

Abstract
The adeno-associated virus type 2 (AAV2) uses heparan sulfate proteoglycan (HSPG) as its primary cellular receptor. In order to identify amino acids within the capsid of AAV2 that contribute to HSPG association, we used biochemical information about heparin and heparin sulfate, AAV serotype protein sequence alignments, and data from previous capsid studies to select residues for mutagenesis. Charged-to-alanine substitution mutagenesis was performed on individual residues and combinations of basic residues for the production and purification of recombinant viruses that contained a green fluorescent protein (GFP) reporter gene cassette. Intact capsids were assayed for their ability to bind to heparin-agarose in vitro, and virions that packaged DNA were assayed for their ability to transduce normally permissive cell lines. We found that mutation of arginine residues at position 585 or 588 eliminated binding to heparin-agarose. Mutation of residues R484, R487, and K532 showed partial binding to heparin-agarose. We observed a general correlation between heparin-agarose binding and infectivity as measured by GFP transduction; however, a subset of mutants that partially bound heparin-agarose (R484A and K532A) were completely noninfectious, suggesting that they had additional blocks to infectivity that were unrelated to heparin binding. Conservative mutation of positions R585 and R588 to lysine slightly reduced heparin-agarose binding and had comparable effects on infectivity. Substitution of AAV2 residues 585 through 590 into a location predicted to be structurally equivalent in AAV5 generated a hybrid virus that bound to heparin-agarose efficiently and was able to package DNA but was noninfectious. Taken together, our results suggest that residues R585 and R588 are primarily responsible for heparin sulfate binding and that mutation of these residues has little effect on other aspects of the viral life cycle. Interactive computer graphics examination of the AAV2 VP3 atomic coordinates revealed that residues which contribute to heparin binding formed a cluster of five basic amino acids that presented toward the icosahedral threefold axis from the surrounding spike protrusion. Three other kinds of mutants were identified. Mutants R459A, H509A, and H526A/K527A bound heparin at levels comparable to that of wild-type virus but were defective for transduction. Another mutant, H358A, was defective for capsid assembly. Finally, an R459A mutant produced significantly lower levels of full capsids, suggesting a packaging defect.
The adeno-associated virus type 2 (AAV2) is a small, nonenveloped parvovirus that has received considerable attention as a gene therapy vector (reviewed in reference 31). The capsid has a diameter of approximately 24 nm and is formed by an icosahedral lattice with T=1 symmetry (60 structurally equivalent subunits) (25, 53). In purified virions, three structural proteins, namely, VP1, VP2, and VP3, with molecular masses of 87, 73, and 62 kDa, respectively, are present in a molar ratio of 1:1:18 (5). mRNAs encoding capsid proteins are synthesized from a single open reading frame and use alternative splicing and start codons to produce three VP proteins that share an identical carboxyl-terminal 532-amino-acid domain (3, 4), with VP2 and VP3 containing successive amino-terminal truncations of VP1.
The atomic structure of the AAV2 capsid has been determined to a resolution of 3.0 Å (53). In this model, 60 copies of VP3 minus 14 amino-terminal residues are present in an icosahedral arrangement. The VP3 protein contains eight antiparallel β-strands that adopt a barreled structure similar to capsid proteins of other nonenveloped viruses (42). Loops of variable length connect the interior β-barrel scaffold and extend outwards to form the capsid surface. Cryoelectron microscopy of empty AAV2 particles generated a surface density map that described depressions, spikes, and canyon features similar to those found in other parvoviruses (25). Before the crystal structure was available, several alternative methods were utilized in attempts to localize specific functional regions of the capsid. Neutralizing antibody screening of peptide sequences derived from VP1 found multiple antigenic determinants distributed on the capsid exterior in both linear and conformation-dependent forms (29). Computer modeling of AAV structure based on the known atomic structure of the related canine parvovirus coupled with genetic modification of the capsid identified several positions that were on the surface of the capsid and could tolerate insertions and substitutions (14, 33, 40, 41, 44, 51, 55).
Cell membrane binding and entry initiate all viral infections. Nonenveloped viruses rely on membrane-bound extracellular receptors for attachment to the cell membrane. AAV2 has evolved a dynamic and multistep infectious entry pathway that utilizes the abundantly expressed heparan sulfate proteoglycan (HSPG) as its primary target (46). Two coreceptors, αVβ5 integrin and basic fibroblast growth factor receptor, have been identified which act as secondary receptors that may stabilize virus attachment or participate during internalization (10, 35, 45). HSPG is a macromolecule expressed by many cell types and is a component of the extracellular matrix of most tissues (reviewed in references 18 and 30). Attached to the core protein are glycosaminoglycan side chains heparin and heparin sulfate (HS). These carbohydrate polymers are formed by disaccharide repeats consisting of alternating N-acetylglucosamine and iduronic acid residues in an α1,4 linkage. The saccharides can be modified by N sulfation as well as 2-O and 6-O sulfation to impart a dense overall negative charge at physiological pH. As a result, HS interacts with an extensive range of proteins, primarily by electrostatic attraction between the electron-dense sulfate groups and a cluster of positively charged amino acids. Two linear HS consensus-binding sequences, XBBXBX and XBBBXXBX, and a conformation-dependent sequence, TXXBXXTBXXXTBB (where B is any basic amino acid, including His, Lys, or Arg, and X is any hydropathic amino acid and T is a turn), have been reported (18). Although HSPG is thought to participate in attachment during the infectious processes of numerous human viruses (26), information about the molecular mechanisms of these interactions is limited. A report describing the atomic structure of the foot-and-mouth disease virus cocrystallized with an HS pentasaccharide is available and serves as the only model defined at the atomic level that describes the molecular interaction between a nonenveloped icosahedral virus and HS (11).
Several laboratories have attempted to retarget AAV vectors to nonpermissive cell types by inserting sequences coding for short foreign peptides into VP3. Interestingly, insertions at position 587, including an L14 integrin binding peptide, a myc tag, an immunoglobulin G binding domain truncation of protein A, and an endothelial cell-targeting peptide, abolished the natural heparin binding ability of virus capsids with these alterations (13, 14, 33, 41, 44). Similarly, an alanine repeat insertion at position 509, an L14 peptide insertion at position 520, a hemagglutinin tag insertion at positions 522 and 591, and peptides derived from the human luteinizing hormone receptor and bovine papillomavirus at inserted positions 520 and 584, respectively, have been reported to disrupt heparin binding (44, 51). Curiously, alanine substitutions of acidic residues between 561 and 565 also reduced heparin binding, suggesting that nearby basic residues were affected (51). Finally, a substitution mutation of two arginines and a glutamine at positions 585, 588, and 587, respectively, binds poorly to heparin-agarose (51). Taken together, these genetic modifications suggested two potential heparin binding loci that cluster between positions 509 to 522 and 561 to 591 (51).
In this study, charged-to-alanine substitution mutants were made to analyze the effects of single and combinatorial mutations in the capsid gene. We have discovered new point mutants that result in assembly, packaging, and receptor binding deficiencies. Importantly, we identified five amino acids, namely, arginines 484, 487, 585, and 588 and one lysine at position 532, that appear to mediate the natural affinity of AAV for HSPG. Our observations contribute to the current map of the AAV capsid and provide a reagent for the discovery of novel, heparin-independent targeting ligands.
MATERIALS AND METHODS
Plasmids.
Plasmid pIM45 (previously called pIM29-45) contains the Rep and Cap coding sequences from AAV, with expression controlled by their natural promoters (27). It was used as the parent template for construction of all the AAV2 mutant vectors.
Plasmid pXX6 supplies the adenovirus helper gene products in trans to allow rAAV production in an adenovirus-free environment (52).
Plasmid pTR2-UF5 supplies the recombinant AAV DNA to be packaged. It contains a cytomegalovirus promoter driving expression of a green fluorescent protein (GFP) reporter gene flanked by AAV2 terminal repeats (23). Plasmid pTR5-UF11 was constructed by using an expression cassette consisting of a strong constitutive CBA promoter (54), GFP reporter gene (57), woodchuck hepatitis virus posttranscriptional regulatory element WPRE (9), and bovine growth hormone gene polyadenylation signal. The cassette was assembled by standard molecular biology techniques and replaced the lacZ cassette in the plasmid backbone of pAAV5RnlacZ containing AAV5 terminal repeats (6).
Plasmids pXYZ1 and pXYZ5 contain the AAV1 and AAV5 Cap coding sequences, respectively, in addition to AAV2 Rep coding sequence with an ACG start codon under control of the AAV2 p5 promoter (58). Plasmid pAAV5-2 contains the AAV5 nucleotides 260 to 4448 without terminal repeats (6).
Construction of mutant capsid plasmids.
QuikChange site-directed mutagenesis (Stratagene) was performed on plasmid pIM45 as per the manufacturer’s instructions. For each AAV2 mutant, two complementary PCR primers were used to introduce changes into pIM45 that contained alanine or lysine substitutions and, in most cases, a silent change for restriction endonuclease screening purposes. The primer pairs are listed in Table 1. For construction of AAV5-HS, pAAV5-2 was used as the parental template. PCR products were digested with DpnI to remove methylated template DNA, phenol:cholorform:isoamyl (25:24:1) extracted, ethanol precipitated, and transformed into electrocompetent JM109 cells. Miniprep DNA was extracted from overnight Luria-Bertani-ampicillin cultures and screened with the appropriate restriction enzyme. All mutants were sequenced prior to use. Transfection-quality plasmid DNA was produced by standard alkaline lysis method of a 1-liter Tris-borate culture followed by polyethylene glycol (PEG) precipitation and cesium chloride gradient purification.
TABLE 1.
Sequences of oligonucleotides used for mutagenesis

Each oligonucleotide is named by its mutant position, the restriction site engineered at the site of the mutation, and the polarity of the oligonucleotide strand (+ or −).
The bold capital letters indicate the specific substitutions made in the parental wild-type sequence.
Cell culture.
Human embryonic kidney 293 cells and cervical carcinoma HeLa C12 cells, a gift from Phil Johnson (8), were grown in Dulbecco's modified Eagle medium (Gibco-BRL) supplemented with 100 U of penicillin/ml, 100 U of streptomycin/ml, 10% bovine calf serum, sodium pyruvate, and l-glutamine. Cells were incubated at 37°C in a 5% CO2 atmosphere.
Production of rAAV2 particles.
To produce AAV2 virions, three 15-cm2 plates were seeded with low-passage 293 cells so that they were approximately 85% confluent at transfection time. A triple plasmid transfection protocol (52) was followed that included pIM45 to supply Rep and mutated capsid genes, pTR2-UF5 (23) to supply recombinant DNA with AAV2 terminal repeats and a cytomegalovirus-driven GFP reporter gene, and pXX6 (52) to supply the adenovirus helper functions in trans. A total of 60 μg of plasmid DNA in a 1:1:1 molar ratio was transfected by Lipofectamine (Invitrogen).
To produce pseudotyped rAAV1 and rAAV5 particles, a total of 60 μg of pXYZ1 or pXYZ5 (58) was cotransfected with pTR2-UF5 plasmid DNA in a 1:1 molar ratio as above. To produce rAAV5 and rAAV5-HS virions, a total of 60 μg of pAAV5 or pAAV5-HS was cotransfected with pTR5-UF11.
Purification of rAAV has been described previously (17, 56, 58). Briefly, 72 h after transfection, cells were harvested and the pellets were resuspended in lysis buffer (0.15 M NaCl, 50 mM Tris-Cl [pH 8.5]). Virus was released by three cycles of freezing and thawing. Benzonase (Sigma) was added to the cell lysate to a final concentration of 140 U/ml and incubated at 37°C for 30 min. Cell debris was pelleted by centrifugation at 3,700 × g for 30 min, and the supernatant was loaded onto an iodixanol {5,5′-[(2-hydroxy-1,3-propanediyl)bis(acetylamino)]bis[N,N′-bis(2,3-dihydroxypropyl)-2,4,6-triiodo-1,3-benzenecarboxamide} step gradient (15, 25, 40, and 60%) (Nycomed). The 40% fraction was collected after centrifugation at 69,000 × g for 1 h and stored at −80°C until further use.
Virus titer determination.
To determine the concentration of intact capsid particles, the A20 enzyme-linked immunosorbent assay (ELISA) (American Research Bioproducts) was used. The A20 antibody detects intact, fully assembled particles, both full and empty (49). Iodixanol-purified stocks were serially diluted and processed by the manufacturer's recommended protocol. Only readings within the linear range of the kit standard were used.
To determine the concentration of DNA-containing particles, we performed real-time (RT)-PCR using a Perkin-Elmer-Applied Biosystems (Foster City, Calif.) Prism 7700 sequence detector system. Equal volumes of iodixanol-purified virus stocks were treated with 600 U of benzonase/ml in 50 mM Tris-Cl (pH 7.5)-10 mM MgCl2-10 mM CaCl2 at 37°C for 30 min. Proteinase K (280 U/ml) was added to the reactions, which were adjusted to 10 mM EDTA and 5% sodium dodecyl sulfate (SDS) and then incubated at 37°C for 30 min. Reactions were extracted with phenol-chloroform-isoamyl alcohol (25:24:1), and undigested DNA was precipitated overnight with ethanol and glycogen carrier. Precipitated DNA pellets were resuspended in 100 μl of water. Five microliters was used for RT-PCR analysis in a reaction mixture that included a 900 nM concentration each of GFP forward (5′-TTCAAAGATGACGGGAACTACAA-3′) and reverse (5′-TCAATGCCCTTCAGCTCGAT-3′) primers, 250 nM Taqman probe (5′-6FAM-CCCGCGCTGAAGTCAAGTTCGAAG-TAMRA-3′), and 1× Taqman universal PCR master mix in a total volume of 50 μl. Cycling parameters were 1 cycle each of 50°C for 5 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Only values within the linear portion of a standard curve having a coefficient of linearity greater than 0.98 were accepted. The average RT-PCR titer was calculated from virus preparations assayed three times.
To determine the infectious titer of the wild-type and mutant virus stocks, we performed a green cell assay (GCA) essentially as previously described (56). Briefly, HeLa C12 cells were seeded in a 96-well plate so that they were approximately 75% confluent at infection time. Cells were infected with 10-fold serial dilutions of iodixanol-purified mutant viruses and Ad5 at a constant multiplicity of infection (MOI) of 10. Cells were incubated at 37°C in a 5% CO2 atmosphere for 24 h and examined by fluorescence microscopy. The average GCA titer was calculated by averaging the number of green cells counted in individual wells from two or three virus preparations assayed three times. Particle-to-infectivity ratios were calculated by dividing the average RT-PCR titer by the average GCA titer. In some figures, this number was expressed as a log10 value with rAAV2 arbitrarily set to one. See the figure legends.
In vitro heparin binding assay.
Bio-Rad microspin columns were treated with silicon dioxide to minimize nonspecific binding of the virus to the column wall. A 500-μl heparin-agarose (Sigma H-6508) gravity column was prepared by washing with 3 column volumes each of 1× TD buffer (137 mM NaCl, 15 mM KCl, 10 mM Na2PO4, 5 mM MgCl2, 2 mM KH2PO4 [pH 7.4]), 1× TD + 2 M NaCl, and 1× TD. Approximately equal numbers of virus particles were added to 1× TD to a final volume of 600 μl and loaded onto the column. The column was washed with 7 column volumes of 1× TD. Bound virus was eluted with 1× TD + 2 M NaCl. The entire volume of the flowthrough, wash, and eluate fractions was pooled separately, denatured by boiling in SDS, and slot blotted onto nitrocellulose for immunoblot analysis. The membrane (Osmonics) was blocked in phosphate-buffered saline-0.05% Tween 20 + 5% dry milk and incubated with B1 antibody (48) at a 1:3,000 dilution for 18 h at 4°C. Anti-mouse immunoglobulin G-horseradish peroxidase was used to detect bands by enhanced chemiluminescence (Amersham-Pharmacia).
FACS.
HeLa C12 cells were seeded in six-well plates so that they were approximately 75% confluent at infection time. Cells were infected with an rAAV MOI of 500 based on the genomic titer as determined by RT-PCR (56). Adenovirus type 5 was used at an MOI of 10 PFU. Twenty-four hours postinfection, cells were washed, trypsinized, and fixed in 2% paraformaldehyde. Fluorescence-activated cell sorting (FACS) analysis for GFP expression was done in the Interdisciplinary Center for Biotechnology Research (ICBR) flow cytometry lab of the University of Florida on a Becton-Dickinson FACScan.
Cell attachment assay.
HeLa C12 cells (106) were infected with rAAV2 or R585A/R588A at a genome-containing-particle MOI of 1,000 as determined by RT-PCR. Cells were incubated at 37°C in a 5% CO2 atmosphere until harvesting. At indicated time points, the infection medium was removed and saved, and the cells were washed four times with phosphate-buffered saline before being scraped. Low-molecular-weight DNA from the infection medium and the cell pellet was extracted by the Hirt procedure (19). DNA pellets were resuspended in 0.2 M NaOH, incubated at 37°C for 20 min, and slot blotted onto nitrocellulose. DNA was UV cross-linked to the nitrocellulose and probed at 65°C for 18 h with [α-32P]dATP-labeled GFP probe in hybridization buffer (7% SDS, 10 mM EDTA, and 0.5 M Na2HPO4). Membranes were washed twice each in 2× SSC-0.1% SDS (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 0.2× SSC-0.1% SDS, and 0.1× SSC-0.1% SDS and then rinsed with water. The membranes were then exposed to film and quantitated using a BAS-1000 phosphorimager (Fuji).
RESULTS
Selection and generation of AAV mutants.
In order to evaluate the role of particular amino acids in receptor binding, we generated a panel of mutants by site-directed mutagenesis of selected residues. We confined our selection primarily to basic amino acids (His, Lys, Arg) in VP3, as AAV-like particles composed only of VP3 proteins have been purified by heparin affinity chromatography in our laboratory (unpublished data) and by others (40). Any basic amino acid substitution mutant that previously had demonstrated capsid instability or efficient purification by heparin affinity chromatography (51) was excluded from our pool of mutants. Additionally, eight AAV serotypes have been reported (1, 12, 20, 34, 43). Of these, rAAV2 and rAAV3 bind efficiently to HS (38, 44, 51). In contrast, rAAV4 and rAAV5 do not bind heparin and instead recognize 2,3 O-linked and 2,6 N-linked sialic acid moieties (21). A single report concerning rAAV1 suggests that it binds with low affinity, if at all, to heparin (38). We reasoned that residues conserved among all five of these serotypes were probably not participating directly in receptor discrimination and binding and were excluded from further consideration. Using a Clustal W algorithm, we then generated a sequence alignment of capsid proteins from serotypes 1 to 5 and identified nine basic residues in AAV2 that were conserved in AAV3 and/or AAV1 but were uncharged or acidic in AAV4 and AAV5 and had not previously been tested for heparin-agarose binding (Table 2). In addition to these nine amino acids, Wu et al. (51) described a virus deficient for heparin binding with alanine substitution mutations at positions 585, 587, and 588. Finally, during the course of these studies, the atomic structure of AAV2 was solved (53) and suggested that residues 484, 513, and 532 might participate in a heparin binding pocket, as they were located close to residues 585, 587, and 588. We included these six extra residues to complete our mutant panel (Table 2).
TABLE 2.
Residues chosen for mutagenesis
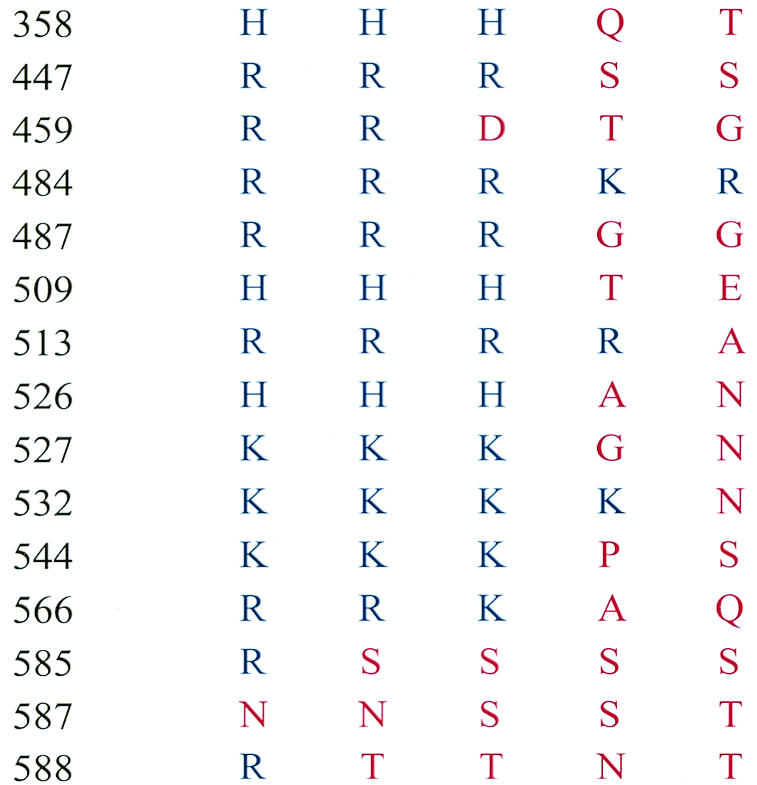
Residues chosen for mutagenesis were generated by a sequence alignment of the VP1 capsid protein from each serotype, using the Clustal W algorithm (Vector NTi 5.2, Informax).
Amino acids are represented by their one-letter abbreviations. Blue letters represent positively charged basic amino acids. Red letters represent any other amino acid.
Mutant virus production and physical characterization.
We generated a series of single and combinatorial capsid mutants from our pool of candidate residues in the AAV2 capsid gene (Table 2). To designate the mutant viruses, we used the number of the mutated amino acid based on its position in VP1. Iodixanol-purified virus stocks were checked by Western blot with the monoclonal antibody B1. The B1 antibody recognizes a linear epitope in the extreme carboxyl terminus of all three VP proteins from AAV serotypes 1, 2, 3, and 5 (38, 50). With the exception of H358A, capsid proteins were detected in all virus stocks (Fig. 1). To confirm that we had purified assembled capsids rather than subunits or assembly intermediates, we measured the particle concentration with an A20 antibody ELISA (Table 3). The A20 antibody recognizes a structural epitope that is found only on assembled capsids with or without packaged DNA (15). Although there was some variability between stocks due to different transfection efficiencies and purification recoveries, only the H358A mutant was negative by A20 ELISA. To determine whether any mutations affected DNA packaging, the titer of DNA containing virions was determined by RT-PCR (7, 47) (Table 3) and confirmed by DNA dot blot hybridization (data not shown). Although there was variation between preparations, the majority of the capsid mutants were able to package detectable DNA (Table 3). As expected, H358A was negative for DNA packaging, as it did not produce virus particles. We concluded that none of the capsids in our mutant panel that made A20-positive particles were completely defective for DNA packaging. However, by comparing the A20 ELISA and PCR titers, we noted that stocks of mutant R459A contained 40-fold more empty particles than wild-type rAAV2. Thus, R459 could have a role in DNA packaging. Although less dramatic, mutants R447A, R566A, R587A, R585K, and R585K/R588K had approximately 10-fold more empty particles than rAAV2. The remainder of the virus preparations packaged DNA at comparable levels to that of wild-type AAV2 (Table 3).
FIG. 1.

Western blot of iodixanol virus stocks. Equal volumes of virus stock were separated by SDS-10% polyacrylamide gel electrophoresis and analyzed by Western blot using the B1 antibody.
TABLE 3.
Titers and heparin binding properties of mutants
| Mutant virusa | Particle titer ofb:
|
Infectious titer (IU/ml)c | Particle-to-infectivity ratiod | Heparin bindinge | Empty/full ratiog | |
|---|---|---|---|---|---|---|
| A20/ml | Genome/ml | |||||
| rAAV2 (WT) | 1.5 × 1012 | 4.6 × 1011 | 1.8 × 1010 | 25 | + | 3.4 |
| H358A | <1.0 × 108 | <1.0 × 106 | <1.0 × 104 | N/Df | N/D | N/D |
| R447A | 1.2 × 1012 | 3.4 × 1010 | 1.3 × 109 | 25 | + | 35.9 |
| R459A | 9.1 × 1010 | 7.2 × 108 | <1.0 × 104 | >72,500 | + | 126.3 |
| R484A | 1.5 × 1011 | 3.0 × 1010 | <1.0 × 104 | >2,976,667 | +/− | 5.1 |
| R487A | 5.4 × 1011 | 2.2 × 1011 | 2.3 × 108 | 954 | +/− | 2.5 |
| H509A | 4.6 × 1010 | 2.3 × 109 | 6.9 × 105 | 3,285 | + | 20.3 |
| R513A | 2.9 × 1011 | 1.7 × 1010 | 1.6 × 108 | 106 | + | 17.9 |
| K532A | 1.1 × 1011 | 3.6 × 1010 | <1.0 × 104 | >3,633,333 | +/− | 3.0 |
| K544A | 2.0 × 1011 | 1.7 × 1010 | 8.3 × 108 | 20 | + | 11.9 |
| R566A | 5.1 × 1011 | 1.6 × 1010 | 7.4 × 108 | 21 | + | 32.6 |
| R585A | 5.0 × 1011 | 4.8 × 1010 | 1.7 × 107 | 2,812 | − | 1.4 |
| N587A | 4.4 × 1011 | 1.3 × 1010 | 7.7 × 107 | 165 | + | 34.7 |
| R588A | 2.4 × 1011 | 5.6 × 1010 | 3.0 × 106 | 18,521 | − | 4.2 |
| H526A, K527A | 1.4 × 1011 | 8.2 × 1010 | 5.5 × 107 | 1,489 | + | 1.8 |
| R585A, R588A | 1.2 × 1012 | 9.2 × 1011 | 1.9 × 107 | 48,421 | − | 1.2 |
| R585K | 1.3 × 1012 | 3.7 × 1010 | 4.0 × 108 | 92 | + | 35.4 |
| R585K, R588K | 1.4 × 1012 | 3.9 × 1010 | 8.9 × 107 | 436 | + | 34.9 |
| AAV1 | N/D | 3.7 × 1010 | 1.1 × 109 | 37 | +/− | N/D |
| AAV5 | N/D | 3.4 × 1010 | 3.2 × 106 | 10,692 | − | N/D |
| AAV5-HS | N/D | 8.0 × 108 | <1.0 × 104 | >80,000 | + | N/D |
Two letters flanking a number designate each mutant. The first letter is the one-letter abbreviation for the wild-type amino acid followed by its numerical position in VP1 followed by the one-letter abbreviation for the amino acid to which it was mutated.
A20 particle titers were determined as described in Materials and Methods using the A20 ELISA. Genomic titers were determined by RT-PCR.
Infectious titers were determined by GCA as described by counting GFP fluorescent cells.
Particle-to-infectivity ratio was calculated by dividing the average genomic titer as determined by RT-PCR by the average GCA titer.
Determined by heparin-agarose binding assay. +, >95% virus recovered in the eluate; +/−, >50% recovered in the eluate; −, <5% of virus recovered in the eluate.
N/D, not determined.
Empty-to-full ratio was determined by dividing the A20 particle titer by the average genomic titer.
In vitro heparin binding of capsid mutants.
To assess the ability of mutant capsids to bind HS, we used a modification of an assay previously described by Wu et al. (51). A representative Western analysis for each mutant is shown in Fig. 2. As expected, wild-type AAV2 was not observed in the flowthrough or wash fractions, and most of the virus bound to the column was recovered at the elution step. Eight other mutants, R447A, R459A, H509A, R513A, K544A, K566A, N587A, and H526A/K527A, had a heparin-agarose binding phenotype indistinguishable from that of the wild type. The results with R513A confirmed a previous report by Wu et al. (51) in which a double mutant at positions 513 and 514 was positive for heparin binding. In marked contrast, we observed that any capsid harboring a nonconservative mutation at position 585 or 588 was detected only in the flowthrough and wash. We also detected intermediate heparin-agarose binding phenotypes in mutants R484A, R487A, and K532A, with approximately equal levels of signal detected in the flowthrough, wash, and eluate. The results with K532A were inconsistent with our previous results in which a mutant containing alanine substitutions at positions 527 to 532 was found to be positive for heparin binding (51). These data suggested that at least five amino acids had the potential to contribute to the electrostatic attraction between AAV and HS. These included predominantly R585 and R588 and, to a lesser but detectable extent, R484, R487, and K532.
FIG. 2.

Heparin-agarose binding profiles of mutant capsids. Approximately 5 × 1010 particles were applied to 500 μl of heparin-agarose affinity matrix at a 100 mM NaCl concentration and washed extensively with the loading buffer, and bound capsids were eluted with 2 M NaCl. Pooled fractions were denatured and slot blotted onto nitrocellulose for immunodetection with monoclonal antibody B1. For each mutant, L is the total amount of iodixanol-purified virus that was loaded onto the heparin agarose column; FT is the total virus that flowed through the column; W is the wash; E is the eluate. See Materials and Methods for details.
To confirm that the charge at R585 and R588 was primarily responsible for heparin interaction, we generated two viruses with conservative mutations, R585K and R585K/R588K, and tested them in the in vitro heparin binding assay. Both lysine and arginine residues are positively charged; however, lysine is slightly larger due to an additional methyl residue in the side group. We found that both of these capsids bound to heparin-agarose almost as well as wild-type virus (Fig. 2). In each case, most of the virus was recovered in the eluate; however, the flowthrough and wash fractions also contained minor amounts of virus. This result suggested that both localized positive surface charge and the relative position of the changes in this region of the capsid are responsible for mediating the interaction with heparin-agarose.
Finally, as a control for our experiments and to validate the heparin binding assay, we compared the ability of wild-type rAAV2, rAAV1, and rAAV5 to bind to heparin-agarose. For this purpose, we produced and purified recombinant viruses by using a pseudotyping protocol developed to package AAV2 terminal-repeat-containing genomes into alternative serotype capsids (Fig. 3A) (38, 58). Approximately equal amounts of input virus as determined by Western blot signal intensity were applied to a heparin-agarose column, and fractions from the column were slot blotted onto nitrocellulose for immunodetection with the B1 antibody (Fig. 3B). As expected, rAAV2 was efficiently retained by heparin-agarose under low-ionic-strength conditions, but the majority of rAAV1 and all of rAAV5 were seen in the flowthrough and wash. A small amount of AAV1 was detected in the eluate. These data were consistent with previous observations by Rabinowitz et al. (38).
FIG. 3.

Production and purification of AAV serotypes. (A) Equivalent amounts of iodixanol-purified AAV1, AAV2, and AAV5 were separated by 10% polyacrylamide gel electrophoresis and analyzed by Western blot using the B1 antibody. (B) Heparin-agarose binding properties of AAV2, AAV1, and AAV5. Abbreviations are as described in the legend for Fig. 2.
Multiple mutations in the AAV2 capsid affect viral transduction.
To determine how the heparin-agarose binding phenotypes correlated to infectivity, iodixanol stocks were tested for their ability to transduce HeLa C12 cells by performing a GCA, and a GCA titer was calculated (Table 3) as described in Material and Methods. The detection limit of this assay was approximately 104 transducing units/ml. The GCA titers were then normalized to genome-containing physical particles by calculating a particle-to-infectivity (P/I) ratio. This ratio is equivalent to the number of genomes required to transduce one cell (Table 3). To get a measure of the relative impact of a particular mutation on viral infectivity, the P/I ratio of each mutant was divided by the wild-type capsid P/I ratio and the log10 of this value was plotted in Fig. 4. This provided a simple comparison of how many genome-containing particles of each mutant were required to achieve the same number of transduced cells as the wild-type virus.
FIG. 4.

Particle-to-infectivity ratios of mutants relative to that of the wild type. The particle-to-infectivity ratio for each mutant was calculated by dividing the average genomic titer by the average GCA titer (see Materials and Methods and Table 3). The P/I ratio of each mutant was then normalized to that of the wild type by dividing the P/I of each mutant by the P/I of wild-type rAAV2, and the log10 value of the ratio was plotted. The wild-type ratio therefore equals zero and is indicated by the dashed line. Grey bars, mutant viruses with infectivity comparable to that of the wild type; black bars, mutant viruses that are heparin binding deficient; white bars, mutant viruses with an undetermined block to infectivity. Asterisks indicate those mutants for which no green cells were scored; for these mutants, the GCA titer used was the limit of detection in the assay. Thus, the log difference is a minimum estimate.
Several phenotypes emerged from this analysis. Mutants R477A, K544A, and K566A were virtually identical to the wild type, and mutants R513A, N587A, R585K, and R585K/R588K were only slightly defective (approximately 1 log). These seven mutants were found previously to bind HS to the same extent as wild-type rAAV2 (Fig. 2).
Three of the mutants, R459A, R484A, and K532A, produced virus that was essentially noninfectious, with P/I ratios between 7.2 × 104 and 3.6 × 106 compared to the wild-type ratio of 25 (Table 3; Fig. 4). The P/I ratios for these mutants were minimum estimates based on the GCA sensitivity of 104 IU/ml. In fact, no transduction events were seen with any of these mutants.
R459A was the most severe example of three mutants (R459A, H509A, and H526A/K527A) that were essentially wild-type for heparin binding but defective for transduction (Fig. 4). These mutants were presumably defective in some late stage of viral infection.
Finally, all five of the mutants that were defective or partially defective for heparin binding (R484A, R487A, K532A, R585A, and R588A) were defective for transduction. However, the loss of infectivity did not correlate completely with the loss of heparin binding (compare Fig. 2 and 4). Two of these mutants (R484A and K532A) were only partially defective for heparin binding but severely defective (>5 logs) for transduction, suggesting that some other step in viral infection was defective in these mutants in addition to heparin binding. The remaining heparin binding mutants (R487A, R585A, and R588A) had defects in transduction that approximately correlated with their ability to bind heparin.
Evaluation of R585A/R588A cell attachment in vivo.
As mentioned earlier, alanine substitutions at either position 585 or 588 were the only mutations that completely abolished binding to HS (Fig. 2), suggesting that these two arginines were primarily responsible for heparin binding. Moreover, the extent to which mutation of either or both of these residues inhibited transduction (1.5 to 3 logs [Fig. 4]) was approximately the same when soluble HS is used to inhibit wild-type rAAV2 infection (reference 16 and data not shown). We therefore examined these mutants in more detail.
To see if the defect in transduction of R585 and R588 mutants could be overcome by using higher input MOIs, cells were coinfected with rAAV2 or the mutant viruses at an MOI of 500 genome-containing particles/cell. At 24 h postinfection, cells were examined by fluorescence microscopy and counted by FACS. The data from three independent experiments and representative histograms are shown in Fig. 5. As expected, the defects in transduction of the single mutants, R585A and R588A, could be overcome by higher MOIs (56 and 25% transduction, respectively). Predictably, the level of recovery of the double mutant, R585A/R588A, was lower (10% transduction). However, it was clear that the fluorescence intensity profile for the heparin binding mutants was quite different from that of the wild type, suggesting a significant delay in the onset of GFP expression by 24 h. In contrast, the level of transduction of the conservative double mutant, R585K/R588K, and the heparin positive mutant, N587A, was indistinguishable from that of the wild type (Fig. 5).
FIG. 5.

GFP transduction ability of mutants in HeLa C12 cells. Cells were infected with wild-type rAAV or mutant virus at an MOI of 500 genomic particles and an Ad5 MOI of 10 PFU per cell. Twenty-four hours postinfection, cells were fixed with 2% paraformaldehyde and the number of GFP-positive cells was determined by FACS analysis.
As a more direct assay for cell attachment, we infected HeLa C12 cells and tracked the location of viral DNA. Cells were infected with rAAV2 or R585A/R588A at an MOI of 1,000 genome-containing particles as determined by RT-PCR. At 1, 4, and 20 h postinfection, the infection medium was removed and saved, and the cells were washed extensively to remove any residual unbound virus. The cells were then harvested, and low-molecular-weight DNA was extracted from both the infection medium (unbound) and the cell pellet (bound) by the Hirt procedure and transferred to nitrocellulose for Southern hybridization with an [α-32P]dATP-labeled GFP probe (Fig. 6A).
FIG. 6.

Binding and uptake of rAAV2 and R585A/R588A in HeLa C12 cells. (A) 106 cells were infected with rAAV2 or R585/R588A at an MOI of 1,000 genome-containing particles per cell. At the indicated times, infection medium was removed and saved. The cells were washed and harvested, and Hirt DNA was extracted from both the infection medium and the cell pellet. Southern analysis was performed using an [α-32P]dATP-labeled GFP probe. (B) The percentage of bound and internalized DNA was calculated by dividing the total DNA present in both the medium and the cell pellet by the amount bound and internalized for each time point. The average of three determinations is shown. Error bars indicate a standard deviation.
We saw that at all time points, rAAV2 DNA was detectable, both bound or internalized and in the infection medium. In contrast, R585A/R588A showed the vast majority of the signal only in the unbound fraction (Fig. 6A). Phosphorimager analysis determined that at each time point, almost one-half of the total rAAV2 DNA was attached or internalized, compared to only 1 to 3% of that of R585A/R588A (Fig. 6B and data not shown). As these infections were performed at 37°C, the process of internalization should not have been prevented. This result demonstrated that the block in infection for mutant R585A/R588A occurred at the cell attachment stage or internalization stage and correlated to HS binding in vitro.
Loop swapping confers heparin binding to AAV5.
Although the primary amino acid sequences are moderately divergent, the architectural position of β-sheets and loops is predicted to be very similar among AAV serotypes (39). We hypothesized that if R585 and R588 were the critical residues involved in HSPG binding, then it should be possible to substitute that region of AAV2 into AAV5 to create a hybrid virus capable of interacting with heparin-agarose. To achieve this, we generated a recombinant virus, designated rAAV5-HS, by using a short loop containing residues 585 through 590 from AAV2 to replace a region predicted to be structurally equivalent in AAV5 (Fig. 7A). Loop substitution rather than point mutagenesis was done to account for the possibility of additional Van der Waals interactions or hydrophobic contributions from nearby amino acids.
FIG. 7.

Modifying the heparin binding properties of AAV5. (A) Alignment of AAV2 amino acid residues 585 through 590 to residues predicted by amino acid alignment to be structurally equivalent in AAV5. (B) Western blot of iodixanol virus stocks. Equal volumes of virus were separated by SDS-10% polyacrylamide gel electrophoresis and analyzed by Western blotting using the B1 antibody. (C) Novel heparin binding properties of AAV5-HS. Heparin-agarose binding was performed as described in the legend for Fig. 2. See the legend for Fig. 2 for explanation of the abbreviations. (D) The log of the particle-to-infectivity ratio of the rAAV5 variants normalized to that of wild-type rAAV2, as described in the legend for Fig. 4.
Production and purification of rAAV5-HS was unaffected by the six-amino-acid substitution (Fig. 7B; Table 3). When we tested rAAV5-HS in the in vitro heparin-agarose binding assay, it was indistinguishable from wild-type rAAV2 (Fig. 7C). These data suggested that this region of AAV5 was surface accessible and that heparin-agarose binding could be artificially conferred by the six amino acids containing R585 and R588.
To compare the infectivity of rAAV5 and rAAV5-HS, we generated packaged viruses that contained a recombinant AAV5 genome in which the GFP reporter gene was flanked by AAV5 terminal repeats (see Materials and Methods). The infectivities of these viruses were compared to that of rAAV2 in a GCA, and particle-to-infectivity ratios were calculated as before (Fig. 7D). rAAV5 was able to transduce HeLa C12 cells at a low efficiency, approximately 2.5 logs lower than AAV2. However, no transduction was seen with AAV5-HS (<1 × 104 IU/ml) (Table 2; Fig. 7D). Given the minimum sensitivity of the GCA, this meant that the P/I ratio of AAV5-HS was at least 3.5 logs higher than that of rAAV2 and at least 1 log higher than that of wild-type rAAV5. We concluded that although substitution of these five heterologous amino acids into the AAV5 capsid restored heparin binding to the level of AAV2 capsids, it was not sufficient to produce AAV2 levels of infectivity in a cell line normally permissive for AAV2.
DISCUSSION
The aim of this study was to identify amino acids in the capsid of AAV2 that mediate binding to HSPG. Several lines of evidence suggest that HSPG serves as the primary receptor for AAV2. Inhibition of AAV2 infection can be demonstrated by competition with soluble analogs, GAG desulfation by sodium chlorate treatment, antibody competition, enzymatic removal of heparin, and use of mutant cell lines that express various levels of HSPG (16, 37, 46, 51). Binding to HS is usually the result of electrostatic charge interactions between basic amino acids (R, K, and H) and negatively charged sulfate residues (18, 30). During the course of previous mutagenesis studies, it was possible to eliminate many of the basic amino acids in the AAV2 capsid that could potentially contribute to HS binding (51). In this study, we examined the remaining basic residues by looking at their conservation in AAV serotypes 1 to 5. Those that were present in all five serotypes were not likely to contribute significantly to heparin binding. Those that were conserved in the heparin binding serotypes, AAV1 to -3, but not in the remaining serotypes, were targeted for mutagenesis. Finally, we also took advantage of the fact that R585 and 588 had been previously identified as potential heparin binding amino acids (51) and that these amino acids were located in a cluster of basic residues at the threefold axis of symmetry (53). All of the basic amino acids in this cluster were also targeted for mutagenesis. This approach yielded a total of 15 amino acids that could have been involved in heparin binding, and we characterized alanine mutations at all of these positions. We note that this approach does not necessarily identify all possible heparin binding amino acids. For example, R484, which is basic in all five serotypes, was tested because of its proximity to R585 and R588 and subsequently proved to be involved in heparin binding.
Heparin binding and infectivity.
Our studies indicated that capsids with a mutation at residue 484, 487, 532, 585, or 588 were partially or completely defective for heparin-agarose binding. The most severe defect was found with mutations in R585 and R588. No binding to HS columns could be detected with either mutant (Fig. 2), and both mutations reduced the particle-to-infectivity ratio by 2 to 3 logs (Table 3). Mutants that contained substitutions at both positions had even lower infectivity.
The phenotypes of R487A, R585A, and R588A were probably due largely to defective heparin binding. For example, the double mutant R585A/R588A was approximately 10- to 50-fold more defective in cell binding and internalization than the wild type (Fig. 6B) at artificially high MOIs (500 to 1,000), and cell binding and internalization were essentially undetectable at lower MOIs (1 to 10) (data not shown). This was consistent with the approximately 2,000-fold-lower infectivity of R585A/R588A (Table 3), as judged by the change in particle-to-infectivity ratio. Another indication that heparin binding was primarily responsible for the defects in R585 and R588 was the fact that conservative mutations at these two positions (R585K and R585K/R588K) produced virus particles with properties similar to those of the wild type (Fig. 2, 4, and 5 and Table 3). Results from the conservative lysine substitutions at R585 and R588 are reasonably consistent with electrostatic attraction being the primary mediator for AAV-heparin interaction. R585K, the least defective heparin binding mutant (Fig. 2), had transduction levels nearly equal to those of rAAV2 (Fig. 4), and R585K/R588K was only slightly more defective for heparin binding (Fig. 2) and transduction (Fig. 4) and was within 1 log of the wild type. Furthermore, when cells were infected at a high MOI, robust transduction was observed for both mutants (Fig. 5). Finally, substitution of a six-amino-acid sequence containing R585 and R588 imparted a heparin binding to AAV5 that was comparable to that seen with AAV2 (Fig. 7). Although similar experiments were not done with the R487 position, it was clear that mutation of R487 produced virus with a more modest defect in heparin binding (Fig. 2) and in infectivity (Fig. 4).
In addition to R487A, R585, and R588, two other mutants were found that were defective for heparin binding, R484A and K532A. R484A and K532A, like R487A, had a more modest effect on binding to HS, but unlike the other heparin binding mutants, these two mutations had a dramatic effect on transduction efficiency. Both R484A and R532A were more than 5 logs less infectious than wild-type capsids (Table 3; Fig. 4). This severe defect is presumably due to a different block in the infection process that is unrelated to heparin binding, but as yet we have not identified the defect. The result from K532A is consistent with our earlier study that identified a mutant (mut 37) that contained six amino acid substitutions that included K532A (51). mut 37 had a phenotype identical to K532A in that it produced full virus particles that were noninfectious and more recently has been shown to have a modest defect (approximately fivefold) in heparin binding and internalization (Xiao and Muzyczka, unpublished). This potentially maps this defect to a single amino acid.
Computer visualization of AAV2 structure.
We took advantage of the recently published atomic structure of AAV2 (PDB ID code: 1LP3) (53) and examined the positions of the heparin binding mutations. Symmetry transformation operations from the original PDB file were applied to generate a VP3 trimer arrangement in the context of an icosahedron. We generated an electrostatic potential surface map of a VP3 trimer (Fig. 8A and B) in which areas of positive and negative charge are represented as blue and red, respectively. When viewed perpendicular to the threefold axis, the five amino acids mapped by these studies appear to contribute collectively to a basic patch on one side of each threefold related spike (Fig. 8B). The charge, clustering, and surface presentation of these residues are all consistent with a model of electrostatic attraction. Residue K527 is surface accessible, unlike its direct neighbor H526, and although it contributes to the base basic cluster at the threefold spike, it does not appear to be involved in heparin binding (Fig. 2). When viewed directly down a threefold axis, residues R484, R487, R532, R585, and R588, represented as balls and sticks, are located in a linear formation lining one side of each threefold related spike (Fig. 8C). When viewed across the top surface of the trimer, residues R585 and R588, which are contributed by one of the peptides in the trimer, are positioned above a linear arrangement of R484, R487, and K532 (Fig. 8D), which are contributed by a second peptide in the trimer. Thus, it appears that a heparin binding motif is formed from some combination of these five amino acids using amino acids from two different polypeptides.
FIG. 8.

Surface and ribbon and diagrams of the atomic model of an AAV2 trimer. (A) Electrostatic surface potential of the VP3 trimer viewed down a threefold axis (black triangle) calculated with GRASP (32) running from −12 (red) to +12 (blue). (B) Electrostatic surface potential of VP3 trimer viewed from the side perpendicular to the threefold axis, indicated by a black line and triangle. Labeled arrows indicate the positions of residues involved in heparin binding. (C) VP3 trimer viewed down a threefold axis. Cα-backbones for individual monomers are rendered as pink, gray, and green ribbons. Residues that contribute to heparin binding are shown in CPK representation with atoms in conventional colors: yellow, carbon; blue, nitrogen; red, oxygen. These images were generated in Bobscript/Raster3D (24, 28). (D) VP3 trimer viewed from approximately the same vantage point as in Fig. 8B. Features are as described above in the legend for panel C.
The five mutations that affected heparin binding were located in the large loop IV region, which among AAV serotypes has low overall sequence conservation and includes all of the previously identified insertion and substitution mutations that affect heparin binding. Interestingly, with the exception of N587, the stretch encompassing amino acids 585 to 590 is unique to AAV2 and is not present in AAV3, which is the other AAV serotype that has been shown to bind efficiently to HS. Mutation of N587 had no effect on heparin-agarose binding and only minor effects on transduction. Conceivably, residues R484, R487, and K532 could be the dominant residues involved in HS binding for AAV3, or the heparin binding region of AAV3 is distinct from the one used by AAV2. The latter explanation is more likely, because both AAV3 (which binds efficiently to heparin) and AAV1 (which binds poorly) have conserved the residues at 484, 487, and 532 (Table 2).
The apparent dissociation constant (Kd) of AAV2 and HS was determined by competition analysis to be 2 × 10−9 M (37). Although this is higher than some heparin-protein interactions, it is sufficiently strong to suggest cooperative binding by one HS glycosaminoglycan chain to multiple attachment points. We were unable to address in this study whether HS could form a bridge between basic residues in one of the threefold spikes to those in another (yellow ovals in Fig. 8A). However, as the average chain length of heparin glycosaminoglycans varies between 50 and 200 disaccharide repeats that adopt a helical conformation of 40 to 160 nm in length, it is conceivable that an HS chain could wrap around the exterior of the capsid through cooperative binding of multiple spikes at the threefold axis of symmetry. Although we did not undertake a rigorous computational docking analysis, we were able to manually superimpose a heparin molecule (PDB ID code: 1NTP) in several orientations that placed multiple reactive sulfate and amine groups within accepted electrostatic attraction distances on pairs of residues spanning the spikes (data not shown). This approach, however, does not account for either the flexibility of heparin or for the possibility of additional molecular interactions. Cocrystallization studies of AAV2 and HS are needed to clarify this issue.
Mutants that bind heparin but are still defective.
Several new mutants were found that bound HS as well as the wild type but still produced defective particles. H358A was defective for particle assembly. There are a number of reported examples of mutations that disrupt AAV2 particle formation, several of which are located in the conserved β-strand regions (40, 44, 51). H358 is neither surface accessible nor in a conserved β-strand; instead, it is internally located in a subunit situated at the base of a loop from another subunit that forms the tall outer peak of the spike. In this position, it may function to stabilize or orient the extensive subunit interdigitation that occurs during capsid assembly in this region. Excluding H358A, we determined a particle concentration range that spanned 1.5 logs and correlated reasonably well with the B1 antibody results (Fig. 1 and Table 3). Several possibilities may account for this range of particle titers, including that capsid subunits containing these mutations (i) form intact particles inefficiently, (ii) are unstable during purification, and (iii) formed a particle with a partially disrupted A20 epitope. Since none of our mutations fell within the antigenic regions that have been mapped for A20 (50), these results suggested that the A20 epitope had probably not been modified but rather that the stability or assembly of some of the mutants was altered so that fewer particles were recovered after iodixanol centrifugation (Fig. 1 and Table 3).
Mutants R459A, H509A, and H526A/K527 bound heparin-agarose efficiently but had particle-to-infectivity ratios that were 2 to more than 3 logs higher than that of the wild type. Like K532A and R484A, these mutants are presumably defective in some stage of the infectious entry pathway between secondary receptor binding and uncoating. H509 is located at the base of the valley between each pair of spikes and does not contribute to the basic heparin binding patch. Ongoing studies in the lab are examining the block in infectivity for these mutants.
DNA packaging.
The process of DNA packaging is thought to occur by an active process requiring NTP consumption coupled to the helicase activity of the small Rep proteins (22). Although none of the mutations that assembled an A20 positive particle were completely deficient for DNA packaging, mutant R459A produced a 40-fold excess of empty capsid particles compared to rAAV2. Other studies have reported that short insertions at positions 323, 339, 466, 520, 540, 595, and 597 that did not interfere with capsid formation still reduced DNA packaging to levels detectable only by PCR amplification (44). In addition, a point mutant, R432A, prevents DNA packaging (51). R459 points away from the threefold axis on the outside of the spike slightly below the level of R585 and R588 and is highly accessible. Although the relationship between these mutations and their mechanism of action is unclear, it is possible that they disrupt protein-capsid or DNA-capsid interactions. It is tempting to speculate that some of these residues might act as a binding site for Rep or a cellular protein. In particular, nucleolin, a 110-kDa nuclear shuttle protein, binds specifically to the AAV2 capsid both in vitro and in vivo and is found colocalized with the capsid in the cytoplasm and nucleus of infected cells (36). It has also been shown to bind single-stranded DNA of a related parvovirus, the minute virus of mice (2).
In summary, we have reported an analysis of the HS binding ability and transduction potential of mutants at fifteen positions within the capsid of AAV2. We identified residues that affect capsid assembly and DNA packaging and mediate HS binding. In particular, mutants with the combined mutations in R585 and R588 should be valuable reagents for the development of heparin-independent retargeted virus vectors.
Acknowledgments
S.R.O. and K.H.W. contributed equally to this report.
We thank the UF vector core members for advice on the production and characterization of rAAV1 and 5. We thank Eric Kolbrenner for technical assistance and the UF ICBR for software and hardware assistance. We also thank Lakshmanan Govindasamy for computer modeling assistance.
This work is supported by NIH grants PO1 HL51811 and P50 HL59412 and the Edwin E. Koger Chair to N.M. K.H.W. is supported by NIH training grant T32 AI 7110.
N.M. is an inventor on patents related to recombinant AAV technology and owns equity in a gene therapy company that is commercializing AAV for gene therapy applications.
Footnotes
This study is dedicated to the memory of Wu Xiao.
REFERENCES
- 1.Bantel Schaal, U., and H. zur Hausen. 1984. Characterization of the DNA of a defective human parvovirus isolated from a genital site. Virology 134:52-63. [DOI] [PubMed] [Google Scholar]
- 2.Barrijal, S., M. Perros, Z. Gu, B. L. Avalosse, P. Belenguer, F. Amalric, and J. Rommelaere. 1992. Nucleolin forms a specific complex with a fragment of the viral (minus) strand of minute virus of mice DNA. Nucleic Acids Res. 20:5053-5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becerra, S. P., F. Koczot, P. Fabisch, and J. A. Rose. 1988. Synthesis of adeno-associated virus structural proteins requires both alternative mRNA splicing and alternative initiations from a single transcript. J. Virol. 62:2745-2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becerra, S. P., J. A. Rose, M. Hardy, B. M. Baroudy, and C. W. Anderson. 1985. Direct mapping of adeno-associated virus capsid proteins B and C: a possible ACG initiation codon. Proc. Natl. Acad. Sci. USA 82:7919-7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buller, R. M., and J. A. Rose. 1978. Characterization of adenovirus-associated virus-induced polypeptides in KB cells. J. Virol. 25:331-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiorini, J. A., F. Kim, L. Yang, and R. M. Kotin. 1999. Cloning and characterization of adeno-associated virus type 5. J. Virol. 73:1309-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark, K. R., X. Liu, J. P. McGrath, and P. R. Johnson. 1999. Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum. Gene Ther. 10:1031-1039. [DOI] [PubMed] [Google Scholar]
- 8.Clark, K. R., F. Voulgaropoulou, and P. R. Johnson. 1996. A stable cell line carrying adenovirus-inducible rep and cap genes allows for infectivity titration of adeno-associated virus vectors. Gene Ther. 3:1124-1132. [PubMed] [Google Scholar]
- 9.Donello, J. E., J. E. Loeb, and T. J. Hope. 1998. Woodchuck hepatitis virus contains a tripartite posttranscriptional regulatory element. J. Virol. 72:5085-5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan, D., Q. Li, A. W. Kao, Y. Yue, J. E. Pessin, and J. F. Engelhardt. 1999. Dynamin is required for recombinant adeno-associated virus type 2 infection. J. Virol. 73:10371-10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fry, E. E., S. M. Lea, T. Jackson, J. W. Newman, F. M. Ellard, W. E. Blakemore, R. Abu-Ghazaleh, A. Samuel, A. M. King, and D. I. Stuart. 1999. The structure and function of a foot-and-mouth disease virus-oligosaccharide receptor complex. EMBO J. 18:543-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao, G. P., M. R. Alvira, L. Wang, R. Calcedo, J. Johnston, and J. M. Wilson. 2002. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 99:11854-11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Girod, A., M. Ried, C. Wobus, H. Lahm, K. Leike, J. Kleinschmidt, G. Deleage, and M. Hallek. 1999. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat. Med. 5:1052-1056. (Erratum, 5:1438.) [DOI] [PubMed]
- 14.Grifman, M., M. Trepel, P. Speece, L. B. Gilbert, W. Arap, R. Pasqualini, and M. D. Weitzman. 2001. Incorporation of tumor-targeting peptides into recombinant adeno-associated virus capsids. Mol. Ther. 3:964-975. [DOI] [PubMed] [Google Scholar]
- 15.Grimm, D., A. Kern, K. Rittner, and J. A. Kleinschmidt. 1998. Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum. Gene Ther. 9:2745-2760. [DOI] [PubMed] [Google Scholar]
- 16.Handa, A., S. Muramatsu, J. Qiu, H. Mizukami, and K. E. Brown. 2000. Adeno-associated virus (AAV)-3-based vectors transduce haematopoietic cells not susceptible to transduction with AAV-2-based vectors. J. Gen. Virol. 81:2077-2084. [DOI] [PubMed] [Google Scholar]
- 17.Hermens, W. T., O. ter Brake, P. A. Dijkhuizen, M. A. Sonnemans, D. Grimm, J. A. Kleinschmidt, and J. Verhaagen. 1999. Purification of recombinant adeno-associated virus by iodixanol gradient ultracentrifugation allows rapid and reproducible preparation of vector stocks for gene transfer in the nervous system. Hum. Gene Ther. 10:1885-1891. [DOI] [PubMed] [Google Scholar]
- 18.Hileman, R. E., J. R. Fromm, J. M. Weiler, and R. J. Linhardt. 1998. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays 20:156-167. [DOI] [PubMed] [Google Scholar]
- 19.Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365-369. [DOI] [PubMed] [Google Scholar]
- 20.Hoggan, M. D., N. R. Blacklow, and W. P. Rowe. 1966. Studies of small DNA viruses found in various adenovirus preparations: physical, biological, and immunological characteristics. Proc. Natl. Acad. Sci. USA 55:1467-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaludov, N., K. E. Brown, R. W. Walters, J. Zabner, and J. A. Chiorini. 2001. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. J. Virol. 75:6884-6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King, J. A., R. Dubielzig, D. Grimm, and J. A. Kleinschmidt. 2001. DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. EMBO J. 20:3282-3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein, R. L., E. M. Meyer, A. L. Peel, S. Zolotukhin, C. Meyers, N. Muzyczka, and M. A. King. 1998. Neuron-specific transduction in the rat septohippocampal or nigrostriatal pathway by recombinant adeno-associated virus vectors. Exp. Neurol. 150:183-194. [DOI] [PubMed] [Google Scholar]
- 24.Kraulis, P. J. 1991. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Cryst. 24:946-950. [Google Scholar]
- 25.Kronenberg, S., J. A. Kleinschmidt, and B. Bottcher. 2001. Electron cryo-microscopy and image reconstruction of adeno-associated virus type 2 empty capsids. EMBO Rep. 2:997-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu, J., and S. C. Thorp. 2002. Cell surface heparan sulfate and its roles in assisting viral infections. Med. Res. Rev. 22:1-25. [DOI] [PubMed] [Google Scholar]
- 27.McCarty, D. M., M. Christensen, and N. Muzyczka. 1991. Sequences required for coordinate induction of adeno-associated virus p19 and p40 promoters by Rep protein. J. Virol. 65:2936-2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merritt, E. A., and D. J. Bacon. 1997. Raster3D photorealistic molecular graphics. Methods Enzymol. 277:505-524. [DOI] [PubMed] [Google Scholar]
- 29.Moskalenko, M., L. Chen, M. van Roey, B. A. Donahue, R. O. Snyder, J. G. McArthur, and S. D. Patel. 2000. Epitope mapping of human anti-adeno-associated virus type 2 neutralizing antibodies: implications for gene therapy and virus structure. J. Virol. 74:1761-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mulloy, B., and R. J. Linhardt. 2001. Order out of complexity—protein structures that interact with heparin. Curr. Opin. Struct. Biol. 11:623-628. [DOI] [PubMed] [Google Scholar]
- 31.Muzyczka, N., and K. I. Berns. 2001. Parvoviridae: the viruses and their replication, p. 2327-2360. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed. Lippincott, Williams and Wilkins, New York, N.Y.
- 32.Nicholls, A., K. A. Sharp, and B. Honig. 1991. Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11:281-296. [DOI] [PubMed]
- 33.Nicklin, S. A., H. Buening, K. L. Dishart, M. de Alwis, A. Girod, U. Hacker, A. J. Thrasher, R. R. Ali, M. Hallek, and A. H. Baker. 2001. Efficient and selective AAV2-mediated gene transfer directed to human vascular endothelial cells. Mol. Ther. 4:174-181. [DOI] [PubMed] [Google Scholar]
- 34.Parks, W. P., M. Green, M. Pina, and J. L. Melnick. 1967. Physicochemical characterization of adeno-associated satellite virus type 4 and its nucleic acid. J. Virol. 1:980-987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qing, K., C. Mah, J. Hansen, S. Zhou, V. Dwarki, and A. Srivastava. 1999. Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 5:71-77. [DOI] [PubMed] [Google Scholar]
- 36.Qiu, J., and K. E. Brown. 1999. A 110-kDa nuclear shuttle protein, nucleolin, specifically binds to adeno-associated virus type 2 (AAV-2) capsid. Virology 257:373-382. [DOI] [PubMed] [Google Scholar]
- 37.Qiu, J., A. Handa, M. Kirby, and K. E. Brown. 2000. The interaction of heparin sulfate and adeno-associated virus 2. Virology 269:137-147. [DOI] [PubMed] [Google Scholar]
- 38.Rabinowitz, J. E., F. Rolling, C. Li, H. Conrath, W. Xiao, X. Xiao, and R. J. Samulski. 2002. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J. Virol. 76:791-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rabinowitz, J. E., and R. J. Samulski. 2000. Building a better vector: the manipulation of AAV virions. Virology 278:301-308. [DOI] [PubMed] [Google Scholar]
- 40.Rabinowitz, J. E., W. Xiao, and R. J. Samulski. 1999. Insertional mutagenesis of AAV2 capsid and the production of recombinant virus. Virology 265:274-285. [DOI] [PubMed] [Google Scholar]
- 41.Ried, M. U., A. Girod, K. Leike, H. Buning, and M. Hallek. 2002. Adeno-associated virus capsids displaying immunoglobulin-binding domains permit antibody-mediated vector retargeting to specific cell surface receptors. J. Virol. 76:4559-4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossmann, M. G., and J. E. Johnson. 1989. Icosahedral RNA virus structure. Annu. Rev. Biochem. 58:533-573. [DOI] [PubMed] [Google Scholar]
- 43.Rutledge, E. A., C. L. Halbert, and D. W. Russell. 1998. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. J. Virol. 72:309-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi, W., G. S. Arnold, and J. S. Bartlett. 2001. Insertional mutagenesis of the adeno-associated virus type 2 (AAV2) capsid gene and generation of aav2 vectors targeted to alternative cell-surface receptors. Hum. Gene Ther. 12:1697-1711. [DOI] [PubMed] [Google Scholar]
- 45.Summerford, C., J. S. Bartlett, and R. J. Samulski. 1999. AlphaVbeta5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat. Med. 5:78-82. [DOI] [PubMed] [Google Scholar]
- 46.Summerford, C., and R. J. Samulski. 1998. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 72:1438-1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veldwijk, M. R., J. Topaly, S. Laufs, U. R. Hengge, F. Wenz, W. J. Zeller, and S. Fruehauf. 2002. Development and optimization of a real-time quantitative PCR-based method for the titration of AAV-2 vector stocks. Mol. Ther. 6:272-278. [DOI] [PubMed] [Google Scholar]
- 48.Wistuba, A., A. Kern, S. Weger, D. Grimm, and J. A. Kleinschmidt. 1997. Subcellular compartmentalization of adeno-associated virus type 2 assembly. J. Virol. 71:1341-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wistuba, A., S. Weger, A. Kern, and J. A. Kleinschmidt. 1995. Intermediates of adeno-associated virus type 2 assembly: identification of soluble complexes containing Rep and Cap proteins. J. Virol. 69:5311-5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wobus, C. E., B. Hugle-Dorr, A. Girod, G. Petersen, M. Hallek, and J. A. Kleinschmidt. 2000. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J. Virol. 74:9281-9293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu, P., W. Xiao, T. Conlon, J. Hughes, M. Agbandje-McKenna, T. Ferkol, T. Flotte, and N. Muzyczka. 2000. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J. Virol. 74:8635-8647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao, X., J. Li, and R. J. Samulski. 1998. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 72:2224-2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie, Q., W. Bu, S. Bhatia, J. Hare, T. Somasundaram, A. Azzi, and M. S. Chapman. 2002. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. USA 99:10405-10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu, L., T. Daly, C. Gao, T. R. Flotte, S. Song, B. J. Byrne, M. S. Sands, and K. Parker Ponder. 2001. CMV-beta-actin promoter directs higher expression from an adeno-associated viral vector in the liver than the cytomegalovirus or elongation factor 1 alpha promoter and results in therapeutic levels of human factor X in mice. Hum. Gene Ther. 12:563-573. [DOI] [PubMed] [Google Scholar]
- 55.Yang, Q., M. Mamounas, G. Yu, S. Kennedy, B. Leaker, J. Merson, F. Wong-Staal, M. Yu, and J. R. Barber. 1998. Development of novel cell surface CD34-targeted recombinant adenoassociated virus vectors for gene therapy. Hum. Gene Ther. 9:1929-1937. [DOI] [PubMed] [Google Scholar]
- 56.Zolotukhin, S., B. J. Byrne, E. Mason, I. Zolotukhin, M. Potter, K. Chesnut, C. Summerford, R. J. Samulski, and N. Muzyczka. 1999. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 6:973-985. [DOI] [PubMed] [Google Scholar]
- 57.Zolotukhin, S., M. Potter, W. Hauswirth, J. Guy, and N. Muzyczka. 1996. A humanized green fluorescent protein cDNA adapted for high-level expression in mammalian cells. J. Virol. 70:4646-4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zolotukhin, S., M. Potter, I. Zolotukhin, Y. Sakai, S. Loiler, T. J. Fraites, Jr., V. A. Chiodo, T. Phillipsberg, N. Muzyczka, W. W. Hauswirth, T. R. Flotte, B. J. Byrne, and R. O. Snyder. 2002. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods 28:158-167. [DOI] [PubMed] [Google Scholar]