

Abstract
The env gene of human immunodeficiency virus type 1 (HIV-1) includes some of the most genetically diverse regions of the viral genome, which are called variable regions 1 through 5 (V1 through V5). We have developed a heteroduplex tracking assay to detect changes in variable regions 1 and 2 of env (V1/V2-HTA). Using sequences from two molecular clones as probes, we have studied the nature of longitudinal virus population changes in a cohort of HIV-1-infected subjects. Viral sequences present in 21 subjects with late-stage HIV-1 infection were initially screened for stability of the virus population by V1/V2-HTA. The virus populations at entry comprised an average of five coexisting V1/V2 genotypic variants (as identified by HTA). Eight of the 21 subjects were examined in detail because of the dynamic behavior of their env variants over an approximately 9-month period. In each of these cases we detected a single discrete transition of V1/V2 genotypes based on monthly sampling. The major V1/V2 genotypes (those present at >10% abundance) from the eight subjects were cloned and sequenced to define the nature of V1/V2 variability associated with a discrete transition. Based on a comparison of V1/V2 genotypic variants present at entry with the newly emerged variants we categorized the newly emerged variants into two groups: variants without length differences and variants with length differences. Variants without length differences had fewer nucleotide substitutions, with the changes biased to either V1 or V2, suggestive of recent evolutionary events. Variants with length differences included ones with larger numbers of changes that were distributed, suggestive of recall of older genotypes. Most length differences were located in domains where the codon motif AVT (V = A, G, C) had become enriched and fixed. Finally, recombination events were detected in two subjects, one of which resulted in the reassortment of V1 and V2 regions. We suggest that turnover in V1/V2 populations was largely driven by selection on either V1 or V2 and that escape was accomplished either through changes focused in the region under selection or by the appearance of a highly divergent variant.
There are several mechanisms for generating genetic variation that are common to all retroviruses: nucleotide substitutions, deletions, insertions, and homologous and nonhomologous recombination (30, 44, 45, 54-56; reviewed in references 74 and 82). These mechanisms have been studied extensively by using different retroviral vectors in cell culture systems. While all of these mechanisms promote genetic diversity within an infected individual, selection and/or stochastic events associated with small population size (e.g., transmission) ultimately determine which genotypic variants predominate.
Human immunodeficiency virus type 1 (HIV-1) exhibits significant genetic diversity, both in cross-sectional analysis of extant, circulating viruses and in longitudinal analysis, where there is evolution of genetically distinct variants over time in an infected individual (reviewed in reference 46). Viral factors that likely contribute to HIV-1 variability in vivo include high levels of replication, an error-prone DNA polymerase (reverse transcriptase [RT]), and efficient recombination (11). This ability to generate genetic diversity allows the virus to escape host immune responses and also to develop resistance to antiretroviral drugs. The role of selection in directing this diversity is implied by the observation that individuals with virus populations displaying higher variability exhibit a slower progression to AIDS (17, 77) and by the fact that drug resistance-associated mutations are largely absent in virus from drug-naïve subjects (2, 37, 39) but rapidly appear with selection.
As a result of selection, genetic variability is not equally distributed across the HIV-1 genome. The most dramatic features of variability are localized to the viral env gene, which encodes the virion surface proteins gp120 and gp41, particularly in the five variable regions of gp120 (25, 71). Variable regions 1 and 2 (V1/V2) are closely linked and form a large loop anchored by disulfide bonds (38). The V1/V2 loop is thought to sit on the protein surface and mask the V3 loop, which is the primary determinant of viral tropism based on coreceptor usage (CCR5 or CXCR4) (38, 70, 72). The V1/V2 loop has been hypothesized to move as the result of CD4 binding (38, 79). In this regard, viruses lacking the V1/V2 loop are hypersensitive to neutralization (6, 32).
Changes in the V1/V2 loop have been associated with evasion from neutralizing antibodies, presumably due to the exposure of the V1/V2 loop on the surface of gp120 (22, 34, 66). Additional studies have highlighted the importance of glycosylation site changes in the V1/V2 loop for neutralization escape (7, 8). Finally, sequences in the region of the V1/V2 loop may play a role in coreceptor use (10, 27, 65). Thus, the highly variable V1/V2 loop represents an important functional determinant on the surface of the Env protein that must also interact with the host immune system.
The heteroduplex mobility assay (HMA) and the heteroduplex tracking assay (HTA) are rapid and sensitive experimental approaches that can be used to study the dynamic behavior of discrete genotypic variants within a virus population over time. Both assays take advantage of the principle that due to induced bends in the DNA duplex, a DNA duplex with mismatches and/or size differences will migrate through a polyacrylamide gel more slowly than a DNA duplex with completely complementary strands. Delwart and colleagues (19) initially used the HMA and HTA to analyze HIV-1 variation. One of the more widely used HIV-1-specific HMAs/HTAs analyzes variability in the C2-V5 region of gp120. This assay was initially developed to subtype HIV-1 variants (19) and has been used to examine longitudinal evolution (17, 18), transmission (12, 14, 47), and compartmentalization (16). In addition, various HMAs and/or HTAs have been developed to address evolutionary issues specific to multiple regions of HIV and simian immunodeficiency virus (SIV) genomes, such as gag (26, 73), the protease and RT coding domains (21, 60, 62), tat (20), the gp120 V3 coding domain (50, 51, 59), and the gp120 V1/V2 coding domain (23, 24, 61, 69, 83).
We have developed a new V1/V2-specific heteroduplex tracking assay (V1/V2-HTA) for HIV-1 to investigate the mechanisms involved in the evolution of V1/V2 in vivo. V1/V2-HTAs and -HMAs have been previously developed for HIV-1, HIV-2, and SIV using homologous probes to study virus transmission (24, 69, 83) and the effect of virus diversity on vaccine protection (61). One study analyzing the effect of virus diversity on neutralizing-antibody susceptibility employed a heterologous, single-stranded probe from a cloned sequence (23). We used a V1/V2-HTA based on molecular clones as probes, rather than isolate-specific probes, to study changes in the virus population from a cohort of HIV-1-infected subjects. We found that these probes were able to detect multiple V1/V2 genotypic variants that represented the large majority of variants present in HIV-1-infected subjects. Using this V1/V2-HTA followed by sequence analysis of molecular clones, we examined the pace and nature of V1/V2 env evolution in longitudinal samples from a cohort of 21 infected subjects. The sensitivity of the V1/V2-HTA to reveal discrete periods of change and the size of the cohort under study allow us to suggest generalizable features of V1/V2 evolution during late-stage infection.
MATERIALS AND METHODS
Subject samples.
Human plasma samples were obtained from Dale Kempf at Abbott Laboratories (Abbott Park, Ill.) from a placebo-controlled trial (M94-247) that investigated the addition of ritonavir to preexisting RT nucleoside inhibitor therapy that was ineffective (5). Blood samples were collected for HIV-1 RNA quantitation and determination of CD4+-T-cell counts every 2 to 4 weeks over approximately 9 months. Virus loads for most of the subject samples used in this study are described elsewhere (50). The average virus loads for the four subjects not described by Nelson et al. (50) are as follows (log10 RNA levels): patient 1007, average = 5.40 (range = 5.23 to 5.58); patient 1025, average = 4.83 (range = 4.63 to 5.02); patient 1079, average = 5.39 (range = 5.23 to 5.58); patient 1103, average = 5.32 (range = 5.11 to 5.64).
Plasmids and probes.
Probes were constructed by amplification using the V1 (strain HXB numbering, nucleotides 6555 to 6584) (5′-TTATGGGATCAAAGCCTAAAGCCATGTGTA-3′ and V2 (HXB numbering, nucleotides 6951 to 6980) (5′-CTTAATTCCATGTGTACATTGTACTGTGCT-3′) primers. The molecular clones used as templates for amplification were as follows: pBa-L from the HIV-1 Ba-L isolate (63), provided by Nathaniel Landau (The Salk Institute, La Jolla, Calif.); pUC112-1, a SacI subclone of JR-FL provided by Irvin Chen (University of California, Los Angeles) (36); a subclone of the NL4-3 genomic molecular clone provided by Malcolm Martin (1); and pYU-2, obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, from Beatrice Hahn and George Shaw (41, 42). The resulting products were 441 bp (Ba-L), 420 bp (JR-FL and NL4-3), and 417 bp (YU-2). All PCR products included 138 bp of the C2 region downstream of V2. PCR products were cloned into the pT7Blue vector by using the Perfectly Blunt cloning kit (Novagen).
All four probes were tested in a V1/V2-HTA with PCR products from each of the clones (Fig. 1) and RT-PCR products from plasma samples derived from several subjects (data not shown). These experiments showed that since V1/V2 sequences typically differ in length, essentially all of the V1/V2 heteroduplexes formed between the probe and PCR product exhibited reduced migration during electrophoresis as a result of the bends formed in the DNA by the insertion/deletion loops. It has been noted that once a heteroduplex has such a bend, its conformation (and thus its migration) becomes more sensitive to smaller sequence changes, i.e., point mutations (15), which enhances the ability of this V1/V2-HTA to resolve multiple genotypic variants. In an initial analysis, the Ba-L and JR-FL probes displayed the highest number of bands for the biological samples analyzed, so these two probes were chosen for the subsequent analyses.
FIG. 1.

V1/V2-HTA analysis using the four probes developed from the molecular clones Ba-L, JR-FL, NL4-3, and YU-2. The probes were annealed with PCR products of all four probes. The samples are identified by a letter indicating which PCR product was used in each lane: B, Ba-L; J, JR-FL; N, NL4-3; Y, YU-2. The position of the radiolabeled, single-stranded probe is indicated by an arrow.
The results with the Ba-L and JR-FL probes were then compared to one another to see if one probe was superior to the other. This analysis was performed by comparing the number of V1/V2 genotypic variants (limited to variants that represented at least 10% of the total population) detected with each probe on a set of 42 subject samples from our study. For 86% of the samples, the two probes detected either the same number of variants or numbers within one of each other. Furthermore, when discordant numbers of V1/V2 variants were detected by the two probes, it was equally likely that the Ba-L or JR-FL probe detected more variants than the other probe. These results suggest that the two probes were on average equivalent and largely overlapping in their ability to detect V1/V2 genotypic variants. We used both probes in the initial screening of each sample set to ensure that the maximum number of variants was detected. For the subjects where the Ba-L and JR-FL probes detected the same number of variants within one, the amount of separation between variants in the gel determined in part which probe was ultimately used.
RNA extraction and RT-PCR.
Viral RNA was extracted from 140 μl of plasma with a QIAamp viral RNA kit (Qiagen) with the final elution in 60 μl of elution buffer. The viral RNA was reverse transcribed by using a Titan One-Tube RT-PCR system (Roche Molecular Biochemicals) with several modifications. Briefly, RT reaction mixtures consisted of 5 μl of the eluted viral RNA, 1× Titan RT-PCR buffer, 5 mM dithiothreitol (DTT), a 1 mM concentration of each deoxynucleoside triphosphate (Amersham Pharmacia), 15 pmol of V2 primer, 10 U of RNase inhibitor (Roche Molecular Biochemicals), and 12 U of avian myeloblastosis virus RT (Roche Molecular Biochemicals) in a total volume of 20 μl. Reaction mixtures were incubated at 42°C for 30 min and then at 95°C for 5 min to inactivate the avian myeloblastosis virus RT. A 30-μl aliquot of PCR mix (1× Titan RT-PCR buffer, 5 mM DTT, 15 pmol of V1 primer, and 0.5 μl of Titan or Expand enzyme mix) was then added to each reaction mixture. PCR was performed in a Stratagene 40 Robocyler with the following program: 1 cycle of 95°C for 2 min 45 s, 55°C for 45 s, and 72°C for 2 min; 40 cycles of 95°C for 45 s, 55°C for 45 s, and 72°C for 2 to 5 min (1-min increase every 10 cycles); and 1 cycle of 95°C for 45 s, 55°C for 45 s, and 72°C for 8 min. To calculate the average number of RNA copies added to the RT-PCR mixture, we assumed an average virus load of 5.0 log10, or 105, copies of RNA/ml. The lowest virus load of an analyzed sample was 4.0 log10, and for samples with a virus load between 4.0 to 4.5 log10, typically virions from 1 ml of plasma were pelleted to increase the amount of input viral RNA and thus enhance sampling. Since 140 μl of plasma was used per RNA extraction, the resulting 60 μl of elution buffer should contain at least 104 copies of RNA. An aliquot of 5 μl of the eluted RNA was used per RT-PCR, which represented at least 800 copies of template RNA per reaction. We created distributions of sampling error values as a function of sample size. Comparisons of these distributions with our observed sampling error suggest that, on average, 200 copies of the template RNA were used as templates in these RT-PCRs. All RT-PCR analyses were independently repeated and analyzed by V1/V2-HTA, and all reaction mixtures appeared by inspection to be equivalent. A subset of these samples was further analyzed by phosphorimaging to quantitate sampling error (see below).
Probe labeling.
Probes were labeled by the following method. An aliquot of 6 μg of probe DNA was digested with NdeI at 37°C for 1 h. The probe in the NdeI reaction buffer was labeled by filling in the NdeI overhangs after adding 10 mM DTT, 50 μM dTTP, 50 μCi of [α-35S]dATP (1,250 Ci/mmol; NEN Life Science Products), and 10 U of Klenow fragment of DNA polymerase I in 70 μl for 15 min at room temperature. The DNA polymerase was inactivated by the addition of 10 mM EDTA followed by a 75°C incubation for 15 min. The unincorporated nucleotides were removed by using a QIAquick PCR purification kit (Qiagen) and eluting in a final volume of 70 μl. The probe was released from the plasmid by digestion with KpnI at 37°C for 1 h. The labeled probe was brought to a final volume of 150 μl.
V1/V2-HTA.
Heteroduplex annealing reaction mixtures consisted of 8 μl of unpurified RT-PCR product, 1 μl of 10× annealing buffer (1 M NaCl, 100 mM Tris-HCl [pH 7.5], 20 mM EDTA), 0.1 μM V1 and V2 primer, and 1 μl of labeled probe in a total volume of 10 μl. Extra primers were added to the annealing reaction mixture to bind unannealed probe and make its migration more uniform. The DNA homoduplexes were denatured at 95°C for 2 min followed by annealing at room temperature for 5 min. The heteroduplexes were separated by electrophoresis in a 6% polyacrylamide gel (acrylamide-bisacrylamide, 37.5:1) in 1× Tris-borate-EDTA buffer by using a model V16-2 vertical gel apparatus (Labrepco) run at 17 mA per gel for 5 h 15 min. The gels were dried and exposed to X-ray film or a phosphorimaging screen (Molecular Dynamics). The relative abundance of each band within a sample was quantitated by using FragmeNT analysis software (Molecular Dynamics). Individual bands (i.e., variants) were identified by the following criteria. Variants that had length differences were well separated and easy to distinguish. For variants with only point mutation differences, the bands were much closer together and thus more difficult to distinguish. Close bands were characterized as discrete when they were visibly separated by one bandwidth.
The difference between any two lanes in an HTA was assessed by three different methods. In each formula below, A and B represent two different lanes (representing, for example, two time points) of an HTA gel containing a total of n distinct bands in the two lanes. Ai and Bi are the frequencies of each band, where ΣAi = ΣBi = 1. A band that is present in only one lane is assigned a frequency of 0 in the lane from which it is absent. The first method used was percent change (50), calculated as:
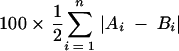 |
(1) |
Here the absolute value of each difference was taken to allow for both increases and decreases in relative abundance. The sum of all differences is divided by 2 to reflect the fact that an increase in the relative abundance of one band results in the concomitant decrease in another. Thus, two lanes with no bands in common (reflecting a complete turnover in the virus population) are assigned a percent change of 100%.
The second method is Nei's standard genetic distance (Ds) (49) for a single locus:
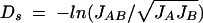 |
(2) |
where
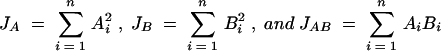 |
(3) |
The third method is Euclidean distance, calculated as
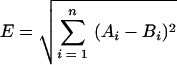 |
(4) |
Sequence analysis.
RT-PCR products were purified with a QIAquick PCR purification kit or a QIAquick gel extraction kit (Qiagen) and then cloned into the pT7Blue or pT7Blue-3 vector using the Perfectly Blunt cloning kit (Novagen). Individual colonies were screened by colony PCR, and the products were analyzed by V1/V2-HTA. Approximately 20% of V1/V2 env clones frm the PCR product did not correspond to the major variants detected by V1/V2-HTA analysis. Plasmids containing V1/V2 inserts of interest (defined by comparing the HTA migration of the clone to the original HTA of the total virus population) were purified with a QIAprep spin miniprep kit (Qiagen), and the inserts were directly sequenced by ABI dye terminator sequencing (Perkin-Elmer Corp.). Each genotypic variant representing at least 10% of the total population (by HTA) was identified between 3 and 31 times (average of 8) among the plasmid clones and sequenced. Sequences were analyzed with the Wisconsin package, version 10.1 (Genetics Computer Group, Madison, Wis.). For most analyses, mutations that appeared only once among the different sequences corresponding to a specific HTA band were not included. These sporadic changes include artifactual errors introduced during the PCR step. On average, the sequence of each clone contained an average of 0.7 such changes over the 400-bp fragment, that were subsequently removed.
All uncorrected pairwise distances were calculated for V1/V2 sequences obtained from each subject. Distance calculations were performed with DISTANCES (Genetics Computer Group Wisconsin package, version 10.1) from codon alignments of nucleotide sequences. Distance scores were classified as either intraband (sequence differences among clones corresponding to the same HTA band) or interband (sequence differences between different HTA bands). Average inter- and intraband distances for each subject were then determined from these scores. Inter- and intraband distances were also calculated after mutations appearing in only one sequence from each subject were replaced by the consensus nucleotide for that subject. For the multiple clones corresponding to each HTA variant, the interband distance was at least five times greater than the intraband distance.
The relationship between the V1/V2-HTA pattern and sequence relatedness within a sample was further examined by using genetic distance and neighbor-joining trees. Length polymorphisms were removed from alignments used in phylogenetic analysis. In most cases, each of the clones identified as being associated with the migration of a specific variant grouped together in a neighbor-joining tree in spite of the minimal amount of phylogenetic information. This result supports the use of the V1/V2-HTA for identifying discrete genetic variants within a single subject. For approximately 20% of the HTA-defined groups (specifically where the genetic distance was small), there were variants that appeared in two different places in the tree due to the presence of polymorphisms. Conversely, in a few cases the V1/V2-HTA distinguished two variants based on single nucleotide differences that were not sufficient to permit distinct branching in the tree. Thus, similar groups were identified by two somewhat independent techniques: V1/V2-HTA, in which migration is greatly influenced by length polymorphism, and neighbor-joining trees, in which clustering is determined by distributed nucleotide polymorphisms.
Some of our analyses relied on consensus sequences derived from all the clones corresponding to one genotypic variant. These analyses included the pairwise comparisons of genotypic variants with the Ba-L and JR-FL probes and the pairwise comparisons of the different genotypic variants within a single subject. The use of consensus sequences did remove additional sequence heterogeneity that was observed in multiple clones representing an individual HTA variant. We examined this heterogeneity to determine how often single HTA variants contained undetected polymorphisms. There were 10 HTA variants for which the number of available clones (≥10) was sufficient to assess sequence variability within a single HTA variant. This analysis focused on the V1 and V2 regions and considered only sequence differences from the consensus sequence that appeared in at least two clones. One set of clones representing a single variant was homogeneous, and the other nine sets of clones contained intraband polymorphisms. In four sets of clones there were polymorphisms that represented 10% of the total population within a subject; in two cases this was at a single position, in one case it represented two positions, and in one set of clones five positions were polymorphic but the variation was unlinked. For example, if an HTA variant represented 50% of the total population within a subject and 2 of 10 clones shared a difference from the consensus sequence of that variant, this would represent a variant that was present in 10% of the total population for that subject that was not resolved by V1/V2-HTA. Thus, when measured at the 10% abundance level, only one variant represented a complex mixture of single polymorphisms. There were 20 further examples of polymorphic nucleotide positions, each of which appeared in ≤20% of the clones from the relevant variant. There was only a single example, involving three positions, where the variation was linked. From this analysis we conclude that undetected heterogeneity within a single V1/V2 HTA variant is largely in the form of single nucleotide polymorphisms. Conversely, HTA is robust in displaying variants that have size differences and in resolving linked nucleotide differences. Furthermore, most of the single-nucleotide polymorphisms within V1/V2 are present in minor abundance and largely unlinked. In cases where single nucleotide differences were resolved, they appeared to be associated with flanking mismatches rather than their proximity to an insertion or deletion.
Database analysis.
A total of 183 full-length subtype B env sequences were obtained from the Los Alamos HIV sequence database (http://hiv-web.lanl.gov) and translated. A subset of the translated sequences was aligned by using CLUSTAL W (75) followed by manual modification. A profile built from this preliminary alignment was used to align the full set of 183 amino acid sequences using hmmbuild and hmmalign (http://hmmr.wustl.edu). Nucleotide alignments were then created by replacing aligned amino acids with the codons from the corresponding nucleotide sequence. Analysis of codon composition was performed with custom software. Alignments, accession numbers, and software are available on request.
Nucleotide sequence accession number.
The GenBank accession numbers of the sequences reported here are AY281386 to AY281744.
RESULTS
V1/V2-HTA development and validation.
In order to develop a versatile V1/V2-HTA capable of detecting as many different genotypic variants as possible, DNA segments spanning the V1 and V2 regions from four HIV-1 molecular clones, Ba-L, JR-FL, NL4-3, and YU-2, were tested as probes. In this approach, the probes were generated from molecular clones and used to analyze viral sequences present in different subjects. Previous approaches have used isolate-specific (i.e., homologous) V1/V2 probes to follow sequence evolution in infected subjects (61, 69, 83). The use of isolate-specific probes allows the detection of sequence evolution away from the starting sequence, but only after the amount of sequence divergence reaches a threshold of 1 to 2% (18, 76), and this approach is limited by the need to make a new probe for the virus isolate in each subject. The use of generic heterologous probes both simplifies the experimental procedure and takes advantage of the increased sensitivity of detecting point mutations as changes in DNA conformation that is gained when the probe and target differ in length (15, 23) (which is a general feature of pairing V1/V2 regions from different isolates). In addition, the generic probes have a greater genetic distance between the probe and the target sequence, further enhancing the detection of small differences among the target sequences. Thus, rather than detecting evolution away from a parental sequence, this approach uses the ability of the HTA to display many DNA conformations to reveal the presence of multiple coexisting genotypic variants and to assess their stability over time. As described in Materials and Methods, clones of the V1/V2 region from Ba-L and JR-FL probes were chosen as probes for our analysis.
We assessed the utility of these heterologous V1/V2 probes for detecting both sequence divergence and length variability in target sequences generated from this study. For this analysis we constructed a consensus sequence corresponding to the predominant sequence representative of each HTA variant (see Materials and Methods). In 112 pairwise comparisons of different intrasubject genotypic variants defined as discrete variants by HTA, 26 (23%) had no length variation with at least one other variant from the same subject, with the mismatch between variants ranging from 0.23 to 2.55%, (median = 1.15%) for the entire 400- to 420-bp fragment. Since a 1 to 2% mismatch is required for changes in migration (18, 76), a significant fraction of these variants likely would have gone undetected if an isolate-specific HTA probe had been used. By contrast, when these genotypic variants were compared to the Ba-L and JR-FL probes, the percent mismatch rose at least fourfold for both Ba-L (range, 6.02 to 14.29%; median, 11.95%) and JR-FL (range, 4.76 to 13.81%; median, 10.48%), without considering length differences between the probes and the genotypic variants. Thus, the use of heterologous molecular clones as probes is predicted to permit more sensitive detection of V1/V2 genotypic variants based on nucleotide mismatches alone.
We then examined the presence of length differences in a comparison of these heterologous V1/V2 probes with the same set of subject-derived sequences. A length difference between two strands in a heteroduplex has been shown to have a greater effect on its electrophoretic mobility than base pair mismatches (3, 15, 18, 28) in addition to increasing the sensitivity to mismatches flanking the length differences (14, 15). Of the 48 variants assessed in this study, 46 differed in length from the Ba-L probe and all 48 differed in length from the JR-FL probe. Therefore, heteroduplexes formed with a heterologous V1/V2 probe have a high likelihood of having length differences between the probe and target strands, which further enhances the sensitivity for resolving highly related genotypes as discrete bands in the gel.
Initial V1/V2-HTA analysis of longitudinal samples.
A cohort of 21 subjects infected with HIV-1 from the placebo arm of a ritonavir clinical trial was studied (5). Subjects had high, stable virus loads (range = 3.96 to 6.21 log10 copies of viral RNA/ml of blood; average = 5.26 log10) and were in the later stages of infection (CD4+-T-cell counts below 100/μl). All subjects were protease inhibitor naïve and on preexisting RT inhibitor therapy that was unchanged over the period analyzed. High and stable virus loads during ineffective RT inhibitor therapy in these subjects suggest that any observed evolution was in the absence of significant pharmacological selective pressure.
The first and last samples from each of the 21 subjects were analyzed by V1/V2-HTA (Fig. 2). The number of V1/V2 genotypes present at a minimum of 3% relative abundance in the entry sample ranged from 1 to 8, with a mean of 4.8. Three different measures were used to quantify the dynamic nature of the V1/V2 genotypes in these subjects when HTA patterns corresponding to the first and last time points were compared: percent change (50), Nei's standard genetic distance (49), and Euclidean distance. All three measures demonstrated that the amount of change in V1/V2 genotypic variants between these two time points varied widely among the subjects (percent change: range = 2 to 100%, mean = 48%; Nei's distance: range = 0 to 4.21, mean = 0.68; Euclidean distance: range = 0.03 to 1.07, mean = 0.51). When these measures were compared, we found that percent change and Euclidean distance exhibited a linear relationship (R2 = 0.9), with the value for Euclidean distance being approximately 10% larger. The Nei standard genetic distance was linearly related to both the percent change and Euclidean distance except at the extreme ends of the change spectrum. In cases where the amount of change in V1/V2 variants over time was very small, the percent change and Euclidean distance formulas were more sensitive in distinguishing the amount of change, whereas in cases where the amount of change was very large, the Nei standard genetic distance formula was more sensitive in distinguishing the amount of change, with values becoming asymptotic as the percent change approached 100%. Over the range of 10 to 70% change, the values were linearly related to Nei standard genetic distance (R2 = 0.81). The percent change formula was selected to analyze these subjects because it was more sensitive in distinguishing small amounts of change and because the values obtained have a more intuitive interpretation.
FIG. 2.

V1/V2-HTA analysis of entry and ending blood plasma samples. V1/V2-HTAs were performed on first and last samples with both the Ba-L and JR-FL probes; the HTAs with the Ba-L probe are shown for 1025, 1036, 1052, 1053, 1073, 1103, 1116, 1124, 1133, and 1146, while the HTAs with the JR-FL probe are shown for 1004, 1007, 1012, 1018, 1024, 1027, 1063, 1066, 1067, 1079, and 1114. Samples are identified by number of days from the start of the trial. The position of the radiolabeled, single-stranded probe is indicated by an arrow, and the probe homoduplexes are not shown. One subject (1025) had a heteroduplex band that migrated above the single-stranded probe, and one subject (1124) had a heteroduplex band that migrated close to the single-stranded probe. The bands above the single-stranded probe in other subjects (including 1004 and 1146) are additional single-stranded probe bands differentially bound to the PCR primer.
The V1/V2-HTAs for two independent RT-PCR products from the same sample were compared to determine the quality of sampling. Sixteen samples were chosen for analysis either in duplicate or triplicate to quantitate sampling error, with the same number of bands observed in each pair. In order to assess the difference in the intensity of the bands between the repeated samples, the percent change between the repeated RT-PCR products was quantitated. The percent change was low for all 26 sets of repeats, with a mean of 7.6%, and only one pair exhibited >20% change. Therefore, we chose the total percent change in V1/V2 genotypic variants between two time points for a subject of >20% as a significant difference, i.e., outside the range of experimental error. Overall, 16 of the 21 subjects (76%) exhibited a >20% change in their V1/V2 variants over the nine-month period of the trial, and in 75% of those subjects the shift in variants included the gain or loss of at least one V1/V2 variant. Nine subjects exhibiting a wide range of percent change in V1/V2 variants (34 to 100%) were selected for further analysis to determine the pace of change and the nature of the sequence changes involved. These nine subjects include most of the cases with shifts in the V1/V2 population of more than 60%.
Detection of V1/V2 discrete transitions by HTA.
V1/V2-HTA analysis was done on monthly samples from eight of the subjects that were selected for further analysis (1027, 1036, 1053, 1066, 1073, 1079, 1124, and 1133) (Fig. 3). Monthly samples were not available for a ninth subject, so further analysis was done on the two samples available (subject 1063 at entry and month 6, 88% change). The pattern of V1/V2 population change for each of the nine subjects was unique, with one subject (1036) only losing V1/V2 genotypes and the other eight both gaining and losing specific genotypes. There was a single discrete transition, which occurred on average over 3 months, in the V1/V2 genotypes of each subject during the time course that was visualized by V1/V2-HTA (denoted by brackets in Fig. 3). These discrete transitions represent the period when at least 85% of the observed change in the V1/V2 genotypes of each subject occurred (data not shown). The transition periods were flanked by periods of relative stability. The change in V1/V2 genotypes had no measurable correlation with any of the subjects' virus loads, which remained constant throughout the time course (data not shown).
FIG.3.

Longitudinal analysis of V1/V2 genotypic variants. V1/V2-HTAs were performed on all monthly samples from eight subjects and shown with either the Ba-L probe (1036, 1053, 1073, 1124, and 1133) or the JR-FL probe (1027, 1066, and 1079). The samples are identified by number of days from the start of the trial (above each lane). Only the heteroduplex region of each gel is shown. The predominant transition period for each subject is indicated by a bracket. SS, single-stranded probe. The small numbers on the sides of the gels are identifiers for the genotypic variants for which sequence information was obtained. Cartoon depictions of the V1 and V2 regions are shown for each sequenced variant. Lines represent nucleotide sequence differences, and dots represent deletions relative to the other variants. Half-lines are used to show the positions of minor sequence variants. These minor variants were linked when displayed attached to the upper line. Each cartoon represents the consensus sequence for each variant. The number to the left of the cartoon corresponds to the identifying number on the gel, and the number to the right indicates the number of clones obtained and sequenced for each variant. The absence of a number to the right of the cartoon indicates that the PCR product was bulk sequenced. An asterisk to the right of the cartoon indicates that the variant has sequence differences in the C2 region, which are not shown.
Sequence analysis of V1/V2 variants present prior to discrete transitions.
Because of the closely spaced (i.e., monthly) sampling in this cohort, we were able to identify discrete transitions in the V1/V2 population, as opposed to the sum of multiple transitions revealed with more widely spaced samples. Having identified the times of these discrete transitions with the V1/V2-HTA, we were interested in defining the extent and nature of the genotypic changes that defined them. RT-PCR products from time points before and after the periods of observed change in V1/V2 were cloned, and between 3 and 31 clones for each major V1/V2 variant (defined as a discrete band in the HTA representing at least 10% of the total population) were sequenced, except for subjects 1063 and 1073, for whom only bulk sequencing was done because of the low complexity of the population. The RT-PCR product from a single time point was analyzed for subject 1036, since major variants were lost and none were gained.
Pairwise comparisons were performed between all of the sequences corresponding to the major genotypes present within a subject prior to the transition. In most cases (75%), the cross-sectional intrasubject genotypes differed from one another by both point mutations and length. As noted earlier, in 23% of the comparisons the two genotypes were the same length, with an average of three nucleotide differences within the V1/V2 region. In only 2% of the comparisons did the two genotypes differ by length alone. Thus, overall, the discrete V1/V2 genotypes within a subject differ by point mutations 98% of the time and by length differences 77% of the time (Fig. 3). The high percentage of intrasubject genotype comparisons with length differences has been noted before (9, 68), with one group finding that deletions in SIV env appeared more frequently than transversions and only two- to sixfold less frequently than transitions (9).
In order to assess the relationship between nucleotide differences and length differences, we divided the pairwise comparisons of the entry variants into two groups based on the absence or presence of a length difference (Fig. 4). Most of the variants without insertions or deletions had small numbers of nucleotide differences clustered in either V1 or V2, as indicated by a mutation ratio greater than or equal to 2:1 between V1 and V2 (i.e., at least twice as many nucleotide differences in one region as in the other) (data not shown). In contrast, variants with insertions or deletions exhibited either one of two patterns: nucleotide differences clustered within either V1 or V2, or differences widely distributed across both V1 and V2 (mutation ratio < 2:1 between V1 and V2) (data not shown). Among variants with length differences, those with clustered sequence differences had significantly fewer changes within the V1/V2 region than those with widely distributed sequence differences (Mann-Whitney rank sum test, P = 0.004). This observation suggests that the coexisting genotypes comprise multiple populations: those with clustered nucleotide differences that more recently evolved from a common ancestor and those with widely distributed differences that were genetically separated from each other at a more distant time, as evidenced by the greater genetic distance and the presence of length variation.
FIG. 4.

V1/V2 variants analyzed based on the absence or presence of length differences. The graph displays pairwise comparisons of the V1/V2 variants present at entry and the newly emerged V1/V2 variants, broken down into variants with and without size differences. The y axis displays the number of nucleotide differences for each variant. Circles, individual variants with a mutation ratio greater than or equal to 2:1 in either V1 or V2 (at least twice as many mutations in one region over the other); squares, individual variants with a mutation ratio less than 2:1 in either V1 or V2; triangles, individual variants with only one mutation across V1/V2.
Sequence analysis of new variants: nucleotide changes and length variation.
In the eight subjects analyzed in detail that gained new variants over time, a total of 15 V1/V2 variants emerged during the observation period. We compared the sequences of these new variants to the closest similar sequence present at entry to determine the nature of the changes that appeared after discrete V1/V2 transitions. As we had done with the entry variants, we grouped 13 of the newly emerged variants based on the absence or presence of a length difference relative to the most similar entry sequence (Fig. 4). Approximately one-half of the newly emerged variants (six) did not have length differences. These variants had small numbers of nucleotide substitutions within V1/V2, with three having only one change (data not shown) and three having between two and eight nucleotide substitutions (Fig. 5). As with the entry variants, the nucleotide substitutions in two of the three variants with multiple nucleotide substitutions were clustered within either the V1 or the V2 region. The presence of clustered nucleotide substitutions suggests localized selection in either the V1 or V2 region.
FIG. 5.

Nucleotide differences and size differences in newly emerged V1/V2 variants. The nucleotide sequence of each newly emerged variant was compared to the nucleotide sequence of an entry variant that was the closest related variant. For each variant, a coding point mutation change is noted by an arrow where the change occurred. Numbers in parentheses next to the subject identification numbers are the total number of coding changes in the V1/V2 region for that variant. Numbers above or below the arrows indicate that more than one coding change occurred within one codon. Size differences are noted by an asterisk, with the change in the number of nucleotides noted above the asterisk.
The other seven newly emerged variants had length differences with respect to the entry variants. The numbers of nucleotide substitutions across V1/V2 varied. Four variants had small numbers of clustered nucleotide substitutions (1 to 9 nucleotide changes, the variant with 1 nucleotide substitution is not shown), and three had larger numbers of widely distributed nucleotide substitutions (18 to 37 nucleotide changes) (Fig. 5). The variants with length differences and clustered mutations had significantly fewer nucleotide changes than those with length differences and widely distributed changes (Mann-Whitney rank sum test, P = 0.02). All of the variants with clustered nucleotide substitutions also had insertions or deletions clustered in the same region. The presence of clustered nucleotide substitutions as well as clustered length differences is again suggestive of the effects of localized selection in either V1 or V2. All of the variants with widely distributed nucleotide substitutions had length differences across V1 and V2, and two of the three variants had multiple length differences across the V1/V2 region. The widely distributed nature of the nucleotide substitutions and length differences across the V1/V2 region in these variants compared to the closest similar sequence at entry is suggestive of a longer period of divergence than for the variants with clustered mutations.
Sequence analysis of new variants: composition of length-variable sequences.
We noted a high percentage of codons in the V1 and V2 regions with the motif AVT (V = A, G, or C) in the length variable regions. The three codons with this motif encode asparagine (Asn, AAT), serine (Ser, AGT), and threonine (Thr, ACT). We examined the distribution of these codons versus the other degenerate codons for these amino acids across the length of gp120 in an alignment of 183 subtype B Env sequences obtained from the Los Alamos HIV-1 sequence database (Fig. 6). There was an enriched representation of these three amino acids encoded by the AVT motif in V1, V2, and V4 but not in V3 or V5. The enriched representation of AVT in the V1 and V2 regions coincides with the regions of V1 and V2 in which we observed length differences in our subjects. A similar observation was previously made by Bosch et al. (4), who suggested that a specific expansion of the AAT codon in V1, V2, V4, and V5 was followed by evolution at the second codon position to generate sequences encoding the glycosylation signal Asn-X-Ser/Thr.
FIG. 6.

Composition of asparagine (N), serine, (S), and threonine (T) codons in an alignment of 183 full-length gp120 coding sequences from the Los Alamos HIV sequence database. Each point in the plot represents the center of a 25-codon sliding window. The percentage of all aligned codons in each window that are AAT, ACT, or AGT is indicated by the gray-filled plot; the heavy black line shows the total percentage of all other asparagine, serine, or threonine codons. The sum of the two plots is therefore equal to the percentage of all asparagine, serine, or threonine codons. Vertical lines demarcate the regions of length polymorphism in V1, V2, V4, and V5 and indicate the positions of the cysteines flanking V3. The percentages of all asparagine, serine, or threonine codons of either category in each region of gp120 are shown at the top of the plot.
Sequence analysis of new variants: recombination.
Retroviruses are able to undergo high levels of recombination (reviewed in references 29 and 31). In two of the nine subjects analyzed, there was evidence that recombination had occurred to create a total of three new viral genotypic variants (Fig. 7). In subject 1133, two preexisting variants (A and B) acquired the same V2 loop by recombination with a third variant (C). Variant C was a minor variant at day 1 that expanded by day 169, possibly because of a lack of selective pressure on its V2 loop sequence. After the population transition, all three predominant V1/V2 variants (C, D, and E) had the same V2 loop. The acquired V2 loop differed from the preexisting V2 loop by five or six point mutations and a three-amino-acid deletion. One of the point mutations caused an N-linked glycosylation site to shift three amino acids towards the N terminus of the loop. We detected a third recombination event in the C2 region of subject 1079 that did not change the makeup of the V1/V2 populations.
FIG. 7.

Example of recombination between V1 and V2 in vivo. The V1/V2-HTA time points from subject 1133 from before and after the recombination events are shown on the left. The cartoon depicts the time points shown in the gel. The V1/V2 variants involved in the recombination event are lettered similarly on the gel and in the cartoon. Major V1/V2 genotypic variants that were not involved in the recombination event are marked on the gel with an asterisk. Different V1 and V2 sequence motifs are distinguished by shading and hatching. SS, single-stranded probe. The V1/V2-HTA lanes are from Fig. 2.
In the two recombinants from subject 1133, the site of recombination was located between the regions encoding V1 and V2. However, because these intervening regions are more conserved, the site of recombination could be narrowed only to an 81-bp region. We speculated that features of RNA secondary structure might promote polymerase stalling and recombination between V1 and V2, allowing them to spread independently through the virus population; however, we found no evidence for such a structure (data not shown).
DISCUSSION
We have developed a V1/V2-HTA that utilizes molecular clones as probes, instead of the less versatile isolate-specific probes. The use of molecular clones as probes increased the amount of sequence divergence between the probe and the different genotypic variants approximately fourfold, which permitted more sensitive detection of coexisting V1/V2 genotypes in cross-sectional analysis. Also, the use of generic probes increased the likelihood that there were size differences between the genotypic variants and the probe, which has a greater effect on heteroduplex mobility than sequence mismatches and also enhances the sensitivity for the detection of mismatches. We have used this more sensitive V1/V2-HTA to explore the nature of V1/V2 sequence change in a cohort of 21 late-stage HIV-1-infected subjects over a 9-month period. The majority of subjects (76%) exhibited detectable changes in their V1/V2 populations over time, with one-half of all subjects displaying a gain or loss of at least one V1/V2 variant. We selected nine subjects with a wide range of genotypic change in V1/V2 populations (34 to 100% change) to further analyze the rate and nature of changes in the V1/V2 region. V1/V2-HTA was performed on monthly samples from eight of the subjects, and the results demonstrated a unique pattern of discrete genotypic changes for each subject, with the majority of subjects both gaining and losing genotypic variants. The observed changes in V1/V2 occurred as discrete transitions, usually within a 3-month period, which were flanked by periods of relative genotypic stability.
Our analysis of both the coexisting genotypic variants present at entry and of new variants that appeared during the discrete transitions shows that multiple V1/V2 lineages can coexist. Within a lineage, variants differ by a small number of nucleotide differences that are largely clustered within either V1 or V2. When length differences are present, they are localized with the clustered nucleotide differences. We interpret these features as evidence that the sequences diverged recently. More distant lineages of V1/V2 variants have larger numbers of nucleotide differences. Also, the differences are dispersed across V1 and V2 and are accompanied by more and larger length differences. We interpret these features as evidence of greater evolutionary distance. Although we do not know how much time is required to accumulate the numbers of nucleotide differences seen with these variants, we suggest that the appearance of the more distant variants during a discrete transition represents the reappearance of older sequences as a result of an altered pattern of selective pressures. These preexisting variants could have been present at a low level in the blood, circulating in another compartment, or archived in proviral DNA and recombined into an actively replicating genome. If this interpretation is correct, then it is analogous to the reappearance of wild-type pro and pol sequences when drug selective pressure is removed from subjects for whom therapy is failing due to drug resistance (13). We found that distant lineages coexisted in all six subjects that had multiple variants present at trial entry. Furthermore, in the three subjects that had multiple new variants emerge, two of the three subjects had combinations of similar and distant variants emerge simultaneously. These results suggest that multiple V1/V2 lineages can evolve independently from one another and be simultaneously maintained.
Bosch et al. (4) proposed that AVT motifs concentrated in V1, V2, V4, and V5 are the result of insertions of an AAT motif followed by selection for glycosylation sites. We considered such a possibility by analyzing glycosylation sites across gp120 among the sequences in the Los Alamos HIV-1 sequence database. Surprisingly, glycosylation sites in the conserved regions of gp120 were more similar to an expansion of an AAT motif than the glycosylation sites in the variable regions. Glycosylation sites in the conserved regions of gp120 differed largely by transitions from AAT in the nine nucleotides of the three codons of the Asn-X-Ser/Thr motif (the one exception was nucleotide 8, which was a C 87% of the time, to encode Thr). By contrast, among glycosylation sites in the variable regions, position 5 in the middle of the second codon was a T 44% of the time and position 6 was a T only 19% of the time. Thus, it is more likely that the glycosylation sites outside the variable regions are the result of AAT expansion than those within the variable regions. We also found that AVT codons are not clustered within V5 as was previously suggested (4). An alternative explanation for the high concentration of AVT motifs in the variable domains is that AVT codons are maintained because of their ability to mediate size variation as well as encode glycosylation sites. While the reason for this concentration of AVT motifs in V1, V2, and V4 remains an open question, there is clearly a strong selective pressure for their presence in these regions, as the AVT motif represents a minority of the codons used to encode these amino acids outside the variable regions (Fig. 6).
Changes in glycosylation have been found to be an important feature in the evolution of V1/V2 in vivo. Studies examining SIV evolution in infected animals have shown that the V1 region gains new N-linked and potential O-linked glycosylation sites as the animals progress to disease (7, 53, 66). Furthermore, studies with both SIV and HIV have shown that alterations in N- and O-linked glycosylation sites in both V1 and V2 can be associated with increased neutralization resistance (8, 43). We observed at least one change in glycosylation sites in 5 of 13 new variants with point mutations. Six of the changes resulted in the gain of a glycosylation site, and three of the changes resulted in the loss of a glycosylation site. Therefore, 8% of the point mutation events appeared to have a direct effect on glycosylation in the V1/V2 region. However, any point mutation or insertion/deletion event resulting in a coding change may have an indirect effect on glycosylation by affecting the angle or position of the sugar groups through adjustments in the polypeptide chain, thus changing the areas that are protected.
Evidence of three independent recombination events was seen in two of the subjects analyzed, 1079 and 1133. Since the breakpoints for two of the three events occurred between V1 and V2, there may be independent selective pressure on each of the domains. Recombination in primate lentiviruses has been demonstrated in cell culture (48) and in vivo (33, 78) and has been inferred to be fairly common given the number of interclade HIV-1 recombinants (reviewed in reference 57). The fact that recombination events occurred in some of these subjects requires that a percentage of cells in these hosts be coinfected and that these coinfections be able to lead to biologically relevant recombinants (33).
Changes in viral populations can occur as the result of a change in selective pressure or by drift due to stochastic processes. There are several lines of evidence that can distinguish between selection and drift due to stochastic processes. Monte Carlo computer simulations and the ratio of synonymous to nonsynonymous point mutations (ds/dn) were both calculated in an effort to determine whether the changes we observed in the V1/V2 populations are consistent with selection or stochastic processes. Computer simulations were done for subject 1133, in whom two variants emerged simultaneously, after which there was a period of genotypic stability. The simulation allowed a determination of the probability of both variants rising from a low frequency to a high frequency over 239 days if stochastic processes were the principal force defining the population structure assuming multiple generation times (0.5 to 2 days) and population sizes (250 to 8,000). The computer simulation results are consistent with the pattern of change in subject 1133 being driven by selection over the range of values tested (D. Nickle and J. Mullins, personal communication). The ds/dn ratios were calculated for the newly emerged V1/V2 variants with clustered point mutations using SNAP (Synonymous/Non-synonymous Analysis Program; www.hiv.lanl.gov). Comparisons of the newly emerged variants with clustered mutations and the presumed parent revealed the overall ds/dn ratio was 1, indicating a neutral mutation rate. However, if one considers only the mutations that became fixed in a majority of sequences, then the ds/dn ratio was less than 1, consistent with positive selection. In addition, the clustering of nucleotide changes and size differences within either V1 or V2 when smaller numbers of nucleotide changes are present is also consistent with positive selection. We believe that the weight of evidence is consistent with a selective pressure, presumably antibody selection, driving these discrete transitions.
Data in the literature are conflicting as to whether evolution of the env gene is driven by positive selection or genetic drift and/or stochastic effects. Several studies have demonstrated that the env gene is under positive selection (52, 64, 67, 80, 81), with some of the studies finding positive selection driving evolution specifically in the V2 region (67, 81). In contrast, other studies have shown that genotypic changes in multiple regions of the env gene, including the V1/V2 region, are driven primarily by genetic drift and/or stochastic effects (35, 40, 58). However, in one of those studies, the argument for a lack of positive selection was weakest for regions in env relative to other regions of the viral genome (35). It is difficult to resolve the different conclusions reached by these previous studies, given the different types of analyses performed on different sequence sets.
In summary, our results suggest a complex structure of V1/V2 populations in vivo composed of both highly related and more divergent sequences. We were able to observe discrete changes in V1/V2 populations with clear examples of evolution through nucleotide substitutions, size differences, and recombination. Newly emerging sequence variants containing clustered nucleotide changes and clustered size differences were suggestive of recent evolution, and variants with high numbers of nucleotide changes and size differences were suggestive of recall of older sequences. Length variation appeared to occur within regions of high AVT motif content. Finally, the periods of discrete transitions flanked by periods of relative stability that we observed suggest that changes in selective pressure, presumably by antibodies, are sporadic in late-stage subjects.
ADDENDUM IN PROOF
Karlsson et al. previously observed the reappearance of founder p17 and V3 sequences in three patients at the time of clinical deterioration and increasing virus load (A. C. Karlsson, H. Gaines, M. Sällberg, S. Lindbäck, and A. Sönnerborg, J. Virol. 73:6191-6196, 1999).
Acknowledgments
This work was supported by NIH grant R01-AI44667 and by the UNC Center for AIDS Research award (P30-AI50410). K.M.K. was supported in part by NIH training grant T32-GM07092, and N.G.H. was supported in part by NIH training grants T32-AI07419 and T32-AI07001.
We thank Dale Kempf for assistance in obtaining samples. The pYU-2 plasmid was obtained from Beatrice Hahn and George Shaw through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. We thank Jim Mullins and Andrew Leigh Brown for comments.
REFERENCES
- 1.Adachi, A., H. E. Gendelman, S. Koenig, T. Folks, R. Willey, A. Rabson, and M. A. Martin. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrie, K. A., E. E. Perez, S. L. Lamers, W. G. Farmerie, B. M. Dunn, J. W. Sleasman, and M. M. Goodenow. 1996. Natural variation in HIV-1 protease, Gag p7 and p6, and protease cleavage sites within gag/pol polyproteins: amino acid substitutions in the absence of protease inhibitors in mothers and children infected by human immunodeficiency virus type 1. Virology 219:407-416. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharyya, A., and D. M. Lilley. 1989. The contrasting structures of mismatched DNA sequences containing looped-out bases (bulges) and multiple mismatches (bubbles). Nucleic Acids Res. 17:6821-6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosch, M. L., A. C. Andeweg, R. Schipper, and M. Kenter. 1994. Insertion of N-linked glycosylation sites in the variable regions of the human immunodeficiency virus type 1 surface glycoprotein through AAT triplet reiteration. J. Virol. 68:7566-7569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cameron, D. W., M. Heath-Chiozzi, S. Danner, C. Cohen, S. Kravcik, C. Maurath, E. Sun, D. Henry, R. Rode, A. Potthoff, and J. Leonard. 1998. Randomised placebo-controlled trial of ritonavir in advanced HIV-1 disease. Lancet 351:543-549. [DOI] [PubMed] [Google Scholar]
- 6.Cao, J., N. Sullivan, E. Desjardin, C. Parolin, J. Robinson, R. Wyatt, and J. Sodroski. 1997. Replication and neutralization of human immunodeficiency virus type 1 lacking the V1 and V2 variable loops of the gp120 envelope glycoprotein. J. Virol. 71:9808-9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chackerian, B., L. M. Rudensey, and J. Overbaugh. 1997. Specific N-linked and O-linked glycosylation modifications in the envelope V1 domain of simian immunodeficiency virus variants that evolve in the host alter recognition by neutralizing antibodies. J. Virol. 71:7719-7727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng-Mayer, C., A. Brown, J. Harouse, P. A. Luciw, and A. J. Mayer. 1999. Selection for neutralization resistance of the simian/human immunodeficiency virus SHIVSF33A variant in vivo by virtue of sequence changes in the extracellular envelope glycoprotein that modify N-linked glycosylation. J. Virol. 73:5294-5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheynier, R., L. Kils-Hutten, A. Meyerhans, and S. Wain-Hobson. 2001. Insertion/deletion frequencies match those of point mutations in the hypervariable regions of the simian immunodeficiency virus surface envelope gene. J. Gen. Virol. 82:1613-1619. [DOI] [PubMed] [Google Scholar]
- 10.Cho, M. W., M. K. Lee, M. C. Carney, J. F. Berson, R. W. Doms, and M. A. Martin. 1998. Identification of determinants on a dualtropic human immunodeficiency virus type 1 envelope glycoprotein that confer usage of CXCR4. J. Virol. 72:2509-2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coffin, J. M. 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science 267:483-489. [DOI] [PubMed] [Google Scholar]
- 12.Contag, C. H., A. Ehrnst, J. Duda, A. B. Bohlin, S. Lindgren, G. H. Learn, and J. I. Mullins. 1997. Mother-to-infant transmission of human immunodeficiency virus type 1 involving five envelope sequence subtypes. J. Virol. 71:1292-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deeks, S. G., T. Wrin, T. Liegler, R. Hoh, M. Hayden, J. D. Barbour, N. S. Hellmann, C. J. Petropoulos, J. M. McCune, M. K. Hellerstein, and R. M. Grant. 2001. Virologic and immunologic consequences of discontinuing combination antiretroviral-drug therapy in HIV-infected patients with detectable viremia. N. Engl. J. Med. 344:472-480. [DOI] [PubMed] [Google Scholar]
- 14.Delwart, E. L., M. P. Busch, M. L. Kalish, J. W. Mosley, and J. I. Mullins. 1995. Rapid molecular epidemiology of human immunodeficiency virus transmission. AIDS Res. Hum. Retrovir. 11:1081-1093. [DOI] [PubMed] [Google Scholar]
- 15.Delwart, E. L., and C. J. Gordon. 1997. Tracking changes in HIV-1 envelope quasispecies using DNA heteroduplex analysis. Methods 12:348-354. [DOI] [PubMed] [Google Scholar]
- 16.Delwart, E. L., J. I. Mullins, P. Gupta, G. H. Learn, Jr., M. Holodniy, D. Katzenstein, B. D. Walker, and M. K. Singh. 1998. Human immunodeficiency virus type 1 populations in blood and semen. J. Virol. 72:617-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delwart, E. L., H. Pan, H. W. Sheppard, D. Wolpert, A. U. Neumann, B. Korber, and J. I. Mullins. 1997. Slower evolution of human immunodeficiency virus type 1 quasispecies during progression to AIDS. J. Virol. 71:7498-7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delwart, E. L., H. W. Sheppard, B. D. Walker, J. Goudsmit, and J. I. Mullins. 1994. Human immunodeficiency virus type 1 evolution in vivo tracked by DNA heteroduplex mobility assays. J. Virol. 68:6672-6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delwart, E. L., E. G. Shpaer, J. Louwagie, F. E. McCutchan, M. Grez, H. Rubsamen-Waigmann, and J. I. Mullins. 1993. Genetic relationships determined by a DNA heteroduplex mobility assay: analysis of HIV-1 env genes. Science 262:1257-1261. [DOI] [PubMed] [Google Scholar]
- 20.Diaz, R. S., C. F. De Oliveira, R. Pardini, E. Operskalski, A. J. Mayer, and M. P. Busch. 1999. HIV type 1 tat gene heteroduplex mobility assay as a tool to establish epidemiologic relationships among HIV type 1-infected individuals. AIDS Res. Hum. Retrovir. 15:1151-1156. [DOI] [PubMed] [Google Scholar]
- 21.Doukhan, L., and E. Delwart. 2001. Population genetic analysis of the protease locus of human immunodeficiency virus type 1 quasispecies undergoing drug selection, using a denaturing gradient-heteroduplex tracking assay. J. Virol. 75:6729-6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Etemad-Moghadam, B., Y. Sun, E. K. Nicholson, G. B. Karlsson, D. Schenten, and J. Sodroski. 1999. Determinants of neutralization resistance in the envelope glycoproteins of a simian-human immunodeficiency virus passaged in vivo. J. Virol. 73:8873-8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon, C. J., and E. L. Delwart. 2000. Genetic diversity of primary HIV-1 isolates and their sensitivity to antibody-mediated neutralization. Virology 272:326-330. [DOI] [PubMed] [Google Scholar]
- 24.Greenier, J. L., C. J. Miller, D. Lu, P. J. Dailey, F. X. Lu, K. J. Kunstman, S. M. Wolinsky, and M. L. Marthas. 2001. Route of simian immunodeficiency virus inoculation determines the complexity but not the identity of viral variant populations that infect rhesus macaques. J. Virol. 75:3753-3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hahn, B. H., M. A. Gonda, G. M. Shaw, M. Popovic, J. A. Hoxie, R. C. Gallo, and F. Wong-Staal. 1985. Genomic diversity of the acquired immune deficiency syndrome virus HTLV-III: different viruses exhibit greatest divergence in their envelope genes. Proc. Natl. Acad. Sci. USA 82:4813-4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heyndrickx, L., W. Janssens, L. Zekeng, R. Musonda, S. Anagonou, G. Van der Auwera, S. Coppens, K. Vereecken, K. De Witte, R. Van Rampelbergh, M. Kahindo, L. Morison, F. E. McCutchan, J. K. Carr, J. Albert, M. Essex, J. Goudsmit, B. Asjo, M. Salminin, A. Buve, and G. van der Groen. 2000. Simplified strategy for detection of recombinant human immunodeficiency virus type 1 group M isolates by gag/env heteroduplex mobility assay. J. Virol. 74:363-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoffman, N. G., F. Seillier-Moiseiwitsch, J. Ahn, J. M. Walker, and R. Swanstrom. 2002. Variability in the human immunodeficiency virus type 1 gp120 Env protein linked to phenotype-associated changes in the V3 loop. J. Virol. 76:3852-3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsieh, C. H., and J. D. Griffith. 1989. Deletions of bases in one strand of duplex DNA, in contrast to single-base mismatches, produce highly kinked molecules: possible relevance to the folding of single-stranded nucleic acids. Proc. Natl. Acad. Sci. USA 86:4833-4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu, W., V. K. Pathak, and H. M. Temin. 1993. Role of reverse transcription in retroviral recombination, p. 251-274. In A. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 30.Hu, W. S., E. H. Bowman, K. A. Delviks, and V. K. Pathak. 1997. Homologous recombination occurs in a distinct retroviral subpopulation and exhibits high negative interference. J. Virol. 71:6028-6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu, W. S., and H. M. Temin. 1990. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. USA 87:1556-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson, W. E., J. Morgan, J. Reitter, B. A. Puffer, S. Czajak, R. W. Doms, and R. C. Desrosiers. 2002. A replication-competent, neutralization-sensitive variant of simian immunodeficiency virus lacking 100 amino acids of envelope. J. Virol. 76:2075-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jung, A., R. Maier, J. Vartanian, G. Bocharov, V. Jung, U. Fischer, E. Meese, S. Wain-Hobson, and A. Meyerhans. 2002. Multiply infected spleen cells in HIV patients. Nature 418:144.. [DOI] [PubMed] [Google Scholar]
- 34.Jurkiewicz, E., G. Hunsmann, J. Schaffner, T. Nisslein, W. Luke, and H. Petry. 1997. Identification of the V1 region as a linear neutralizing epitope of the simian immunodeficiency virus SIVmac envelope glycoprotein. J. Virol. 71:9475-9481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kils-Hutten, L., R. Cheynier, S. Wain-Hobson, and A. Meyerhans. 2001. Phylogenetic reconstruction of intrapatient evolution of human immunodeficiency virus type 1: predominance of drift and purifying selection. J. Gen. Virol. 82:1621-1627. [DOI] [PubMed] [Google Scholar]
- 36.Koyanagi, Y., S. Miles, R. T. Mitsuyasu, J. E. Merrill, H. V. Vinters, and I. S. Y. Chen. 1987. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science 236:819-822. [DOI] [PubMed] [Google Scholar]
- 37.Kozal, M. J., N. Shah, N. Shen, R. Yang, R. Fucini, T. C. Merigan, D. D. Richman, D. Morris, E. Hubbell, M. Chee, and T. R. Gingeras. 1996. Extensive polymorphisms observed in HIV-1 clade B protease gene using high-density oligonucleotide arrays. Nat. Med. 2:753-759. [DOI] [PubMed] [Google Scholar]
- 38.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648-659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lech, W. J., G. Wang, Y. L. Yang, Y. Chee, K. Dorman, D. McCrae, L. C. Lazzeroni, J. W. Erickson, J. S. Sinsheimer, and A. H. Kaplan. 1996. In vivo sequence diversity of the protease of human immunodeficiency virus type 1: presence of protease inhibitor-resistant variants in untreated subjects. J. Virol. 70:2038-2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leigh Brown, A. J. 1997. Analysis of HIV-1 env gene sequences reveals evidence for a low effective number in the viral population. Proc. Natl. Acad. Sci. USA 94:1862-1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li, Y., H. Hui, C. J. Burgess, R. W. Price, P. M. Sharp, B. H. Hahn, and G. M. Shaw. 1992. Complete nucleotide sequence, genome organization, and biological properties of human immunodeficiency virus type 1 in vivo: evidence for limited defectiveness and complementation. J. Virol. 66:6587-6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li, Y., J. C. Kappes, J. A. Conway, R. W. Price, G. M. Shaw, and B. H. Hahn. 1991. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J. Virol. 65:3973-3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ly, A., and L. Stamatatos. 2000. V2 loop glycosylation of the human immunodeficiency virus type 1 SF162 envelope facilitates interaction of this protein with CD4 and CCR5 receptors and protects the virus from neutralization by anti-V3 loop and anti-CD4 binding site antibodies. J. Virol. 74:6769-6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mansky, L. M., and H. M. Temin. 1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087-5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mansky, L. M., and H. M. Temin. 1994. Lower mutation rate of bovine leukemia virus relative to that of spleen necrosis virus. J. Virol. 68:494-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGrath, K. M., N. G. Hoffman, W. Resch, J. A. E. Nelson, and R. Swanstrom. 2001. Using HIV-1 sequence variability to explore virus biology. Virus Res. 76:137-160. [DOI] [PubMed] [Google Scholar]
- 47.Menu, E., F. X. M'bopi-Kéou, S. Lagaye, S. Pissard, P. Mauclère, G. Scarlatti, J. Martin, M. Goossens, G. Chaouat, F. Barré-Sinoussi, and the European Network for In Utero Transmission of HIV-1. 1999. Selection of maternal human immunodeficiency virus type 1 variants in human placenta. J. Infect. Dis. 179:44-51. [DOI] [PubMed] [Google Scholar]
- 48.Moutouh, L., J. Corbeil, and D. D. Richman. 1996. Recombination leads to the rapid emergence of HIV-1 dually resistant mutants under selective drug pressure. Proc. Natl. Acad. Sci. USA 93:6106-6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nei, M. 1972. Genetic distance between populations. Am. Nat. 106:283-291. [Google Scholar]
- 50.Nelson, J. A. E., F. Baribaud, T. Edwards, and R. Swanstrom. 2000. Patterns of changes in human immunodeficiency virus type 1 V3 sequence populations late in infection. J. Virol. 74:8494-8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nelson, J. A. E., S. A. Fiscus, and R. Swanstrom. 1997. Evolutionary variants of the human immunodeficiency virus type 1 V3 region characterized by using a heteroduplex tracking assay. J. Virol. 71:8750-8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nielsen, R., and Z. Yang. 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929-936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Overbaugh, J., and L. M. Rudensey. 1992. Alterations in potential sites for glycosylation predominate during evolution of the simian immunodeficiency virus envelope gene in macaques. J. Virol. 66:5937-5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Parthasarathi, S., A. Varela-Echavarria, Y. Ron, B. D. Preston, and J. P. Dougherty. 1995. Genetic rearrangements occurring during a single cycle of murine leukemia virus vector replication: characterization and implications. J. Virol. 69:7991-8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pathak, V. K., and H. M. Temin. 1990. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: deletions and deletions with insertions. Proc. Natl. Acad. Sci. USA 87:6024-6028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pathak, V. K., and H. M. Temin. 1990. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: substitutions, frameshifts, and hypermutations. Proc. Natl. Acad. Sci. USA 87:6019-6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peeters, M. 2000. HIV sequence compendium 2000. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, N.Mex.
- 58.Pelletier, E., W. Saurin, R. Cheynier, N. L. Letvin, and S. Wain-Hobson. 1995. The tempo and mode of SIV quasispecies development in vivo calls for massive viral replication and clearance. Virology 208:644-652. [DOI] [PubMed] [Google Scholar]
- 59.Ping, L.-H., J. A. E. Nelson, I. F. Hoffman, J. Schock, S. L. Lamers, M. Goodman, P. Vernazza, P. Kazembe, M. Maida, D. Zimba, M. M. Goodenow, J. J. Eron, Jr., S. A. Fiscus, M. S. Cohen, and R. Swanstrom. 1999. Characterization of V3 sequence heterogeneity in subtype C HIV-1 isolates from Malawi: underrepresentation of X4 variants. J. Virol. 73:6271-6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinones-Mateu, M. E., J. L. Albright, A. Mas, V. Soriano, and E. J. Arts. 1998. Analysis of pol gene heterogeneity, viral quasispecies, and drug resistance in individuals infected with group O strains of human immunodeficiency virus type 1. J. Virol. 72:9002-9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Radaelli, A., G. Kraus, A. Schmidt, P. Badel, J. McClure, S. L. Hu, W. Morton, C. De Giuli Morghen, F. Wong-Staal, and D. Looney. 1998. Genetic variation in a human immunodeficiency virus type 2 live-virus Macaca nemestrina vaccine model. J. Virol. 72:7871-7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Resch, W., P. N., E. L. Stuelke, and R. Swanstrom. 2000. A multiple site-specific heteroduplex tracking assay as a new tool for the study of viral population dynamics. Proc. Natl. Acad. Sci. USA 98:176-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robert-Guroff, M., M. Popovic, S. Gartner, P. Markham, R. C. Gallo, and M. S. Reitz. 1990. Structure and expression of tat-, rev-, and nef-specific transcripts of human immunodeficiency virus type 1 in infected lymphocytes and macrophages. J. Virol. 64:3391-3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ross, H. A., and A. G. Rodrigo. 2002. Immune-mediated positive selection drives human immunodeficiency virus type 1 molecular variation and predicts disease duration. J. Virol. 76:11715-11720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ross, T. M., and B. R. Cullen. 1998. The ability of HIV type 1 to use CCR-3 as a coreceptor is controlled by envelope V1/V2 sequences acting in conjunction with a CCR-5 tropic V3 loop. Proc. Natl. Acad. Sci. USA 95:7682-7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rudensey, L. M., J. T. Kimata, E. M. Long, B. Chackerian, and J. Overbaugh. 1998. Changes in the extracellular envelope glycoprotein of variants that evolve during the course of simian immunodeficiency virus SIVMne infection affect neutralizing antibody recognition, syncytium formation, and macrophage tropism but not replication, cytopathicity, or CCR-5 coreceptor recognition. J. Virol. 72:209-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seibert, S. A., C. Y. Howell, M. K. Hughes, and A. L. Hughes. 1995. Natural selection on the gag, pol, and env genes of human immunodeficiency virus 1 (HIV-1). Mol. Biol. Evol. 12:803-813. [DOI] [PubMed] [Google Scholar]
- 68.Simmonds, P., P. Balfe, C. A. Ludlam, J. O. Bishop, and A. J. Brown. 1990. Analysis of sequence diversity in hypervariable regions of the external glycoprotein of human immunodeficiency virus type 1. J. Virol. 64:5840-5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sodora, D. L., F. Lee, P. J. Dailey, and P. A. Marx. 1998. A genetic and viral load analysis of the simian immunodeficiency virus during the acute phase in macaques inoculated by the vaginal route. AIDS Res. Hum. Retrovir. 14:171-181. [DOI] [PubMed] [Google Scholar]
- 70.Stamatatos, L., M. Wiskerchen, and C. Cheng-Mayer. 1998. Effect of major deletions in the V1 and V2 loops of a macrophage-tropic HIV type 1 isolate on viral envelope structure, cell entry, and replication. AIDS Res. Hum. Retrovir. 14:1129-1139. [DOI] [PubMed] [Google Scholar]
- 71.Starcich, B. R., B. H. Hahn, G. M. Shaw, P. D. McNeely, S. Modrow, H. Wolf, E. S. Parks, W. P. Parks, S. F. Josephs, R. C. Gallo, and F. Wong-Staal. 1986. Identification and characterization of conserved and variable regions in the envelope gene of HTLV-III/LAV, the retrovirus of AIDS. Cell 45:637-648. [DOI] [PubMed] [Google Scholar]
- 72.Sullivan, N., Y. Sun, Q. Sattentau, M. Thali, D. Wu, G. Denisova, J. Gershoni, J. Robinson, J. Moore, and J. Sodroski. 1998. CD4-induced conformational changes in the human immunodeficiency virus type 1 gp120 glycoprotein: consequences for virus entry and neutralization. J. Virol. 72:4694-4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tatt, I. D., K. L. Barlow, and J. P. Clewley. 2000. A gag gene heteroduplex mobility assay for subtyping HIV-1. J. Virol. Methods 87:41-51. [DOI] [PubMed] [Google Scholar]
- 74.Temin, H. M. 1993. Retrovirus variation and reverse transcription: abnormal strand transfers result in retrovirus genetic variation. Proc. Natl. Acad. Sci. USA 90:6900-6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Upchurch, D. A., R. Shankarappa, and J. I. Mullins. 2000. Position and degree of mismatches and the mobility of DNA heteroduplexes. Nucleic Acids Res. 28:E69.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wolinsky, S. M., B. T. Korber, A. U. Neumann, M. Daniels, K. J. Kunstman, A. J. Whetsell, M. R. Furtado, Y. Cao, D. D. Ho, and J. T. Safrit. 1996. Adaptive evolution of human immunodeficiency virus-type 1 during the natural course of infection. Science 272:537-542. [DOI] [PubMed] [Google Scholar]
- 78.Wooley, D. P., R. A. Smith, S. Czajak, and R. C. Desrosiers. 1997. Direct demonstration of retroviral recombination in a rhesus monkey. J. Virol. 71:9650-9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wyatt, R., J. Moore, M. Accola, E. Desjardin, J. Robinson, and J. Sodroski. 1995. Involvement of the V1/V2 variable loop structure in the exposure of human immunodeficiency virus type 1 gp120 epitopes induced by receptor binding. J. Virol. 69:5723-5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yamaguchi, Y., and T. Gojobori. 1997. Evolutionary mechanisms and population dynamics of the third variable envelope region of HIV within single hosts. Proc. Natl. Acad. Sci. USA 94:1264-1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yamaguchi-Kabata, Y., and T. Gojobori. 2000. Reevaluation of amino acid variability of the human immunodeficiency virus type 1 gp120 envelope glycoprotein and prediction of new discontinuous epitopes. J. Virol. 74:4335-4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang, J., L. Tang, T. Li, Y. Ma, and C. M. Sapp. 2000. Most retroviral recombinations occur during minus-strand DNA synthesis. J. Virol. 74:2313-2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu, T., N. Wang, A. Carr, D. S. Nam, R. Moor-Jankowski, D. A. Cooper, and D. D. Ho. 1996. Genetic characterization of human immunodeficiency virus type 1 in blood and genital secretions: evidence for viral compartmentalization and selection during sexual transmission. J. Virol. 70:3098-3107. [DOI] [PMC free article] [PubMed] [Google Scholar]