

Abstract
Recent studies have identified the white adipose tissue (WAT) as an important endocrine organ that regulates energy and glucose metabolism via a number of secreted factors. Mice lacking acyl CoA:diacylglycerol acyltransferase 1 (DGAT1), a key enzyme in mammalian triglyceride synthesis, are protected against diet-induced obesity and glucose intolerance because of increased energy expenditure and enhanced insulin sensitivity. Because DGAT1 is highly expressed in WAT, we hypothesized that DGAT1 deficiency affects the expression of adipocyte-derived factors that regulate energy and glucose metabolism. Here we show that the transplantation of DGAT1-deficient WAT decreases adiposity and enhances glucose disposal in wild-type mice. Analysis of DGAT1-deficient WAT revealed a twofold increase in the expression of adiponectin, a molecule that enhances fatty acid oxidation and insulin sensitivity, and this increase may account in part for the transplantation-induced metabolic changes. Our results highlight the importance of the endocrine function of WAT and suggest that an alteration in this function contributes to the increased energy expenditure and insulin sensitivity in DGAT1-deficient mice.
Introduction
Recent studies have altered conceptions of the adipose tissue as an inert tissue that serves merely to store energy. Instead, it plays a vital role in regulating energy and glucose homeostasis (1, 2), in part by secreting factors such as leptin (3), adiponectin (4, 5), TNF-α (6), and resistin (7) that modulate the storage, mobilization, and utilization of lipids and glucose. The importance of adipose tissue in normal physiology is demonstrated by the insulin resistance and abnormal lipid metabolism in mice that lack fat (8, 9) or that do not secrete adipocyte-derived factors such as leptin (3). In addition, the transplantation of normal adipose tissue into A-ZIP/F1 fatless (lipoatrophic) mice improves their metabolic function (10), in part by restoring the expression of adipocyte-derived factors.
A number of genetic alterations in mice result in a leanness phenotype, and several of these models also have increased insulin sensitivity (reviewed in ref. 11). One such model is mice that lack acyl CoA:diacylglycerol acyltransferase 1 (DGAT1) (12), one of two known enzymes that catalyze the final step in mammalian triglyceride synthesis (13, 14). DGAT1-deficient (Dgat1–/–) mice synthesize triglycerides through the actions of DGAT2 (15) and possibly other mechanisms (14). However, these mice are protected against diet-induced obesity and insulin resistance (12) because of increased energy expenditure (12) and enhanced insulin sensitivity (16). Because DGAT1 is highly expressed in the white adipose tissue (WAT) (13), we hypothesized that DGAT1 deficiency affects the endocrine function of WAT and alters the secretion of adipocyte-derived factors that act systemically to increase energy expenditure and insulin sensitivity.
To test this hypothesis, we transplanted WAT from Dgat1–/– mice into mice prone to obesity and insulin resistance and assessed its effects on glucose disposal and the response to a high-fat diet. We also examined the expression of several adipocyte-derived factors that regulate energy and glucose metabolism in Dgat1–/– WAT. Our results indicate that DGAT1 deficiency alters the endocrine function of WAT and suggest that this alteration contributes to the increased energy expenditure and insulin sensitivity in Dgat1–/– mice.
Methods
Mice.
Dgat1–/– mice (∼99% C57BL/6, 1% 129/SvJae background) were generated previously (12). Wild-type (Dgat1+/+), Agouti yellow (AY/a), and heterozygous leptin-deficient (ob/+) mice (all on the C57BL/6 background) were from The Jackson Laboratory (Bar Harbor, Maine, USA). AY/a mice are insulin- and leptin-resistant (17), reflecting the antagonism of melanocortin receptors (18), whereas leptin-deficient (ob/ob) mice are obese and diabetic because of leptin deficiency (3). Genotyping was performed as previously described (12, 19). Mice were housed in a pathogen-free barrier facility (12-hour light/12-hour dark cycle) and fed rodent chow (Ralston Purina Co., St. Louis, Missouri, USA). For high-fat diet experiments, mice were fed a Western-type diet (21% fat by weight; Harlan Teklad, Madison, Wisconsin, USA). All experiments were approved by the Committee on Animal Research of the University of California, San Francisco.
Fat transplantation.
Fat transplantation was performed as previously described (10). Donor and recipient mice were age- and sex-matched to minimize rejection. Reproductive fat pad (500 mg) from the donor was inserted into the subcutaneous space on the back of the recipient. The amount of WAT transplanted represented about 30% of the total fat pad weight in chow-fed C57BL/6 mice and less than 10% of the total fat pad weight in AY/a and ob/ob mice. At the end of experiments, the grafts were evaluated visually and fixed for histological analysis.
Fat pad measurement.
Unless stated otherwise, reproductive, inguinal, mesenteric, and perirenal fat pads were isolated and weighed 2 weeks after transplantation for chow experiments and 10 weeks after transplantation for high-fat experiments.
Serum protein measurements.
Leptin and TNF-α levels were measured with enzyme immunoassay kits (R&D Systems Inc., Minneapolis, Minnesota, USA). Adiponectin levels were measured by AniLytics (Gaithersburg, Maryland, USA).
Triglycerides, protein, and lean body mass measurements.
For mice fed a high-fat diet for 5–6 weeks, total body triglycerides and muscle triglycerides were measured as previously described (12). Muscle samples comprised the quadriceps, soleus, and gastrocnemius muscles from the hind legs. Total body protein was measured with a colorimetric kit (Bio-Rad Laboratories Inc., Hercules, California, USA). For mice fed a high-fat diet for 12 weeks, body triglycerides and lean body mass were measured by dual-energy x-ray absorptiometry (GE Medical Systems; General Electric Co., Waukesha, Wisconsin, USA).
Energy metabolism studies.
Food consumption and energy expenditure were measured in metabolic chambers as previously described (12). Experiments were performed after 12 weeks of high-fat feeding.
Glucose metabolism studies.
Glucose (1 g/kg body weight) or bovine insulin (1 U/kg body weight; Sigma Chemical Co., St. Louis, Missouri, USA) was injected intraperitoneally 1 hour after fasting, and glucose concentrations were measured with a glucometer (Accu-chek; Roche Diagnostics Corp., Indianapolis, Indiana, USA). Experiments were performed 1 week after transplantation.
Real-time PCR.
Real-time PCR was performed as previously described (20). Primer and probe sequences (Table 1) were selected with Primer Express (Perkin-Elmer Applied Biosystems, Foster City, California, USA).
Table 1.
Real-time PCR primer and probe sequences

Leptin administration.
Mice were infused with recombinant murine leptin (a gift from Amgen Inc., Thousand Oaks, California, USA) for 5 days with a micro-osmotic pump (Alza model 1002; DURECT Corp., Cupertino, California, USA) inserted subcutaneously into the interscapular region.
Tissue culture.
Reproductive fat pads (500 mg) from chow-fed male AY/a Dgat1+/+ and AY/a Dgat1–/– mice were sliced into 50-mg pieces, washed twice with PBS, placed in DMEM (Invitrogen Corp., Carlsbad, California, USA) supplemented with 1% FBS (HyClone Laboratories, Logan, Utah, USA), and incubated at 37°C and 5% CO2. The conditioned media were collected after 10–12 hours. Total protein was measured with a colorimetric kit (Bio-Rad Laboratories Inc.), FFAs were measured as previously described (12), leptin and TNF-α were measured with enzyme immunoassay kits (R&D Systems Inc.), and adiponectin was measured with an ELISA kit (B-Bridge International Inc., San Jose, California, USA). These factors were also measured in growth media not conditioned by adipose tissue, and the basal levels were subtracted from the reported results.
Immunoblots.
Serum samples (1 μl) were loaded onto a 10% polyacrylamide gel, transferred to a nitrocellulose membrane, and incubated with a polyclonal primary antibody that recognizes the globular head region of murine adiponectin (a gift from Genset Corp., San Diego, California, USA). Binding was detected by an anti-IgG antibody–peroxidase conjugate (Santa Cruz Biotechnology Inc., Santa Cruz, California, USA) and enhanced chemiluminescence (Amersham Pharmacia Biotech, Buckinghamshire, United Kingdom).
Statistical methods.
Data are shown as mean ± SD unless stated otherwise. Measurements were compared with the two-tailed t test or Mann-Whitney rank-sum test. Energy expenditure and lean body mass measurements were analyzed by linear regression. Weight curves were compared with repeated-measures ANOVA, and serum adiponectin and TNF-α results were compared with ANOVA. Both comparisons were followed by post hoc Tukey-Kramer test, if appropriate, to determine the effect of genotype.
Results
Fat transplantation.
We transplanted WAT from Dgat1–/– mice into Dgat1+/+ mice (WATDgat1–/–→Dgat1+/+). We also transplanted a similar amount of WAT from Dgat1+/+ mice into Dgat1+/+ mice as controls (WATDgat1+/+→Dgat1+/+). Ninety mice received transplants during the study, and 72 had viable grafts, as judged by visual inspection, at sacrifice. The transplanted fat pads retained their characteristic histological appearance (16); that is, Dgat1–/– WAT contained smaller adipocytes than Dgat1+/+ WAT (Figure 1). The transplanted fat pads decreased slightly in weight 20 weeks after the procedure (410 ± 36 mg for Dgat1–/– WAT and 423 ± 33 mg for Dgat1+/+ WAT).
Figure 1.
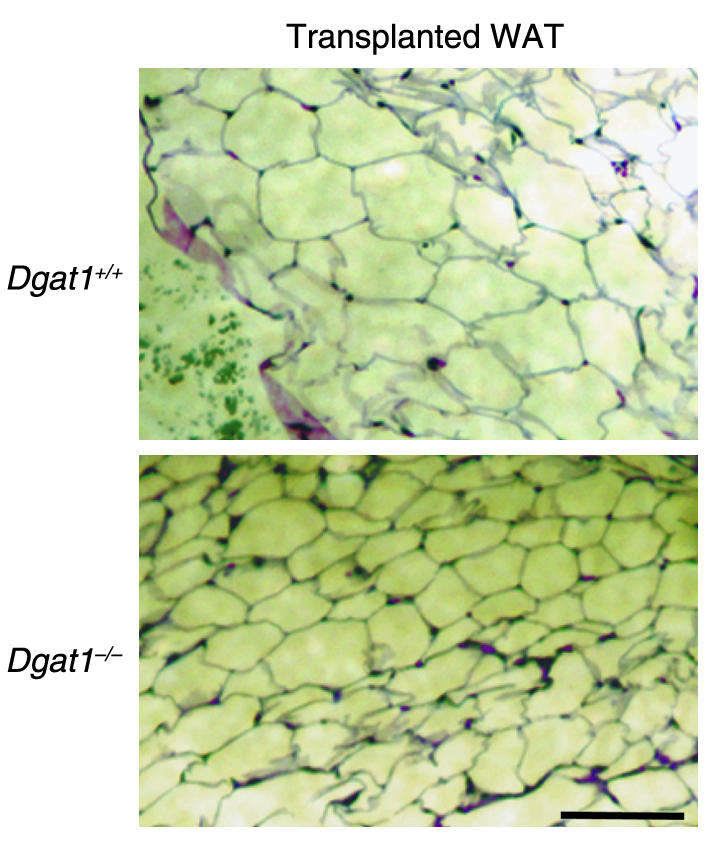
Histology of transplanted Dgat1+/+ and Dgat1–/– WAT. Representative results are shown. Bar, 100 μm.
Resistance to diet-induced obesity in wild-type mice transplanted with Dgat1–/– WAT.
WATDgat1–/–→Dgat1+/+ mice gained less weight than WATDgat1+/+→Dgat1+/+ mice in response to a Western-style high-fat diet (Figure 2a). The difference in weight curves became apparent after 4 weeks of high-fat feeding. WATDgat1–/–→Dgat1+/+ mice with nonviable grafts at sacrifice had weight curves similar to those of WATDgat1+/+→Dgat1+/+ mice (46.2 ± 2.3 vs. 47.5 ± 1.6 g, P > 0.05), indicating that obesity resistance in these mice required viable grafts. WATDgat1–/–→Dgat1+/+ mice with viable grafts also had an approximately 50% reduction in total fat pad content when fed either a chow or a high-fat diet (Figure 2b). In contrast, transplantation of Dgat1+/+ WAT into Dgat1–/– mice did not affect their resistance to diet-induced obesity (Figure 2c) or total fat pad content (Figure 2d).
Figure 2.

Effects of WAT transplantation on body weight and total fat pad content. (a) Decreased weight gain in WATDgat1–/–→Dgat1+/+ mice fed a high-fat diet. n = 8–9 male mice per group. (b) Decreased total fat pad content (excluding transplanted fat pads) in WATDgat1–/–→Dgat1+/+ mice. n = 4–6 male mice per group. (c) Lack of effect of transplanted Dgat1+/+ WAT on obesity resistance of Dgat1–/– mice. n = 3–4 female mice per group. (d) Lack of effect of transplanted Dgat1+/+ WAT on total fat pad content in Dgat1–/– mice. n = 4 male mice per group. *P < 0.05.
To further characterize the effects of transplanted Dgat1–/– WAT on obesity resistance, we measured serum leptin levels as a marker of adiposity. WATDgat1–/–→Dgat1+/+ and WATDgat1+/+→Dgat1+/+ mice had similar serum leptin levels after 2 weeks of high-fat feeding (Figure 3a). However, after 5 weeks of high-fat feeding, WATDgat1–/–→Dgat1+/+ mice had lower serum leptin levels than WATDgat1+/+→Dgat1+/+ mice. This difference in serum leptin levels corresponded to the divergence in weight curves. WATDgat1–/–→Dgat1+/+ mice had lower levels of body triglycerides (Figure 3b) than WATDgat1+/+→Dgat1+/+ mice, after either 5 or 12 weeks of high-fat feeding. WATDgat1–/–→Dgat1+/+ mice also had decreased levels of muscle triglycerides (Figure 3c) after 5 weeks of high-fat feeding. In contrast, WATDgat1–/–→Dgat1+/+ and WATDgat1+/+→Dgat1+/+ mice had similar amounts of total body protein or lean body mass (Figure 3d). These results indicate that transplanted Dgat1–/– WAT decreased adiposity without affecting lean body mass in the recipient mice.
Figure 3.

Effects of transplanted Dgat1–/– WAT on adiposity in mice fed a high-fat diet. (a) Decreased serum leptin levels in WATDgat1–/–→Dgat1+/+ mice. n = 4–5 male mice per group. (b) Decreased body triglyceride content in WATDgat1–/–→Dgat1+/+ mice. n = 4 male mice per group. (c) Decreased muscle triglyceride content in WATDgat1–/–→Dgat1+/+ mice. n = 3 male mice per group. (d) Similar body-protein contents in WATDgat1–/–→Dgat1+/+ and WATDgat1+/+→Dgat1+/+ mice. n = 4 male mice per group. Mice were fed a high-fat diet for 2, 5, or 12 weeks. *P < 0.05.
To investigate the mechanism for this resistance to diet-induced obesity in WATDgat1–/–→Dgat1+/+ mice, we fasted the mice for 16 hours and used the fasting-associated weight loss as a surrogate of energy expenditure. WATDgat1–/–→Dgat1+/+ mice lost more weight than WATDgat1+/+→Dgat1+/+ mice after the fast (3.4 ± 0.1 vs. 2.9 ± 0.3 g, n = 4, P < 0.05) and ate more during the refeeding (6.7 ± 0.5 vs. 6.0 ± 0.1 g/24 h, P < 0.05). The enhanced weight loss in WATDgat1–/–→Dgat1+/+ mice suggested an increase in energy expenditure. To further investigate this, we conducted energy-balance studies on transplanted mice fed a high-fat diet. When adjusted for total body weight, WATDgat1–/–→Dgat1+/+ and WATDgat1+/+→Dgat1+/+ mice consumed similar amounts of calories (Figure 4a); however, WATDgat1–/–→Dgat1+/+ mice had increased energy expenditure. Plotting of the daily metabolic rate versus lean body mass for each mouse revealed that values for WATDgat1–/–→Dgat1+/+ mice were on or above the regression line for WATDgat1+/+→Dgat1+/+ mice (Figure 4b), consistent with increased energy expenditure.
Figure 4.

Effects of transplanted Dgat1–/– WAT on energy intake and expenditure in mice fed a high-fat diet. (a) Energy intake and expenditure in WATDgat1–/–→Dgat1+/+ and WATDgat1+/+→Dgat1+/+ mice. n = 3–4 male mice per group. Error bars represent SEM. *P < 0.05. (b) Relationship between energy expenditure and lean body mass in WATDgat1–/–→Dgat1+/+ and WATDgat1+/+→Dgat1+/+ mice. Regression lines for both groups are shown.
Enhanced glucose disposal in wild-type mice transplanted with Dgat1–/– WAT.
To assess the effects of the WAT transplantation on glucose metabolism, we conducted glucose and insulin tolerance tests. Chow-fed WATDgat1–/–→Dgat1+/+ mice had lower blood glucose concentrations than chow-fed WATDgat1+/+→Dgat1+/+ mice after either a glucose load (Figure 5a) or an insulin injection (Figure 5b), suggesting increased insulin sensitivity in Dgat1+/+ mice transplanted with Dgat1–/– WAT. In contrast, WATDgat1+/+→Dgat1–/– and WATDgat1–/–→Dgat1–/– mice had similar glucose concentrations after either a glucose load (Figure 5c) or an insulin injection (Figure 5d), suggesting that Dgat1+/+ WAT did not affect the glucose disposal of Dgat1–/– mice.
Figure 5.

Effects of WAT transplantation on glucose metabolism. (a and b) Enhanced glucose disposal in WATDgat1–/–→Dgat1+/+ mice. n = 5–6 chow-fed male mice per group. (c and d) Lack of effect of transplanted Dgat1+/+ WAT on glucose disposal in Dgat1–/– mice. n = 4 chow-fed male mice per group. *P < 0.05.
Transplantation of Dgat1–/– WAT decreases adiposity and enhances glucose disposal in Agouti yellow but not ob/ob mice.
We performed WAT transplantation experiments in two genetic models of obesity and insulin resistance, the Agouti yellow (AY/a) and the leptin-deficient (ob/ob) mice. We had introduced DGAT1 deficiency into both models by genetic crosses in previous studies (16, 20). One week after transplantation, AY/a Dgat1+/+ mice transplanted with AY/a Dgat1–/– WAT (WATAY/aDgat1–/–→AY/a Dgat1+/+) and control mice (WATAY/aDgat1+/+→AY/a Dgat1+/+) had similar mean body weights (32.7 ± 1.2 vs. 33.6 ± 2.4 g, n = 4–5, P > 0.05). WATAY/aDgat1–/–→AY/a Dgat1+/+ mice, however, had lower blood glucose concentrations than control mice during both glucose tolerance (Figure 6a) and insulin tolerance (Figure 6b) tests, suggesting increased insulin sensitivity. After 6 weeks of a high-fat diet, WATAY/aDgat1–/–→AY/a Dgat1+/+ mice had a lower mean body weight (47.0 ± 0.9 vs. 50.3 ± 1.9 g, n = 4, P < 0.05) and total fat pad content (8.7% ± 0.6% vs. 9.6% ± 0.1% of total body weight, n = 3, P < 0.05) than controls. Furthermore, muscle triglyceride content was about 25% lower in WATAY/aDgat1–/–→AY/a Dgat1+/+ mice than in control mice (14.7 ± 2.1 vs. 20.4 ± 2.1 mg/g tissue weight, n = 3, P < 0.05).
Figure 6.

Metabolic effects of transplanted Dgat1–/– WAT in AY/a and ob/ob mice. (a and b) Enhanced glucose disposal in WATAY/aDgat1–/–→AY/a Dgat1+/+ mice. n = 4–5 chow-fed male mice per group. (c and d) Lack of effect of transplanted ob/ob Dgat1–/– WAT on glucose disposal in ob/ob Dgat1+/+ mice. n = 4 chow-fed male mice per group. *P < 0.05.
In contrast, transplantation of ob/ob Dgat1–/– WAT into ob/ob (WATob/obDgat1–/–→ob/ob Dgat1+/+) mice fed a chow diet did not significantly affect their blood glucose concentrations after a glucose load (Figure 6c) or an insulin injection (Figure 6d). Chow-fed WATob/obDgat1–/–→ob/ob Dgat1+/+ and control mice also had similar mean body weights (69.2 ± 6.2 vs. 66.4 ± 3.9 g, n = 3, P > 0.05), total fat pad content (12.9% ± 0.6% vs. 13.4% ± 2.0% of total body weight, n = 3, P > 0.05), and muscle triglyceride content (7.0 ± 2.3 vs. 6.7 ± 1.9 mg/g tissue weight, n = 3, P > 0.05).
Increased adiponectin mRNA expression in Dgat1–/– mice.
The WAT transplantation results suggested that Dgat1–/– WAT secretes factors that increase energy expenditure and enhance insulin sensitivity. We therefore examined the gene expression of several adipocyte-derived molecules in WAT of Dgat1–/– mice. Dgat1–/– and Dgat1+/+ mice had similar levels of expression of resistin and TNF-α (Figure 7a), molecules implicated in the development of obesity-induced insulin resistance (6, 7). In contrast, the mRNA levels of adiponectin, a molecule that enhances insulin sensitivity and fatty acid oxidation (4, 5, 21), trended higher in chow-fed Dgat1–/– mice than in chow-fed Dgat1+/+ mice, and the increase was statistically significant in Dgat1–/– mice fed a high-fat diet. DGAT1 deficiency also increased adiponectin expression twofold in AY/a mice. Furthermore, 1 week after transplantation, adiponectin expression was twofold greater in the transplanted AY/a Dgat1–/– WAT than in the native AY/a Dgat1+/+ WAT (Figure 7b), indicating that the increase in adiponectin mRNA expression in Dgat1–/– WAT was maintained after transplantation.
Figure 7.

Expression of adipocyte-derived molecules. (a) Resistin, TNF-α, and adiponectin expression in WAT of Dgat1–/– mice. n = 7–10 male mice per group. Error bars represent SEM. *P < 0.05 vs. Dgat1+/+ mice fed a high-fat diet for 20 weeks. (b) Comparison of adiponectin expression in transplanted AY/a Dgat1–/– WAT and native AY/a Dgat1+/+ WAT obtained from transplanted mice. n = 4 male mice per group. *P <0.05 vs. naive WAT. (c) Effect of leptin infusion on adiponectin expression in ob/ob Dgat1–/– mice. n = 3–5 male mice per group. Expression of chow-fed Dgat1+/+ mice = 1.0. *P < 0.05 vs. ob/ob Dgat1+/+ mice without leptin treatment. (d) Effect of leptin infusion on adiponectin expression in Dgat1–/– and Dgat1+/+ mice. n = 8–10 male mice per group.
To determine the effects of leptin and DGAT1 deficiency on adiponectin expression, we measured adiponectin mRNA levels in ob/ob mice. Adiponectin expression levels were low in ob/ob Dgat1+/+ mice (Figure 7c), consistent with previous findings (22). In contrast to the results in high-fat fed mice and AY/a mice, DGAT1 deficiency had no effect on the low expression levels of adiponectin in ob/ob mice. Five days of leptin infusion (6 or 12 μg/d) significantly increased adiponectin expression in ob/ob Dgat1–/– mice but had a minimal effect on adiponectin expression in ob/ob Dgat1+/+ mice.
To investigate whether leptin directly increases the expression of adiponectin in mice with an intact leptin pathway, we infused leptin subcutaneously into Dgat1–/– and Dgat1+/+ mice. Leptin infusion (12 μg/d) did not increase the expression of adiponectin in either genotype (Figure 7d). These results suggest that leptin plays a permissive rather than a direct regulatory role in the expression of adiponectin.
Correlation of secreted-protein levels with mRNA expression in Dgat1–/– WAT.
To determine whether WAT mRNA expression correlated with the amounts of proteins secreted by Dgat1–/– WAT, we analyzed growth media conditioned by either AY/a Dgat1–/– or AY/a Dgat1+/+ WAT. Media conditioned by either AY/a Dgat1–/– or AY/a Dgat1+/+ WAT had similar levels of total protein (Figure 8a) and FFAs (Figure 8b). Medium conditioned by AY/a Dgat1–/– WAT had less leptin than medium conditioned by AY/a Dgat1+/+ WAT (Figure 8c). This result is consistent with our previous in vivo finding that Dgat1–/– WAT has decreased expression of leptin (16). TNF-α levels were similar in the two media (Figure 8d), whereas adiponectin levels were increased twofold in medium conditioned by AY/a Dgat1–/– WAT (Figure 8e).
Figure 8.

Concentrations of adipocyte-derived factors in growth media conditioned by AY/a Dgat1–/– or AY/a Dgat1+/+ WAT. (a) Total protein. (b) FFAs. (c) Leptin. (d) TNF-α. (e) Adiponectin. n = 6–10 mice per group. *P < 0.05, **P < 0.01.
Increased serum adiponectin levels in mice transplanted with Dgat1–/– WAT.
To determine whether the ex vivo finding of increased adiponectin secretion by Dgat1–/– WAT correlated with increased serum adiponectin levels in the transplant model, we measured serum adiponectin concentrations in WATDgat1–/–→Dgat1+/+ and WATDgat1+/+→Dgat1+/+ mice fed a high-fat diet. After 2 weeks of high-fat feeding, serum adiponectin levels trended higher in WATDgat1–/–→Dgat1+/+ mice than in WATDgat1+/+→Dgat1+/+ mice (Figure 9a). After 5 weeks of high-fat feeding, the difference was statistically significant and persisted after 20 weeks of high-fat feeding (Figure 9b).
Figure 9.

Increased serum adiponectin levels in WATDgat1–/–→Dgat1+/+ mice fed a high-fat diet. (a) After 2 or 5 weeks of high-fat feeding. n = 4–5 male mice per group. *P < 0.05. (b) After 20 weeks of high-fat feeding. Each lane represents a serum sample from an individual mouse. The experiment was performed twice, and representative results are shown.
Serum adiponectin and TNF-α levels in Dgat1–/– mice.
We measured serum levels of adiponectin and TNF-α in nontransplanted Dgat1–/– mice. After 8 weeks of high-fat feeding, Dgat1+/+, Dgat1+/–, and Dgat1–/– mice had similar serum adiponectin levels (Figure 10a). However, after adjustment of these values for total fat pad weight (1.0 ± 0.3 g for Dgat1–/–, 1.7 ± 0.3 g for Dgat1+/–, and 4.4 ± 0.5 g for Dgat1+/+ mice, n = 5–7, P < 0.05 for all comparisons), Dgat1–/– mice had the highest relative adiponectin levels, and Dgat1+/+ mice had the lowest. We also examined serum adiponectin levels in chow-fed AY/a mice. AY/a Dgat1+/+ and AY/a Dgat1–/– mice had comparable serum adiponectin levels (18.1 ± 2.5 vs. 19.3 ± 4.5 μg/ml, n = 5–6, P > 0.05). However, AY/a Dgat1–/– mice had higher serum adiponectin levels than AY/a Dgat1+/+ mice after adjustment for fat pad weight (Figure 10b).
Figure 10.

Serum adiponectin and TNF-α levels in Dgat1–/– mice. (a) Serum adiponectin levels in Dgat1+/+, Dgat1+/–, and Dgat1–/– mice fed a high-fat diet. n = 5–7 male mice per group. P < 0.01 for all comparisons for adjusted serum concentrations. (b) Adjusted serum adiponectin levels in AY/a Dgat1+/+ and AY/a Dgat1–/– mice fed a chow diet. n = 5–6 male mice per group. *P < 0.01. (c) Serum TNF-α levels in Dgat1–/– mice fed a high-fat diet. n = 5–7 male mice per group. *P < 0.05 vs. Dgat1+/+ mice.
In contrast to adiponectin, serum TNF-α levels were lower in Dgat1–/– mice than in Dgat1+/+ mice after 8 weeks of high-fat feeding (Figure 10c). TNF-α levels also trended lower in Dgat1+/– mice than in Dgat1+/+ mice, although the difference was not statistically significant. After adjustment for total fat pad weight, however, all three groups of mice had similar relative TNF-α levels.
Because TNF-α may antagonize the actions of adiponectin (23), the serum adiponectin/TNF-α ratio may be an important determinant of energy metabolism and systemic insulin sensitivity. The mean adiponectin/TNF-α ratio was twofold higher in high-fat fed Dgat1–/– mice than in high-fat fed Dgat1+/+ mice (2.4 vs. 1.2), correlating with their increased energy expenditure and enhanced insulin sensitivity.
Discussion
In this study, we show that the transplantation of Dgat1–/– WAT decreased adiposity and enhanced glucose disposal in Dgat1+/+ mice. Our results suggest that DGAT1 deficiency alters the endocrine function of WAT in a manner that promotes obesity resistance and enhances insulin sensitivity. Although multiple secretory factors may be involved, we identified one possible contributing factor — the increased production of adiponectin by Dgat1–/– WAT. Our findings show that the technique of fat transplantation described by Gavrilova et al. (10) and Colombo et al. (24) can be used to study the metabolic effects of genetically engineered WAT.
The transplantation of Dgat1–/– WAT conferred several aspects of the DGAT1-deficiency phenotype on Dgat1+/+ mice. Chow-fed WATDgat1–/–→Dgat1+/+ mice had enhanced glucose disposal. WATDgat1–/–→Dgat1+/+ mice gained less weight than control mice in response to a high-fat diet, similar to what occurs in Dgat1–/– mice (12). WATDgat1–/–→Dgat1+/+ mice lost more weight during prolonged fasting and consumed more food than controls during refeeding, again mirroring the responses of Dgat1–/– mice (25). These findings, along with the results from the energy-balance studies, suggest that the transplantation of Dgat1–/– WAT confers obesity resistance primarily by increasing energy expenditure.
The transplantation of Dgat1–/– WAT decreased adiposity and enhanced glucose disposal in AY/a mice but had no apparent effects in ob/ob mice. One possible explanation for this lack of effect is that the amount of transplanted WAT may be insufficient to overcome the severe obesity and insulin resistance in ob/ob mice. Alternatively, the effects of DGAT1 deficiency on WAT endocrine functions may require leptin. This explanation would be consistent with our previous findings that the effects of DGAT1 deficiency on obesity resistance (16), increased expression of uncoupling protein 1 (25, 26), and sebaceous gland atrophy and hair loss (20) were present in AY/a mice but not in ob/ob mice.
Our transplantation results suggest that DGAT1 deficiency alters the expression of adipocyte-derived factors that confer obesity resistance and enhance glucose disposal. The adipose tissue has been recognized as an important endocrine organ, secreting several molecules that modulate energy and glucose metabolism (1, 2). These molecules include adiponectin (also known as Acrp30 [ref. 27], apM1 [ref. 28], and AdipoQ [ref. 22]), which enhances fatty acid oxidation and increases insulin sensitivity, and TNF-α, which induces insulin resistance. Mice treated with adiponectin have decreased adiposity, decreased muscle triglyceride content, and increased insulin sensitivity (4, 5, 21). Recent studies suggest that TNF-α suppresses adiponectin expression and antagonizes its effects on energy and glucose metabolism (23). Thus, the relative levels of adiponectin and TNF-α in the serum may be an important determinant of energy expenditure and systemic insulin sensitivity (23).
Several of our findings indicate that Dgat1–/– WAT produced more adiponectin than an equivalent amount of Dgat1+/+ WAT. These findings include the increased mRNA expression in Dgat1–/– WAT, increased protein concentration in growth medium conditioned by AY/a Dgat1–/– WAT, and increased serum levels in WATDgat1–/–→Dgat1+/+ mice. These results imply that increased adiponectin secretion by transplanted Dgat1–/– WAT may contribute to the obesity resistance and enhanced glucose metabolism in WATDgat1–/–→Dgat1+/+ mice. However, our data do not exclude the possibility that alterations in the expression of additional adipocyte-derived factors, including nonpeptides such as lysophosphatidic acid (29), may also contribute to the findings in WATDgat1–/–→Dgat1+/+ mice.
Interestingly, despite increased adiponectin production by Dgat1–/– WAT, serum adiponectin levels were similar in nontransplanted Dgat1–/– and Dgat1+/+ mice. This lack of difference most likely reflects the 50% reduction in adipose mass in Dgat1–/– mice (12). The decreased adiposity also helps to explain the 50–60% reduction in serum TNF-α levels in Dgat1–/– mice, even though TNF-α expression was similar in Dgat1–/– and Dgat1+/+ WAT. Although Dgat1–/– and Dgat1+/+ mice had similar serum levels of adiponectin, increased adiponectin action may still contribute to the increased energy expenditure (12) and enhanced insulin sensitivity (16) in Dgat1–/– mice. Because of the decreased serum TNF-α levels in Dgat1–/– mice, their adiponectin/TNF-α ratio was increased twofold. This increase may correlate with enhanced adiponectin action, or increased adiponectin sensitivity, in Dgat1–/– mice. Such an effect would be similar to the effect of DGAT1 deficiency on increasing leptin sensitivity (16).
It is unclear how DGAT1 deficiency alters the endocrine functions of the adipocyte. Adiponectin expression is increased by insulin (30) and correlates inversely with adipocyte size (31). Two characteristics of Dgat1–/– mice, therefore, may contribute to their increased adiponectin expression: increased insulin sensitivity and small adipocyte size (16). It would be of interest to determine whether adiponectin expression is increased in other murine knockout models of obesity resistance, enhanced insulin sensitivity, and decreased adipocyte size (reviewed in ref. 11).
In conclusion, we show that the transplantation of Dgat1–/– WAT confers obesity resistance and enhances glucose disposal in mouse models of obesity and insulin resistance. These results highlight the importance of WAT as an endocrine organ that regulates energy and glucose metabolism through secreted factors. In addition, our study demonstrates that fat transplantation may provide a useful tool for studying the endocrine and metabolic functions of genetically engineered WAT.
Acknowledgments
We thank M. Reitman and O. Gavrilova for advice on fat transplantation, J. Fruebis, S. Javorschi, and C. Vaisse for helpful discussions, B. Taylor for manuscript preparation, S. Ordway and G. Howard for editorial assistance, and M. Schambelan, R. Mahley, J. Herz, K. Weisgraber, and R. Streeper for comments on the manuscript. This work was supported by the NIH (K08-DK61363 to H.C. Chen, R01-DK26356 to R.H. Eckel, and R01-DK56084 to R.V. Farese), the Sandler Family Supporting Foundation, and the J. David Gladstone Institutes.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Nonstandard abbreviations used: acyl CoA:diacylglycerol acyltransferase 1 (DGAT1); white adipose tissue (WAT).
References
- 1.Flier JS. The adipocyte: storage depot or node on the energy information superhighway? Cell. 1995;80:15–18. doi: 10.1016/0092-8674(95)90445-x. [DOI] [PubMed] [Google Scholar]
- 2.Frühbeck G, Gómez-Ambrosi J, Muruzábal FJ, Burrell MA. The adipocyte: a model for integration of endocrine and metabolic signaling in energy metabolism regulation. Am. J. Physiol. Endocrinol. Metab. 2001;280:E827–E847. doi: 10.1152/ajpendo.2001.280.6.E827. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 4.Yamauchi T, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001;7:941–946. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 5.Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001;7:947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 6.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor–α: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 7.Steppan CM, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 8.Shimomura I, et al. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes Dev. 1998;12:3182–3194. doi: 10.1101/gad.12.20.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moitra J, et al. Life without white fat: a transgenic mouse. Genes Dev. 1998;12:3168–3181. doi: 10.1101/gad.12.20.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gavrilova O, et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J. Clin. Invest. 2000;105:271–278. doi: 10.1172/JCI7901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen HC, Farese RV., Jr Turning WAT into BAT gets rid of fat. Nat. Med. 2001;7:1102–1103. doi: 10.1038/nm1001-1102. [DOI] [PubMed] [Google Scholar]
- 12.Smith SJ, et al. Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking DGAT. Nat. Genet. 2000;25:87–90. doi: 10.1038/75651. [DOI] [PubMed] [Google Scholar]
- 13.Cases S, et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. U. S. A. 1998;95:13018–13023. doi: 10.1073/pnas.95.22.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farese RV, Jr, Cases S, Smith SJ. Triglyceride synthesis: insights from the cloning of diacylglycerol acyltransferase. Curr. Opin. Lipidol. 2000;11:229–234. doi: 10.1097/00041433-200006000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Cases S, et al. Cloning of DGAT2, a second mammalian diacylglycerol acyltransferase, and related family members. J. Biol. Chem. 2001;276:38870–38876. doi: 10.1074/jbc.M106219200. [DOI] [PubMed] [Google Scholar]
- 16.Chen HC, et al. Increased insulin and leptin sensitivity in mice lacking acyl CoA:diacylglylcerol acyltransferase 1. J. Clin. Invest. 2002;109:1049–1055. doi:10.1172/JCI200214672. doi: 10.1172/JCI14672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Halaas JL, et al. Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc. Natl. Acad. Sci. U. S. A. 1997;94:8878–8883. doi: 10.1073/pnas.94.16.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 19.Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat. Genet. 1996;12:318–320. doi: 10.1038/ng0396-318. [DOI] [PubMed] [Google Scholar]
- 20.Chen HC, Smith SJ, Tow B, Elias PM, Farese RV., Jr Leptin modulates the effects of acyl CoA:diacylglycerol acyltransferase deficiency on murine fur and sebaceous glands. J. Clin. Invest. 2002;109:175–181. doi:10.1172/JCI200213880. doi: 10.1172/JCI13880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fruebis J, et al. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci. U. S. A. 2001;98:2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J. Biol. Chem. 1996;271:10697–10703. doi: 10.1074/jbc.271.18.10697. [DOI] [PubMed] [Google Scholar]
- 23.Maeda N, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat. Med. 2002;8:731–737. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- 24.Colombo C, et al. Transplantation of adipose tissue lacking leptin is unable to reverse the metabolic abnormalities associated with lipoatrophy. Diabetes. 2002;51:2727–2733. doi: 10.2337/diabetes.51.9.2727. [DOI] [PubMed] [Google Scholar]
- 25.Chen HC, Ladha Z, Farese RV., Jr Deficiency of acyl CoA:diacylglycerol acyltransferase 1 increases leptin sensitivity in murine obesity models. Endocrinology. 2002;143:2893–2898. doi: 10.1210/endo.143.8.8941. [DOI] [PubMed] [Google Scholar]
- 26.Chen HC, Ladha Z, Smith SJ, Farese RV., Jr Analysis of energy expenditure at different ambient temperatures in mice lacking DGAT1. Am. J. Physiol. Endocrinol. Metab. 2003;284:E213–E218. doi: 10.1152/ajpendo.00248.2002. [DOI] [PubMed] [Google Scholar]
- 27.Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 1995;270:26746–26749. doi: 10.1074/jbc.270.45.26746. [DOI] [PubMed] [Google Scholar]
- 28.Maeda K, et al. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (adipose most abundant gene transcript 1) Biochem. Biophys. Res. Commun. 1996;221:286–289. doi: 10.1006/bbrc.1996.0587. [DOI] [PubMed] [Google Scholar]
- 29.Valet P, et al. α2-Adrenergic receptor–mediated release of lysophosphatidic acid by adipocytes. A paracrine signal for preadipocyte growth. J. Clin. Invest. 1998;101:1431–1438. doi: 10.1172/JCI806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halleux CM, et al. Secretion of adiponectin and regulation of apM1 gene expression in human visceral adipose tissue. Biochem. Biophys. Res. Commun. 2001;288:1102–1107. doi: 10.1006/bbrc.2001.5904. [DOI] [PubMed] [Google Scholar]
- 31.Yamauchi T, et al. The mechanisms by which both heterozygous peroxisome proliferator-activated receptor γ (PPARγ) deficiency and PPARγ agonist improve insulin resistance. J. Biol. Chem. 2001;276:41245–41254. doi: 10.1074/jbc.M103241200. [DOI] [PubMed] [Google Scholar]