probed with antibodies to M2AP (Affipure RabbitαrM2AP; Rabenau et al., 2001) and MIC4 (RabbitαrMIC4; Brecht et al., 2001). Candidate knockout clones that were positive for MIC4 and negative for M2AP were expanded and tested by immunofluorescence (see below) for M2AP expression.
m2apKO was genetically complemented by co-transfection with 100 µg of pM2AP and 10 µg of pTUB5CATSag1. A drug-resistant population of transformants was selected using 20 µM chloramphenicol and parasite clones were isolated by limiting dilution as described above. Clones were analyzed by western blotting and immunofluorescence for MIC2 expression. Clone 1C4 was selected for phenotypic analysis because it appeared to express normal levels of MIC2. Since MIC2 expression was stable in the absence of drug selection, 1C4 was routinely cultured without chloramphenicol.
Western blotting and Southern blotting
Western blotting was performed as described previously (Wan et al., 1997). Southern blotting was performed essentially as described (Hoff et al., 2001) except for the following. The HXGPRT probe was PCR amplified from pminiHXGPRT using primers HXG.661.F (5′-GAGAACTTACTTCGGCGAG-3′) and HXG.978.R (5′-ATCGACTTCGGACCGACG-3′), the M2AP probe from pM2AP using M2AP.527.F (5′-CTAGCGACGGGTGGAAAGTGA-3′) and M2AP.879.R (5′-CGAATCTGGGTTCTCACCATC-3′), and the CAT probe from pCAT using CAT.1225.F (5′-CCGTTGATATATCCCAATCG-3′) and CAT1591.R (5′-GGCAGGTTTTCACCGTAAC-3′). Genomic DNA was digested with NcoI and NaeI. The M2AP probe contains a single NaeI site, whereas the HXGPRT and CAT probes do not contain NcoI or NaeI sites.
Immunolocalization experiments
Indirect immunofluorescence assays were performed on intracellular tachyzoites grown overnight in HFF cells in eight-well chamber slides (Nunc). Infected monolayers were washed in PBS and fixed in 4% formaldehyde/0.02% glutaraldehyde for 20 min. After three washes with PBS, monolayers were permeabilized with 0.1% Triton X-100/PBS for 10 min, blocked with 10% fetal bovine serum (FBS) for 30 min, incubated with primary antibody (Affipure RabbitαrM2AP, mAb 6D10 versus MIC2 or UVT59 versus AMA1) diluted in 1% FBS/1% normal goat serum (NGS)/PBS for 1 h, washed five times with PBS and incubated with secondary antibody (Oregon Green goat anti-mouse, Oregon Green goat anti-rabbit, Texas Red goat anti-mouse or Texas Red goat anti-rabbit) for 1 h. After five washes with PBS, slides were mounted in Mowiol and viewed by phase contrast and epifluorescence using a Nikon Eclipse E800 equipped with an RT Spot Slider CCD camera. Images were digitally refined, assembled and annotated using Photoshop (Adobe).
For immunolocalization at the electron microscopy level, infected cells were fixed in 4% paraformaldehyde/0.25% glutaraldehyde in 100 mM PIPES/0.5 mM MgCl2 pH 7.2 for 1 h at 4°C. Samples were then embedded in 10% gelatin and infiltrated overnight with 2.3 M sucrose/20% polyvinyl pyrrolidone in PIPES/MgCl2 at 4°C. Samples were trimmed, frozen in liquid nitrogen and sectioned with a RMC MT7/CR21 cryo-ultramicrotome. Sections (70 nm) were blocked with 5% FBS/5% NGS for 30 min and subsequently incubated with rabbit anti-MIC2 (C domain) antibody for 1 h. After washing with block buffer, samples were probed with 18 nm colloidal gold-conjugated anti-rabbit secondary antibody for 1 h. Parallel controls omitting the primary antibody were consistently negative at the concentration of colloidal gold-conjugated secondary antibodies used in these studies. Sections were washed in PIPES buffer followed by a water rinse, and stained with 0.3% uranyl acetate/2% polyvinyl alcohol. Samples were viewed with a JEOL 1200EX transmission electron microscope.
Secretion assays
Secretion assays were performed by prewarming to 37°C 100 µl of filter-purified tachyzoites (1 × 108/ml in invasion medium: DMEM/20 mM HEPES/3% FBS) in a 96-well plate for 0.5 min followed by addition of 11 µl of invasion medium and 30 min incubation for constitutive secretion or addition of 11 µl of 2 M ethanol (final concentration 200 mM) or 2 µM A23187 (final concentration 200 nM) and 2 min incubation for induced secretion. Secretion was arrested by placing the plate on ice for 5 min and culture supernatants were collected after removing parasites by centrifugation (1000 g, 3 min, 4°C, twice). A 0.25 vol of 5× SDS– PAGE sample buffer was added to culture supernatants and samples were boiled for 3 min then loaded on SDS–PAGE gels and western blotted as described above.
Invasion and attachment assays
Red/green invasion assays were performed as described for indirect immunofluorescence with the following changes. Monolayers were infected with 1 × 107 parasites/chamber for 15 min, fixed, and external (attached) parasites were stained with RabbitαP30 (SAG1) before Triton X-100 permeabilizeation and detection of internal (invaded) parasites with mAb G11-9 (Argene), which recognizes SAG1. Secondary antibodies were Oregon Green goat anti-mouse (Molecular Probes) and Texas Red goat anti-rabbit (Molecular Probes). DAPI (5 µg/ml; Sigma) was added to the secondary antibody solution to stain host nuclei. Data were compiled from four independent experiments, each from counting six fields/clone at 600× total magnification. Fields were randomly selected (operator moved the microscope stage without viewing the sample) in the same pattern for all samples and cell counts were made in a blinded fashion.
The attachment assay was performed as described previously, except that parasites (1 × 107/chamber) were allowed to attach for 15 min and attached parasites were observed by calcein-AM fluorescence (parasites preloaded by treatment with 1 µM calcein-AM, 30 min).
Replication/growth rate assay
Parasites were harvested from one T25 as per the invasion assay with the exception that HFF monolayers were inoculated with 1.25 × 105 parasites/chamber of an eight-well chamber slide and allowed to invade and replicate for 26 h. The slides were then fixed, permeabilized, immunolabeled with mAb G11-9 (SAG1) and Oregon Green goat anti-mouse secondary antibody, and the numbers of vacuoles containing 1, 2, 4, 8 or 16 parasites/vacuole were enumerated.
Statistical analyses
Statistical analysis was performed using Prism software for a Mann–Whitney test of the immunoelectron microscopy data and a two-sided Student’s t-test for the invasion data. Differences were considered significant if P values were <0.05.
Acknowledgments
Acknowledgements
We thank Gale Sherman and Jeff Diffenderfor for technical support, Gary Ward, Marie-France Cesbron-Delauw, Lloyd Kasper and Dominique Soldati for providing antibodies, James Ajioka for providing cosmid clones containing M2AP, Dominique Soldati for many helpful discussions, Nirbhay Kumar for critically reading this paper prior to submission, and all members of the Sibley and Carruthers labs for their comments and suggestions for this work. We also gratefully thank Mr Ted Hanf for his generous support. This study was supported in part by grants from the National Institutes of Health (AI34036 to L.D.S. and AI46675 to V.B.C.). L.D.S. is the recipient of a Scholar Award in Molecular Parasitology from the Burroughs Wellcome Fund. V.B.C. is a Burroughs Wellcome Fund New Investigator in Molecular Parasitology.
References
- Arvan P., Zhao,X., Ramos-Castaneda,J. and Chang,A. (2002) Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic, 3, 771–780. [DOI] [PubMed] [Google Scholar]
- Black M.W. and Boothroyd,J.C. (2000) Lytic cycle of Toxoplasma gondii. Microbiol. Mol. Biol. Rev., 64, 607–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blond-Elguindi S., Cwirla,S.E., Dower,W.J., Lipshutz,R.J., Sprang,S.R., Sambrook,J.F. and Gething,M.J. (1993) Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell, 75, 717–728. [DOI] [PubMed] [Google Scholar]
- Brecht S., Carruthers,V.B., Ferguson,D.J., Giddings,O.K., Wang,G., Jaekle,U., Harper,J.M., Sibley,L.D. and Soldati,D. (2001) The Toxoplasma micronemal protein MIC4 is an adhesin composed of six conserved apple domains. J. Biol. Chem., 276, 4119–4127. [DOI] [PubMed] [Google Scholar]
- Brossier J., Jewett,T.J., Lovett,J.L. and Sibley,L.D. (2003) C-terminal processing of the Toxoplasma protein MIC2 is essential for invasion of host cells. J. Biol. Chem., 278, 6229–6234. [DOI] [PubMed] [Google Scholar]
- Carruthers V.B. (2002) Host cell invasion by the opportunistic pathogen Toxoplasma gondii. Acta Trop., 81, 111–122. [DOI] [PubMed] [Google Scholar]
- Carruthers V.B. and Sibley,L.D. (1997) Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur. J. Cell Biol., 73, 114–123. [PubMed] [Google Scholar]
- Carruthers V.B. and Sibley,L.D. (1999) Mobilization of intracellular calcium stimulates microneme discharge in Toxoplasma gondii. Mol. Microbiol., 31, 421–428. [DOI] [PubMed] [Google Scholar]
- Carruthers V.B., Giddings,O.K. and Sibley,L.D. (1999a) Secretion of micronemal proteins is associated with Toxoplasma invasion of host cells. Cell. Microbiol., 1, 225–235. [DOI] [PubMed] [Google Scholar]
- Carruthers V.B., Moreno,S.N.J. and Sibley,L.D. (1999b) Ethanol and acetaldehyde elevate intracellular calcium and stimulate microneme discharge in Toxoplasma gondii. Biochem. J., 342, 379–386. [PMC free article] [PubMed] [Google Scholar]
- Carruthers V.B., Hakansson,S., Giddings,O.K. and Sibley,L.D. (2000a) Toxoplasma gondii uses sulfated proteoglycans for substrate and host cell attachment. Infect. Immun., 68, 4005–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers V.B., Sherman,G.D. and Sibley,L.D. (2000b) The Toxoplasma adhesive protein MIC2 is proteolytically processed at multiple sites by two parasite-derived proteases. J. Biol. Chem., 275, 14346–14353. [DOI] [PubMed] [Google Scholar]
- Cerede O., Dubremetz,J.F., Bout,D. and Lebrun,M. (2002) The Toxoplasma gondii protein MIC3 requires pro-peptide cleavage and dimerization to function as adhesin. EMBO J., 21, 2526–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski J.M. and Sibley,L.D. (1996) Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell, 84, 933–939. [DOI] [PubMed] [Google Scholar]
- Donahue C.G., Carruthers,V.B., Gilk,S.D. and Ward,G.E. (2000) The Toxoplasma homolog of Plasmodium apical membrane antigen-1 (AMA-1) is a microneme protein secreted in response to elevated intracellular calcium levels. Mol. Biochem. Parasitol., 111, 15–30. [DOI] [PubMed] [Google Scholar]
- Donald R.G. and Roos,D.S. (1998) Gene knock-outs and allelic replacements in Toxoplasma gondii: HXGPRT as a selectable marker for hit-and-run mutagenesis. Mol. Biochem. Parasitol., 91, 295–305. [DOI] [PubMed] [Google Scholar]
- Dzierszinski F., Mortuaire,M., Cesbron-Delauw,M.F. and Tomavo,S. (2000) Targeted disruption of the glycosylphosphatidylinositol-anchored surface antigen SAG3 gene in Toxoplasma gondii decreases host cell adhesion and drastically reduces virulence in mice. Mol. Microbiol., 37, 574–582. [DOI] [PubMed] [Google Scholar]
- Fourmaux M.N., Achbarou,A., Mercereau-Puijalon,O., Biderre,C., Briche,I., Loyens,A., Odberg-Ferragut,C., Camus,D. and Dubremetz,J.F. (1996) The MIC1 microneme protein of Toxoplasma gondii contains a duplicated receptor-like domain and binds to host cell surface. Mol. Biochem. Parasitol., 83, 201–210. [DOI] [PubMed] [Google Scholar]
- Garcia-Réguet N., Lebrun,M., Fourmaux,M.-N., Mercereau-Puijalon,O., Mann,T., Beckers,C.J.M., Samyn,B., Van Beeumen,J., Bout,D. and Dubremetz,J.-F. (2000) The microneme protein MIC3 of Toxoplasma gondii is a secretory adhesin that binds to both the surface of the host cells and the surface of the parasite. Cell. Microbiol., 2, 353–364. [DOI] [PubMed] [Google Scholar]
- Grimwood J. and Smith,J.E. (1992) Toxoplasma gondii: the role of a 30-kDa surface protein in host cell invasion. Exp. Parasitol., 74, 106–111. [DOI] [PubMed] [Google Scholar]
- Grimwood J. and Smith,J.E. (1996) Toxoplasma gondii: the role of parasite surface and secreted proteins in host cell invasion. Int. J. Parasitol., 26, 169–173. [DOI] [PubMed] [Google Scholar]
- Grimwood J., Mineo,J.R. and Kasper,L.H. (1996) Attachment of Toxoplasma gondii to host cells is host cell cycle dependent. Infect. Immun., 64, 4099–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakansson S., Morisaki,H., Heuser,J. and Sibley,L.D. (1999) Time-lapse video microscopy of gliding motility in Toxoplasma gondii reveals a novel, biphasic mechanism of cell locomotion. Mol. Biol. Cell, 10, 3539–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hehl A.B., Lekutis,C., Grigg,M.E., Bradley,P.J., Dubremetz,J.F., Ortega-Barria,E. and Boothroyd,J.C. (2000) Toxoplasma gondii homologue of Plasmodium apical membrane antigen 1 is involved in invasion of host cells. Infect. Immun., 68, 7078–7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoff E.F., Cook,S.H., Sherman,G.D., Harper,J.M., Ferguson,D.J., Dubremetz,J.F. and Carruthers,V.B. (2001) Toxoplasma gondii: molecular cloning and characterization of a novel 18-kDa secretory antigen, TgMIC10. Exp. Parasitol., 97, 77–88. [DOI] [PubMed] [Google Scholar]
- Kappe S., Bruderer,T., Gantt,S., Fujioka,H., Nussenzweig,V. and Menard,R. (1999) Conservation of a gliding motility and cell invasion machinery in Apicomplexan parasites. J. Cell Biol., 147, 937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenco E.V., Pereira,S.R., Faca,V.M., Coelho-Castelo,A.A., Mineo,J.R., Roque-Barreira,M.C., Greene,L.J. and Panunto-Castelo,A. (2001) Toxoplasma gondii micronemal protein MIC1 is a lactose-binding lectin. Glycobiology, 11, 541–547. [DOI] [PubMed] [Google Scholar]
- Lovett J.L., Howe,D.K. and Sibley,L.D. (2000) Molecular characteriz ation of a thrombospondin-related anonymous protein homologue in Neospora caninum. Mol. Biochem. Parasitol., 107, 33–43. [DOI] [PubMed] [Google Scholar]
- Luft B.J. and Remington,J.S. (1992) Toxoplasmic encephalitis in AIDS. Clin. Infect. Dis., 15, 211–222. [DOI] [PubMed] [Google Scholar]
- Martin S. (2001) Congenital toxoplasmosis. Neonatal Netw., 20, 23–30. [DOI] [PubMed] [Google Scholar]
- Meissner M., Reiss,M., Viebig,N., Carruthers,V., Toursel,C., Tomavo,S., Ajioka,J. and Soldati,D. (2002a) A family of transmembrane microneme proteins of Toxoplasma gondii contain EGF-like domains and function as escorters. J. Cell Sci., 115, 563–574. [DOI] [PubMed] [Google Scholar]
- Meissner M., Schluter,D. and Soldati,D. (2002b) Role of Toxoplasma gondii myosin A in powering parasite gliding and host cell invasion. Science, 298, 837–840. [DOI] [PubMed] [Google Scholar]
- Mordue D.G., Desai,N., Dustin,M. and Sibley,L.D. (1999) Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. J. Exp. Med., 190, 1783–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki J.H., Heuser,J.E. and Sibley,L.D. (1995) Invasion of Toxoplasma gondii occurs by active penetration of the host cell. J. Cell Sci., 108, 2457–2464. [DOI] [PubMed] [Google Scholar]
- Opitz C. and Soldati,D. (2002) ‘The glideosome’: a dynamic complex powering gliding motion and host cell invasion by Toxoplasma gondii. Mol. Microbiol., 45, 597–604. [DOI] [PubMed] [Google Scholar]
- Ortega-Barria E. and Boothroyd,J.C. (1999) A Toxoplasma lectin-like activity specific for sulfated polysaccharides is involved in host cell infection. J. Biol. Chem., 274, 1267–1276. [DOI] [PubMed] [Google Scholar]
- Rabenau K.E., Sohrabi,A., Tripathy,A., Reitter,C., Ajioka,J.W., Tomley,F.M. and Carruthers,V.B. (2001) TgM2AP participates in Toxoplasma gondii invasion of host cells and is tightly associated with the adhesive protein TgMIC2. Mol. Microbiol., 41, 1–12. [DOI] [PubMed] [Google Scholar]
- Reiss M., Viebig,N., Brecht,S., Fourmaux,M.N., Soete,M., Di Cristina,M., Dubremetz,J.F. and Soldati,D. (2001) Identification and characteriz ation of an escorter for two secretory adhesins in Toxoplasma gondii. J. Cell Biol., 152, 563–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinai A.P. and Joiner,K.A. (1997) Safe haven: the cell biology of nonfusogenic pathogen vacuoles. Annu. Rev. Microbiol., 51, 415–462. [DOI] [PubMed] [Google Scholar]
- Soldati D., Dubremetz,J.F. and Lebrun,M. (2001) Microneme proteins: structural and functional requirements to promote adhesion and invasion by the Apicomplexan parasite Toxoplasma gondii. Int. J. Parasitol., 31, 1293–1302. [DOI] [PubMed] [Google Scholar]
- Sultan A.A., Thathy,V., Frevert,U., Robson,K.J., Crisanti,A., Nussenzweig,V., Nussenzweig,R.S. and Menard,R. (1997) TRAP is necessary for gliding motility and infectivity of Plasmodium sporozoites. Cell, 90, 511–522. [DOI] [PubMed] [Google Scholar]
- Suss-Toby E., Zimmerberg,J. and Ward,G.E. (1996) Toxoplasma invasion: the parasitophorous vacuole is formed from host cell plasma membrane and pinches off via a fission pore. Proc. Natl Acad. Sci. USA, 93, 8413–8418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan K.L., Carruthers,V.B., Sibley,L.D. and Ajioka,J.W. (1997) Molecular characterization of an expressed sequence tag locus of Toxoplasma gondii encoding the micronemal protein MIC2. Mol. Biochem. Parasitol., 84, 203–214. [DOI] [PubMed] [Google Scholar]