

Abstract
It has been proposed that γ-secretase-mediated release of the amyloid precursor protein (APP) intracellular domain (AICD) results in nuclear translocation and signaling through a complex with the adaptor protein Fe65 and the histone acetyltransferase Tip60. Here, we show that APP and Fe65 activate transcription through a Gal4-Tip60 reporter in presenilin-1/2-deficient cells lacking generation of AICD. APP and Fe65 also activated transcription in the presence of γ-secretase inhibitors that prevent amyloid β-peptide production in human embryonic kidney 293 and SH-SY5Y cells. In contrast to the transcriptionally active Notch intracellular domain, expression of AICD did not activate transcription. An alternative mechanism for APP signal transduction is suggested by the identification of essential cyclin-dependent kinase (CDK) phosphorylation sites in Tip60. Mutation of these Tip60 phosphorylation sites or treatment with the CDK inhibitor roscovitine blocked the ability of APP to signal through Tip60. Moreover, APP stabilized Tip60 through CDK-dependent phosphorylation. Subcellular fractionation and confocal immunofluorescence showed that APP recruited Tip60 to membrane compartments. Thus, APP may signal to the nucleus by a γ-secretase-independent mechanism that involves membrane sequestration and phosphorylation of Tip60.
The amyloid precursor protein (APP)2 has played a central role in Alzheimer disease research ever since pathogenic mutations were discovered within APP over a decade ago. APP is a type 1 transmembrane protein that resembles a cell-surface receptor (1). Studies on APP biology have focused primarily on its proteolytic processing. APP undergoes two separate proteolytic pathways that have been termed the non-amyloidogenic and amyloidogenic pathways (reviewed in Ref. 2). In the non-amyloidogenic pathway, APP is first cleaved by γ-secretase, generating the C-terminal transmembrane fragment C83. The amyloidogenic pathway involves cleavage by the γ-secretase BACE, generating the C-terminal fragment C99, which is then cleaved by γ-secretase to generate the amyloid β-peptide (Aβ) and the C50/APP intracellular domain (AICD) fragment. This series of proteolytic events of ectodomain shedding followed by intramembrane cleavage is reminiscent of the processing of the Notch receptor following binding to its ligands (reviewed in Ref. 3). Both APP and Notch require the presenilins for γ-secretase cleavage within the membrane (4–7).
The pathogenic autosomal dominant mutations in APP and the presenilins lead to increased generation of the fibrillogenic peptide Aβ42, which forms amyloid plaques, one of the hallmark pathogenic lesions of Alzheimer disease (reviewed in Refs. 8 and 9). Moreover, Aβ is neurotoxic both in vitro and in vivo (10, 11). These observations have led to the amyloid hypothesis, which proposes that the accumulation of Aβ in the brain causes neurodegeneration and the dementia of Alzheimer disease. However, despite the significant effort spent investigating the properties of APP, its biological function and the reason for its cleavage remain enigmatic. There is evidence that APP plays a role in cell adhesion, axonal transport, neurite outgrowth, and, recently, signal transduction (12–18).
It has been suggested that APP may signal by a mechanism similar to that of the Notch receptor, in which the intracellular tail released by presenilin/γ-secretase translocates to the nucleus and activates transcription. This idea was supported by the observation that APP, together with the adaptor protein Fe65, could activate the transcriptional activity of the histone acetyltransferase Tip60 (18). Thus, presenilins may mediate the regulated intramembrane proteolysis of a number of transmembrane proteins that release soluble intracellular domains that carry out signaling functions (reviewed in Refs. 19 and 20).
The adaptor protein Fe65 contains three protein interaction domains: the WW domain, which binds proline-rich sequences, and two C-terminal phosphotyrosine-binding domains, which interact with Tip60 and APP, respectively (18, 21). Fe65 and APP have been placed within a biological pathway in Caenorhabditis elegans, as the respective mutants have similar pharyngeal pumping phenotypes (22). Expression of Fe65 can lead to the stabilization and nuclear translocation of AICD, where it may induce apoptosis through Tip60 (23–25, 48). In addition, Tip60 and its histone acetyltransferase activity have been implicated in the regulation of gene expression and DNA repair (26–28). However, the mechanism underlying the ability of APP to regulate Tip60-mediated transcription has not been determined. In this study, we provide evidence that, in contrast to Notch, APP is able to activate transcription through Tip60 independently of γ-secretase cleavage by recruiting Tip60 to the membrane, leading to Tip60 activation through cyclin-dependent kinase-mediated phosphorylation. This is followed by nuclear translocation of Tip60 and Fe65 and activation of transcription.
MATERIALS AND METHODS
Cell Lines and Treatments
Embryonic stem cells were cultured at 37 °C in a 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium supplemented with 15% fetal bovine serum, 292 μg/ml l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, 100 μm nonessential amino acids, 1 mm sodium pyruvate, 100 units of leukemia inhibitory factor, and 0.008% 2-mercaptoethanol. Human embryonic kidney 293 (HEK-293) and SH-SY5Y cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 292 μg/ml l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 1 mm sodium pyruvate. Cells were treated with 10 μm roscovitine (Sigma), 20 μm benzyloxycarbonyl-VAD-fluoromethyl ketone (Sigma), or 20 nm leptomycin B (LC Laboratories). γ-Secretase inhibitors WPE-II-72 and WPE-III-31c were obtained from Michael Wolfe, and cells were treated at 500 nm and 5 μm, respectively, which are 25 times their IC50 values. In the dephosphorylation assay, 5 μl of calf intestinal alkaline phosphatase (New England Biolabs Inc.) or H2O was added to 40 μl of sample plus 5 μl of NE Buffer 3, followed by incubation for 1 h at 37 °C. The reaction was stopped by the addition of 50 μl of 2× sample buffer.
Transfection and Plasmids
Cells were transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocol. Basically, the cell medium was replaced with Opti-MEM I, followed by the addition of a mixture of Opti-MEM I, DNA, and Lipofectamine 2000. The cells were then incubated at 37 °C for 3–5 h, at which time an equivalent volume of the medium was added along with appropriate drug treatments. The plasmids used were as follows: pM-Tip60 (for Gal4-Tip60), pCMV-Fe65, and pCMV-Myc-Fe65-(128–711) (obtained from T. C. Sudhof); pCMV-APP695wt; pcDNA-C50; pcDNA-C50-Myc; pcDNA-HA-Tip60 and pcDNA-HA-Tip60s86/90a (obtained from S. Khochbin); pM-Gal4-Tip60wt (generated by subcloning hemagglutinin (HA)-Tip60 by cutting the pM and HA-Tip60 vectors with EcoRI and XbaI); pM-Gal4-Tip60s86/90a (generated by subcloning HA-Tip60 by cutting the pM and HA-Tip60 vectors with EcoRI and XbaI); pRK-PS1wt; pFR-luciferase (Stratagene); pRL-TK (Renilla; Promega); HES1-luciferase; mNotchΔE; pcDNA-NICD; and pcDNA3 empty vector (to normalize total DNA).
Transactivation Assays
Cells were plated onto 24-well plates and transfected when ~70–80% confluent. Three wells were transfected per treatment. We optimized DNA concentrations for the signaling assay in 293 cells: 0.2 μg of pFR-luciferase, 0.05 μg of pRL-TK, 0.2 μg of pM-Gal4-Tip60, 0.2 μg of pCMV-Myc-Fe65-(128–711), and 0.1 μg of pCMV-APP695wt per well. The pCMV-Myc-Fe65-(128–711) plasmid was used because it was shown previously to act similarly to full-length Fe65 (18) and, in our hands, required a lower amount of DNA to allow activation of the reporter by Tip60. Additionally, this construct has a Myc tag, which facilitated confirmation of equivalent expression by Western blotting between different conditions. Approximately 20–24 h after transfection, the cells were lysed with Promega passive lysis buffer. Assays were then performed (Dual-Luciferase kit, Promega) according to the protocol recommended by the manufacturer. Luciferase activity was read with a Wallac VICTOR Model 1420 multilabel counter. To control for transfection efficiency, the activity of the firefly luciferase reporter driven by the GAL4 promoter was normalized to the activity of the Renilla luciferase reporter (PRL-TK). The -fold induction was then determined as the average of three wells per treatment normalized to the average signal of the reporter-alone wells.
Antibodies
Anti-HA antibody was obtained from Sigma. To detect Tip60, we used antibody N1 (a kind gift of John Lough) (29). Paul Greengard generously provided anti-Fe65 antibody (30). We used rabbit antibody C8, which is directed against the APP C terminus. Anti-Aβ antibodies 159 and 6E10 were used for immunoprecipitation and immunoblotting, respectively. Anti-lamin B2 antibody was obtained from Zymed Laboratories Inc. We purchased anti-actin antibody from Oncogene Science. Mouse monoclonal antibody 56C6 (Lab Vision Corp.) was used for neprilysin. All secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc.
Subcellular Fractionation
Cells were collected in phosphate-buffered saline when ~90% confluent and pelleted by centrifugation at 1000 × g for 10 min. They were then washed with buffer A (10 mm HEPES-KOH, 1.5 mm MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 1 mM EDTA, 1 mm EGTA, 1 μm microcystin, 1 mm NaVO4, and Complete protease inhibitor mixture (Roche Applied Science)) and centrifuged at 1000 × g for 10 min. The cells were swelled for 10 min in buffer A, followed by 20× Dounce homogenization with a glass pestle. Nuclei were then pelleted by centrifugation at 1000 × g for 10 min. The pellet and supernatant were separated, and the pellet was lysed with deoxycholate-containing radioimmune precipitation assay buffer (RIPA-DOC) plus phosphatase inhibitors (50 mm NaF, 5 mm Na2P2O7, 1 μM microcystin, and 1 mm NaVO4) and Complete protease inhibitor mixture and saved as the nuclear fraction. The supernatant was then combined with 0.11 volume of buffer B (0.3 m HEPES-KOH, 1.4 m KCl, 0.03 m MgCl2, 1 mM EDTA, 1 mM EGTA, 1 μM microcystin, 1 mM NaVO4, and Complete protease inhibitor mixture), followed by ultracentrifugation at 100,000 × g for 1 h. The pellet was resuspended in RIPA-DOC with the same phosphatase and protease inhibitors and saved as the membrane fraction. The supernatant was then saved as the cytoplasmic fraction.
Transgenic Mice
The cortices and hippocampi were dissected from 3-month-old and 2-year-old Tg2576 APP transgenic mice. We used two animals of each genotype and age. The brain samples were then subjected to fractionation as described above. Briefly, the brain samples were washed with phosphate-buffered saline and swelled in buffer A for 10 min, followed by Dounce homogenization with a glass pestle. Nuclei were pelleted by centrifugation at 1000 × g for 10 min. The supernatant was then combined with 0.11 volume of buffer B and ultracentrifuged at 100,000 × g for 1 h. The pellet was resuspended in RIPA-DOC and used as the membrane fraction for Western blot analysis.
Immunoprecipitation and Western Blotting
For the co-immunoprecipitation experiments, protein lysates were precleared with protein G-Sepharose (Amersham Biosciences) for 1 h at 4 °C, followed by an overnight incubation with the immunoprecipitating antibodies. The next morning, fresh protein G-Sepharose was added and rocked at 4 °C for 2 h, followed by three washes with RIPA-DOC for the anti-HA antibody immunoprecipitations and with phosphate-buffered saline for the antibody 159 immunoprecipitations. Proteins were then eluted from beads by boiling in 2× sample buffer for 5 min. In the Aβ immunoprecipitation/Western blot experiments, HEK-293 and SH-SY5Y cells transiently transfected with Tip60, Fe65, and APP and the 20E2 APPsw-expressing HEK-293 stable cell line were incubated for 48 or 24 h, respectively, to condition the medium. The γ-secretase inhibitor was added at the beginning of the conditioning at a final concentration of 500 nm, which is 25 times the IC50 value. For transient transfection, additional γ-secretase inhibitor was added after 24 h at 12.5 times the IC50 value to compensate for turnover of the inhibitor. The conditioned medium was collected and spun at 1000 rpm for 10 min to remove cellular debris. Anti-Aβ antibody 159 was added to the conditioned medium and rocked overnight at 4 °C. The next morning, protein A-Sepharose (Amersham Biosciences) was added and rocked 4 °C for 2 h. The protein A-Sepharose was then washed three times with phosphate-buffered saline, and Aβ was eluted from the beads with 2× sample buffer and boiled for 5 min. In the experiments on APP stabilization of Tip60, the cells were collected 24 h post-transfection using Promega passive lysis buffer. The lysates were clarified by centrifugation at 10,000 × g for 10 min, and equivalent cell lysates were loaded onto the gel for Western blotting.
Immunocytochemistry
SH-SY5Y cells were plated onto coverslips in 24-well plates 2 days prior to transfection. The cells were transfected with HA-Tip60 alone or with Fe65 and APP as described above and then incubated for ~24 h. The cells were then fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.2% Triton X-100 for 10 min, and blocked with 10% donkey serum for 1 h, followed by overnight incubation with anti-HA antibody and antibody C8 to detect Tip60 and APP, respectively. The next day, we used Cy3-conjugated anti-mouse antibody to label Tip60 and Cy2-conjugated anti-rabbit antibody to label APP. Finally, Hoechst stain was included in the first wash to label the nuclei. Images were then taken with a Zeiss confocal microscope.
RESULTS
APP Signaling in Presenilin-deficient Cells
Activation of the Notch signaling pathway requires γ-secretase cleavage by the presenilins to release the Notch intracellular domain (NICD), which translocates to the nucleus and activates target genes. To determine whether APP could signal to the nucleus by a similar mechanism, we utilized embryonic stem (ES) cells deficient in both presenilin-1 and presenilin-2 (presenilin-1/2 knockout (PS-KO) cells). These cells are deficient in γ-secretase cleavage of APP and do not secrete Aβ (6). To address the potential signaling function of APP, we used the Gal4-Tip60 fusion construct, previously shown to activate a Gal4-luciferase reporter upon cotransfection of both Fe65 and APP (18). This is a more physiological system for measuring the signaling ability of APP compared with APP-Gal4 assays, as it does not rely on the fusion of an artificial DNA-binding domain to APP itself. As reported previously (18), significant transactivation by Gal4-Tip60 occurred only upon coexpression of both Fe65 and APP (Fig. 1A). APP signaling was reduced by ~45% in PS-KO cells relative to wild-type ES cells and was rescued upon expression of presenilin-1 (PS1) (Fig. 1A). A γ-secretase inhibitor reduced APP signaling in wild-type cells to a level comparable with that in PS-KO cells (data not shown). PS-KO cells were resistant to the γ-secretase inhibitor, consistent with the lack of γ-secretase activity in these cells. However, APP retained the ability to activate Tip60 in the absence of presenilins, suggesting that γ-secretase cleavage of APP is not required for activation of Tip60. This contrasts with Notch signaling, which was completely inhibited in PS-KO cells (Fig. 1B). These results were confirmed in two independently derived wild-type and presenilin-deficient cell lines, indicating that presenilin-independent APP signaling is not a unique property of an individual presenilin-deficient cell line. These results suggest that APP may signal through a mechanism that is distinct from that of the Notch pathway, despite similar mechanisms of proteolytic processing.
FIGURE 1. APP signaling does not require γ-secretase cleavage, in contrast to Notch signaling.
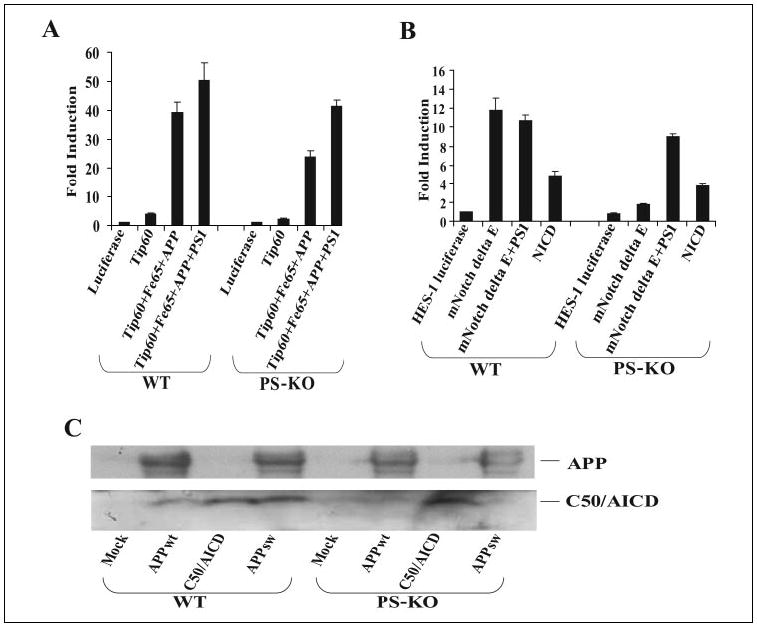
A, wild-type (WT) ES or PS-KO cells were cotransfected in triplicate with the pFR-luciferase, pRL-TK, Gal4-Tip60, Fe65, APP, and PS1 plasmids as indicated. Approximately 24 h after transfection, the cells were collected, and dual-luciferase reporter assays were performed. B, wild-type ES or PS-KO cells were cotransfected in triplicate with HES1-luciferase, pRL-TK, mNotchΔE, PS1, and NICD as indicated, and luciferase assays were performed at 24 h post-transfection. C, wild-type ES or PS-KO cells were transfected with the pcDNA (Mock), APPwt, C50/AICD, and APPsw plasmids. The cells were harvested 24 h after transfection, separated on a 10 –20% Tris/Tricine gel, and Western-blotted with antibody C8 (directed against the APP C-terminal domain).
We then confirmed that presenilin-deficient cells fail to generate detectable levels of the C50 fragment corresponding to AICD. We probed lysates from either wild-type ES or PS-KO cells transfected with plasmids encoding wild-type APP (APPwt), C50/AICD, or APPsw. Immunoblot analyses of wild-type cells expressing APPwt or APPsw with an antibody raised against the APP C terminus showed a band at ~6 kDa that comigrated with transfected C50/AICD (Fig. 1C). However, PS-KO cells expressing APPwt or APPsw did not show a clear 6-kDa band, although this band appeared in PS-KO cells transfected with the C50/AICD plasmid. Thus, APP is able to activate Tip60 by a mechanism that does not require γ-secretase generation of C50/AICD.
We then confirmed that presenilins are required for Notch signaling, in contrast to APP signaling, which did not require presenilins. We expressed the activated Notch construct mNotchΔE, which encodes a truncated Notch-1 protein retaining the transmembrane domain, together with a Notch-1-responsive HES1-luciferase reporter. mNotchΔE was able to drive significant transactivation of the HES1 reporter in wild-type cells, but this signaling ability was absent in PS-KO cells (Fig. 1B), consistent with previous reports (6, 7). Expression of presenilin in the PS-KO cells rescued the ability of mNotchΔE to activate the HES1 reporter (Fig. 1B). This shows that the lack of Notch signaling in presenilin-deficient cells is due solely to the lack of presenilin-dependent γ-secretase cleavage of mNotchΔE. Further illustrating this point, a similar level of transactivation was obtained upon expression of soluble NICD in both wild-type and presenilin-deficient cells (Fig. 1B), demonstrating that both cell lines are similarly competent in signal transduction. These results suggest that presenilin-deficient cells cannot carry out signaling that requires γ-secretase activity, yet these cells can support APP-mediated activation of Tip60. Thus, APP may signal through an alternative mechanism.
γ-Secretase Independence of APP Signaling
To confirm this surprising γ-secretase-independent mode of APP signaling, we performed similar assays in HEK-293 and SH-SY5Y cells. As expected, we observed significant transactivation only upon coexpression of Fe65 and APP (Fig. 2, A and B). Treatment with the γ-secretase inhibitor WPE-II-72 at 25 times the IC50 value did not significantly affect APP-Tip60 signaling in HEK-293 or SH-SY5Y cells (Fig. 2, A and B). This concentration of the γ-secretase inhibitor greatly decreased Aβ production (Fig. 2C) and markedly increased accumulation of APP C-terminal fragments (Fig. 2A, lower panel). The γ-secretase independence of APP signaling was further confirmed using another γ-secretase inhibitor, WPE-III-31c, in both HEK-293 and SH-SY5Y cells (data not shown). These results confirm that APP nuclear signaling through Tip60 can occur independently of γ-secretase cleavage.
FIGURE 2. γ-Secretase inhibitors do not affect APP signaling, and AICD does not have transcriptional activity.

A (upper panel) and B, HEK-293 and SH-SY5Y cells, respectively, were cotransfected in triplicate with pFR-luciferase, pRL-TK, Gal4-Tip60, Fe65, APP, and C50-Myc as indicated. After 3 h, the γ-secretase inhibitor WPE-II-72 (I-72, I72; 500 nm) was added. Luciferase assays were performed on cell lysates at 24 h after transfection. A (lower panel), cell lysates were Western-blotted for APP with antibody C8 and for actin as a loading control. C, HEK-293 cells stably expressing APPsw or HEK-293 and SH-SY5Y cells transiently transfected with Tip60, Fe65, and APPwt were incubated in the presence or absence (Control) of the γ-secretase inhibitor WPE-II-72 (500 nm). Aβ was immunoprecipitated with antibody 159 and Western-blotted with antibody 6E10. D, HEK-293 cells were transfected with wild-type Tip60 (Tip60wt) or the Tip60(S86A/S90A) mutant (Tip60s86/90a) together with Fe65 or APP as indicated. Tip60 was immunoprecipitated (IP) with anti-HA antibody, and the co-immunoprecipitated APP was detected by Western blotting with antibody C8.
To further examine the role of AICD in this signal transduction pathway, we replaced APP in the signaling assay with a C50 construct in HEK-293 cells. C50 had essentially no signaling ability (Fig. 2A, upper panel), in contrast to the signaling-competent Notch-derived NICD (Fig. 1B). Moreover, we observed that C50 had a dominant-negative effect in which it decreased APP signaling when coexpressed with APP (Fig. 2A, upper panel). Immunoblot analysis of lysates showed equivalent levels of APP expression (Fig. 2A, lower panel). Similar results were obtained in APP signaling assays in wild-type ES and PS-KO cells (data not shown). These results suggest that C50/AICD does not activate transcription through Tip60, but rather acts as a dominant-negative protein in this signaling pathway.
It has been suggested that APP may be able to carry out signal transduction through cleavage by caspases at a cleavage site within its cytoplasmic C-terminal domain (31, 32). To address this possibility, we used the pan-caspase inhibitor benzyloxycarbonyl-VAD-fluoromethyl ketone in another series of luciferase assays in HEK-293 cells. Inhibiting caspase activity with benzyloxycarbonyl-VAD-fluoromethyl ketone failed to significantly affect the ability of APP to activate Tip60 (Fig. 3A). Notably, there was still no effect on APP signaling when both γ-secretase and caspase inhibitors were added together. Similar results were observed in the wild-type ES and PS-KO cells (Fig. 3B). Thus, APP retains the ability to activate Tip60 signaling in the absence of cleavage by either γ-secretase or caspases.
FIGURE 3. APP signaling is independent of caspase cleavage and does not regulate neprilysin levels.
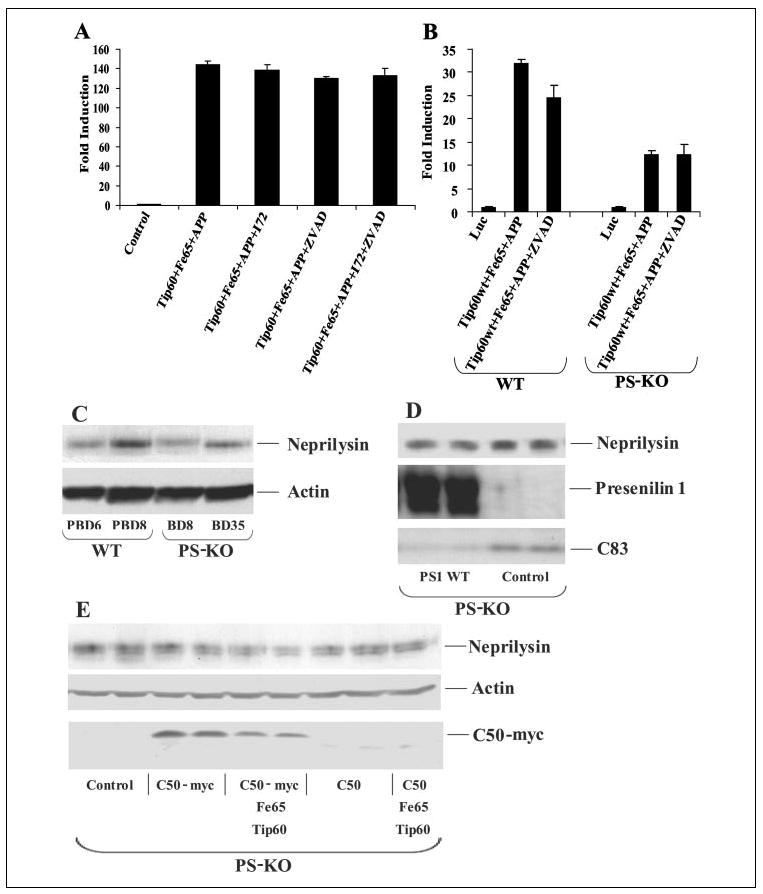
A and B, HEK-293 cells and wild-type (WT) ES and PS-KO cells, respectively, were cotransfected with pFR-luciferase (Luc), pRL-TK, Gal4-Tip60, Fe65, and APP as indicated. Cells were treated 4 h post-transfection with the γ-secretase inhibitor WPE-II-72 (I72; 500 nm) and with the caspase inhibitor benzyloxycarbonyl-VAD-fluoromethyl ketone (ZVAD; 20 μm). Luciferase assays were performed 24 h after transfection. C, whole cell lysates from two independent wild-type ES (PBD6 and PBD8) and two PS-KO (BD8 and BD35) cell lines were probed from neprilysin and then reprobed for actin. D, BD8 PS-KO cells were transfected with PS1, and lysates were collected 24 h later and Western-blotted in duplicate for neprilysin, presenilin, and APP C-terminal fragments. E, BD8 PS-KO cells were transfected with either C50 or C50-Myc alone or with Tip60 and Fe65. After 24 h, the cells were harvested and Western-blotted for neprilysin, actin, and C50-Myc expression. (C50 expression was confirmed on a darker exposure (data not shown).)
A recent study identified the Aβ-degrading enzyme neprilysin as a potential transcriptional target of C50/AICD (33). We attempted to confirm these results using our wild-type ES and PS-KO cell lines. We observed significant cell line variability in the levels of endogenous neprilysin that did not correlate with presenilin genotype (Fig. 3C), indicating that the variation is not related to presenilin expression. Furthermore, we attempted to elevate neprilysin levels in PS-KO cells by overexpression of PS1, C50, or C50-Myc. We did not observe any reproducible changes in neprilysin levels upon expression of any of the plasmids 24 h after transfection (Fig. 3, D and E). We confirmed that both PS1 and C50-Myc were expressed by Western blotting (Fig. 3, D and E). Moreover, we confirmed that transfected PS1 was functional, as it rescued the accumulation of APP C-terminal fragments that was observed in the absence of γ-secretase cleavage of APP (Fig. 3D). Similar results were observed in HEK-293 cells transfected with C50 or C50-Myc (data not shown). These results suggest that neprilysin may not be a transcriptional target of C50/AICD.
Complex Formation of Tip60, Fe65, and APP
Having shown that holo-APP or the membrane-spanning C-terminal fragments rather than C50/AICD may mediate signaling, we investigated whether Tip60 forms a complex with holo-APP. Tip60 co-immunoprecipitated holo-APP (Fig. 2D). The presence of holo-APP suggests that Tip60 may form a complex with APP in a membrane compartment rather than in the nucleus as suggested previously (18). Even with longer exposures, we were unable to detect significant levels of APP C-terminal fragments or AICD in the Tip60 immunoprecipitates, suggesting that Tip60 forms a complex with holo-APP. Coexpression of Fe65 along with Tip60 and APP increased complex formation between Tip60 and APP (Fig. 2D). These results suggest that Tip60 forms a complex with holo-APP, presumably at the membrane, and support the hypothesis that APP may signal through a mechanism that involves membrane recruitment of Tip60 rather than nuclear translocation of AICD.
Role of Cyclin-dependent Kinase (CDK) Phosphorylation of Tip60 in Signaling
Because APP can activate Tip60 through a pathway that does not require γ-secretase cleavage of APP, we considered whether APP signaling involves the activation of kinase cascades. Two CDK sites were recently identified in Tip60 at serines 86 and 90 (34). To examine the role of these phosphorylation sites in APP-Tip60 signaling, we fused the Gal4 DNA-binding domain to a Tip60 mutant in which serines 86 and 90 were converted to alanines. We then assessed the activity of the Tip60 mutant in luciferase reporter assays of transfected HEK-293 cells. The Tip60 mutant could not be activated by coexpression of Fe65 and APP, in contrast to the strong transactivation observed with wild-type Tip60 (Fig. 4A). To further confirm the requirement of CDK phosphorylation of Tip60 in APP signal transduction, we tested the CDK inhibitor roscovitine. CDK inhibition with roscovitine significantly decreased the ability of APP to activate Tip60 (Fig. 4A). Similar results were observed in the ES cells (Fig. 4B). Thus, phosphorylation of Tip60 at serines 86 and 90 by CDKs contributes to transcriptional signaling. The Tip60 phosphorylation mutant was able to co-immunoprecipitate APP as efficiently as wild-type Tip60 when coexpressed with Fe65 (Fig. 2D). Thus, the inability of APP to activate the Tip60 mutant is unlikely to be due to an effect on complex formation between APP and Tip60.
FIGURE 4. CDK phosphorylation of Tip60 is required for APP-dependent transactivation and stabilization of Tip60.
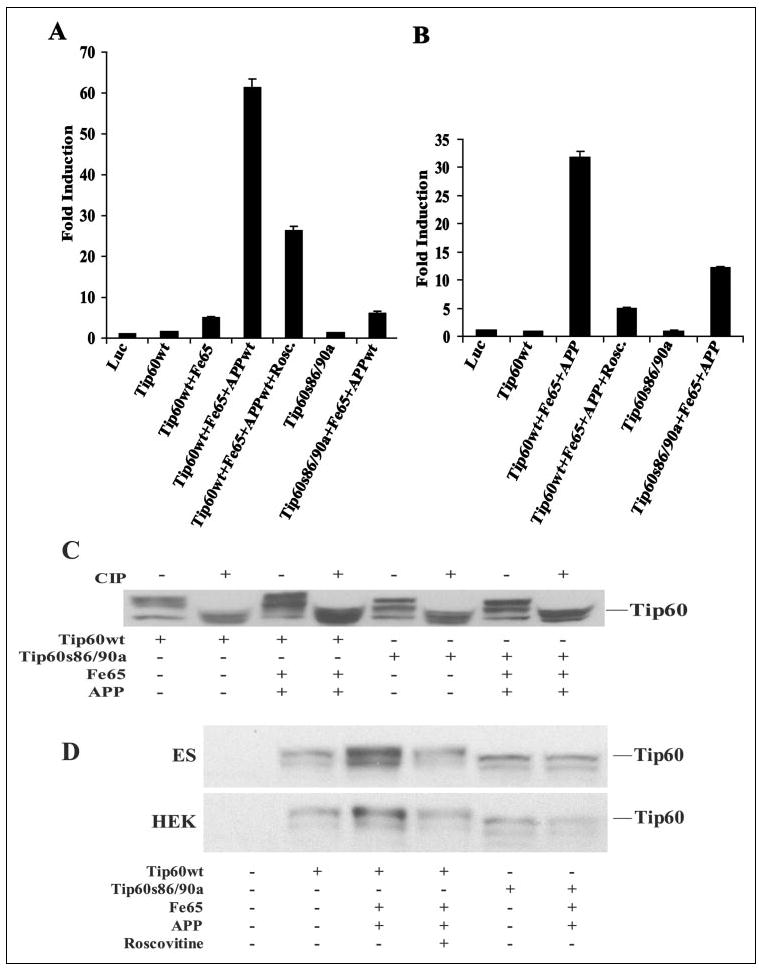
A and B, luciferase assays were performed on HEK-293 and wild-type ES cells, respectively, transfected with Gal4-Tip60 (Tip60wt) or Gal4-Tip60(S86A/S90A) (Tip60s86/ 90a) together with Fe65 and APP. Cells were treated with the CDK inhibitor roscovitine (Rosc.; 20 μm) as indicated for 24 h. Luc, luciferase. C, shown is the phosphorylation of Tip60. HEK-293 cells were cotransfected with wild-type Tip60 (Tip60wt) or Tip60(S86A/S90A) (Tip60s86/90a) alone or with Fe65 and APP. Cells were harvested at 24 h post-transfection, and equivalent amounts of lysate protein were incubated in the absence or presence of calf intestinal alkaline phosphatase (CIP) and then Western-blotted with anti-Tip60 antibody N1. D, APP-dependent stabilization of Tip60 requires CDK phosphorylation. Wild-type ES and HEK-293 cells were cotransfected with wild-type Tip60 or Tip60(S86A/S90A), Fe65, and APP and treated with roscovitine (20 μm).
Stabilization of Phosphorylated Forms of Tip60 by APP and Fe65
We then determined whether APP facilitates Tip60 phosphorylation. The wild-type or phosphorylation mutant Tip60 construct was cotransfected with Fe65 and APP in HEK-293 cells (Fig. 4C). Treatment of the cell lysates with calf intestinal alkaline phosphatase led to a condensation of the multiple Tip60 bands into a single faster migrating form, suggesting that the slower migrating forms are phosphorylated forms of Tip60. The Tip60 mutant migrated at a lower apparent molecular mass compared with wild-type Tip60, consistent with lack of phosphorylation of Tip60 at the CDK sites. Alkaline phosphatase treatment also led to the condensation of the Tip60 mutant bands into a single faster migrating form that comigrated with dephosphorylated wild-type Tip60, indicating that there are additional phosphorylation sites yet to be identified. Coexpression of Tip60 with Fe65 and APP led to markedly enhanced levels of the slowest migrating phosphorylated forms of wild-type Tip60 (Fig. 4C). Densitometric analysis of the blot showed an ~73% increase in the slowest migrating band relative to the fastest migrating form of Tip60. Fe65 and APP did not affect the Tip60 CDK phosphorylation mutant, suggesting that they act to specifically increase phosphorylation at the CDK sites. Coexpression of APP and Fe65 also led to significant phosphorylation of Tip60 at the CDK sites in ES cells (Fig. 4D). To confirm that this phosphorylation event is mediated by CDKs, we tested the CDK inhibitor roscovitine. The increase in phosphorylation of Tip60 mediated by Fe65 and APP was completely blocked by the CDK inhibitor roscovitine (Fig. 4D). These results indicate that APP and Fe65 potentiate CDK-mediated phosphorylation of Tip60, thereby activating nuclear signaling.
APP Recruits Tip60 to Membrane Compartments
We then determined the subcellular localization of the Tip60-Fe65-APP complex by performing subcellular fractionation of HEK-293 cells expressing Tip60. A substantial portion of Tip60 was present in the nuclear fraction whether or not Fe65 and APP were coexpressed (Fig. 5A), as expected based on previous studies (18). However, upon expression of Fe65 and APP, there was a marked redistribution of Tip60 to the membrane fraction (Fig. 5A). The nuclear protein lamin B2 was present in the nuclear fraction, but was not detectable in the membrane fraction, suggesting that membrane-associated Tip60 is not due to nuclear contamination. We then determined whether endogenous Tip60 can be recruited to the membrane by APP. For this purpose, we utilized control HEK-293 cells and HEK-293 cells stably expressing APPwt. As observed with overexpressed Tip60, most of the endogenous Tip60 immunoreactivity was in the nuclear fraction in both the control and APP-expressing cells (Fig. 5B). However, in APP-expressing cells, there was substantial Tip60 immunoreactivity in the membrane fraction that was not observed in the control cells. The APPwt-expressing cells showed an ~10-fold increase in membrane-associated Tip60 compared with control cells that did not overexpress APP and an ~2-fold increase in total cellular Tip60. Thus, endogenous Tip60 can be recruited to the membrane by APP. Once again, the lack of lamin B2 signal in the membrane fractions suggests that Tip60 immunoreactivity in the membrane fraction is not due to nuclear contamination.
FIGURE 5. APP recruits Tip60 to a membrane compartment in vitro and in vivo.
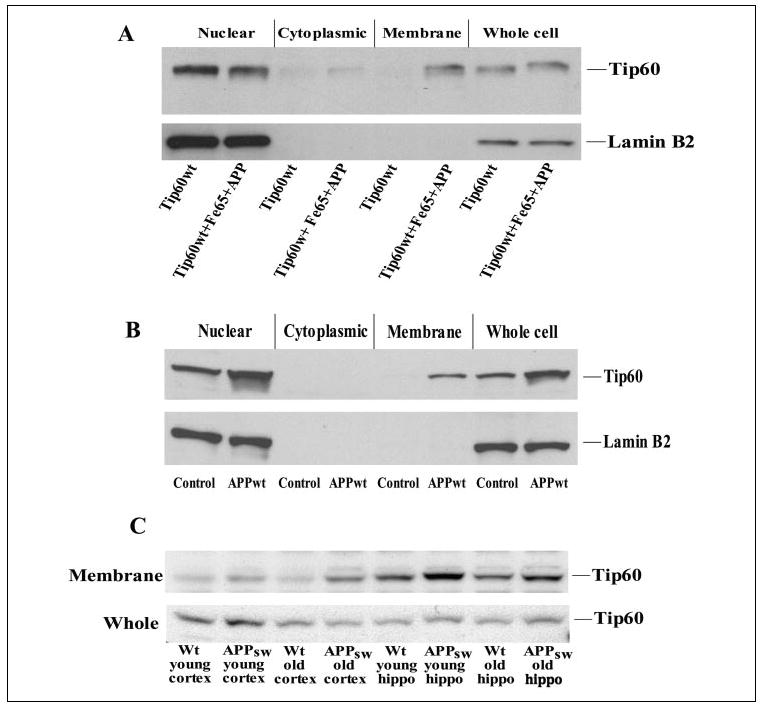
A, HEK-293 cells were transfected with wild-type Tip60 (Tip60wt) alone or with Fe65 and APP; and 24 h after transfection, the cells were subjected to sub-cellular fractionation into nuclear, cytoplasmic, and membrane fractions. The fractions were Western-blotted for Tip60, which showed membrane recruitment by coexpression of Fe65 and APP. Western blotting for the nuclear protein lamin B2 confirmed the lack of nuclear contamination of the membrane fraction. B, similar results were observed with endogenous Tip60 in HEK-293 cells stably expressing APP. C, cortices and hippocampi were isolated from APP transgenic and littermate control (wild-type (Wt)) mice that were either 3 months (young) or 2 years (old) of age. Cortical and hippocampal samples were then fractionated, and Western blotting was performed for Tip60.
We then determined whether APP recruits Tip60 to the membrane in vivo by performing subcellular fractionation of wild-type and APP transgenic mouse brains. Tip60 levels were compared in membrane fractions from the cortices and hippocampi of wild-type and Tg2576 APP transgenic mice that were either 3 months old (prior to plaque formation) or 2 years old (when the amyloid plaque load is high). Significantly more Tip60 appeared in the membrane fractions of all APP transgenic samples compared with age-matched wild-type control samples (Fig. 5C). However, Tip60 was also detected in the membrane fractions of wild-type mice, particularly in the hippocampus, suggesting that membrane sequestration occurs under physiological conditions. The purity of these membrane fractions was once again confirmed by the absence of the nuclear protein lamin B2. These results suggest that APP recruits Tip60 to membrane compartments, consistent with the formation of a Tip60 complex with the transmembrane form of APP.
Colocalization of Tip60 and APP
The effect of APP on Tip60 localization was also examined by confocal immunofluorescence microscopy. Neuroblastoma SH-SY5Y cells were transfected with HA-Tip60 alone (Fig. 6, A–C) or with Fe65 and APP (D–F). Tip60 was labeled with anti-HA antibody, and APP was labeled with antibody C8 (raised against the APP C-terminal domain). The nucleus was visualized by staining with Hoechst dye. When Tip60 was expressed alone, it localized to the nucleus, as shown by colocalization with the nuclear Hoechst dye (Fig. 6C). Coexpression of Tip60 with Fe65 and APP led to a dramatic change in the localization of Tip60, with Tip60 now appearing outside of the nucleus (Fig. 6F). Moreover, there was substantial colocalization of Tip60 and APP on the cell surface (Fig. 6F). APP was excluded from the nucleus as expected (Fig. 6D). The presence of non-transfected cells that were not labeled indicated the specificity of Tip60 and APP labeling. These results are consistent with the formation of a Tip60-Fe65-APP complex at the membrane and confirm the results of subcellular fractionation.
FIGURE 6. Colocalization of APP and Tip60.
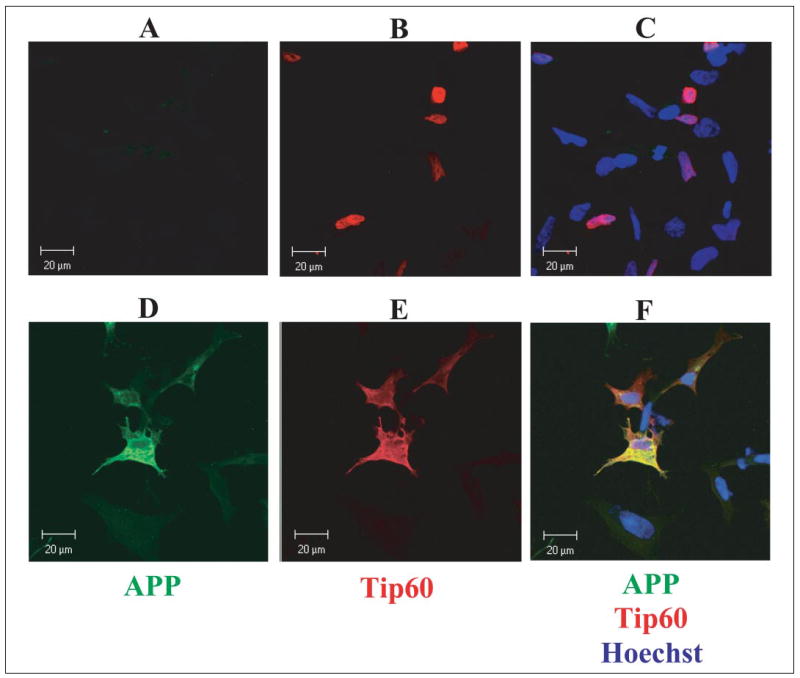
SH-SY5Y cells were cotransfected with Tip60 alone (A–C) or with Fe65 and APP (D–F) and then fixed and permeabilized at 24 h post-transfection. Tip60 was visualized with an antibody against the HA tag and a Cy3-conjugated secondary antibody (red), and APP was visualized with antibody C8 and a Cy2-conjugated secondary antibody (green). The nucleus was labeled with Hoechst dye.
Shuttling of Tip60 between the Nucleus and Cytoplasm
Our results suggest that APP recruits Tip60 to the membrane, which activates it through phosphorylation and complex formation with Fe65. We investigated whether Tip60 contains nuclear localization and export sequences that could mediate Tip60 shuttling between the nucleus and cytoplasm. The PSORTII program identified two potential nuclear localization sequences within Tip60. These were the short basic nuclear localization sequence starting at amino acid 184 (PGRKRKS) and the type 1 bipartite nuclear localization sequence beginning at amino acid 295 (RKGTISFFEIDGRKNKS). Both of these nuclear localization sequences are completely conserved in the mouse, rat, and human Tip60 proteins. We also identified a potential nuclear export sequence (LTTLPVLYL) that conforms to the classic LX2–3(L/I/V/F/M)X2–3LX(L/I) motif, which is completely conserved in the mouse, rat, and human Tip60 proteins (35). This raises the possibility that active transport of Tip60 out of the nucleus may be required for the interaction of Tip60 with APP, which may then be followed by nuclear translocation and transcriptional activation. To determine whether nuclear export is required for Tip60 activation, we used the CRM1 nuclear export inhibitor leptomycin B in the Gal4-Tip60 luciferase assay. Treatment with leptomycin B greatly suppressed the ability of APP to activate Tip60 (Fig. 7B). Thus, nuclear export of Tip60 or some other factor is required for APP to be able to activate transcription through Tip60. These results support a model in which Tip60 actively shuttles between the cytoplasm and nucleus, enabling its activation by APP and Fe65 at the membrane.
FIGURE 7. Shuttling of Tip60 between the nucleus and cytoplasm and APP signaling.
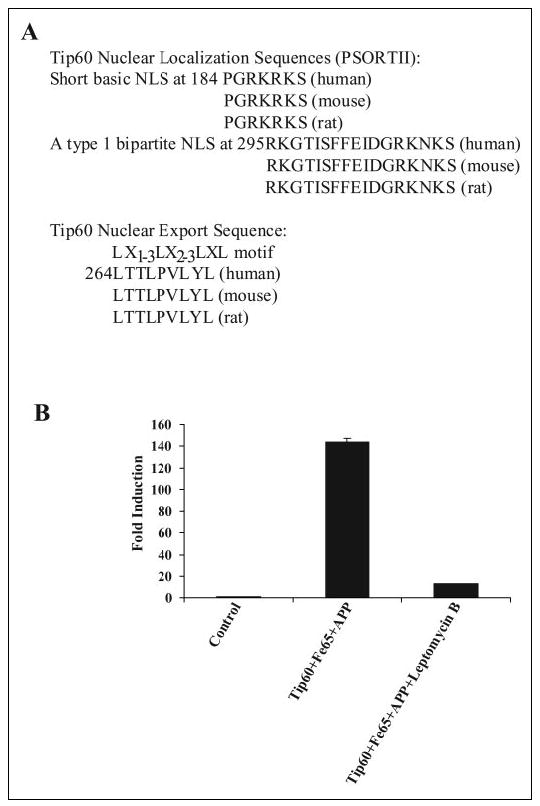
A, identification of two potential nuclear localization sequences (NLS) within Tip60 are conserved in the mouse, rat, and human orthologs. Additionally, a conserved potential nuclear export sequence is indicated. B, HEK-293 cells were cotransfected with pFR-luciferase, pRL-TK, Gal4-Tip60, Fe65, and APP. Cells were treated with the nuclear export inhibitor leptomycin B (20 nm), and luciferase assays were performed 24 after transfection.
DISCUSSION
These experiments indicate that APP can activate Tip60-mediated transcription in the absence of presenilin-mediated γ-secretase cleavage, contrary to current dogma. This contrasts with Notch signaling, in which γ-secretase cleavage is required to activate the transcription of target genes. These results were obtained in ES cells from PS-KO mice lacking detectable γ-secretase activity. Additionally, we have shown that, in other cell lines, APP retained the ability to activate Tip60 in the presence of γ-secretase inhibitors at concentrations that prevent production of Aβ and generation of the C50/AICD fragment. Our findings suggest an alternative model of APP signaling in which holo-APP recruits Tip60 to the membrane to induce Tip60 phosphorylation and complex formation with Fe65, leading to activation of signal transduction (Fig. 8). This phosphorylation event appears to be carried out by a CDK, as signaling could be blocked by mutating the known CDK sites in Tip60 or by the CDK inhibitor roscovitine. A key aspect of this model is that the signaling role of APP may lie in its ability to recruit proteins to the microdomain of the membrane, where particular kinases, such as CDK5, are active.
FIGURE 8. Model of APP-mediated activation of Tip60.

APP recruits Tip60 to the membrane through the adaptor protein Fe65. This facilitates the phosphorylation and stabilization of Tip60 by CDKs, resulting in Tip60 activation. This is followed by dissociation of the complex and nuclear translocation of Tip60 and Fe65, leading to the transcriptional activation of Tip60 target genes. PTB, phosphotyrosine-binding domain.
The model of APP signaling that we propose differs from that of the canonical Notch signaling pathway in that APP signals via activation of cytoplasmic proteins through post-translational modifications rather than by providing an intracellular domain to activate transcription. This difference is critical, as both we and another group (36) found that the C50/AICD fragment is unable to activate transcription, in contrast to the transcriptional activity of NICD. Moreover, our results indicate that overexpressed C50/AICD acts as a dominant-negative protein to inhibit APP-Tip60 signaling. However, γ-secretase cleavage appears to facilitate signaling in ES cells, presumably through the release of the Fe65-Tip60 complex from the membrane. One potential γ-secretase-independent mechanism for the dissociation of Fe65 and Tip60 from APP and the membrane could be phosphorylation of APP at threonine 668, which has been shown to decrease the interaction between Fe65 and APP (37). Interestingly, CDK5 has been shown to phosphorylate APP at this site (38), raising the possibility that CDK5 could play the dual role of activating Tip60 and releasing the complex from the membrane.
One difference between Notch and APP processing is that APP undergoes constitutive ectodomain shedding and γ-secretase cleavage, whereas Notch undergoes ectodomain shedding only following ligand binding. Thus, it is difficult to understand how APP signaling could be regulated if it occurs constitutively. Our findings indicate that one mechanism of regulation of this pathway is through the activity of CDKs. This is particularly intriguing, as altered activity of CDK5 has already been implicated in Alzheimer disease (39, 40). Furthermore, the CDK5 activator p35 is membrane-associated, suggesting that CDK5 could specifically phosphorylate Tip60 when it is recruited to the membrane by APP.
A number of potential target genes of C50/AICD have been reported recently. One intriguing potential target is the Aβ-degrading enzyme neprilysin, which was reported to be responsive to C50/AICD in presenilin-deficient cells (33). We did not observe reproducible regulation of neprilysin expression by PS1 or C50. Moreover, we observed considerable variation in neprilysin levels between ES cell lines that did not strictly correlate with presenilin genotype. We also attempted to verify two other proposed target genes of Tip60, KAI1 and manganese-super-oxide dismutase (27, 41). In our hands, reporter constructs containing the promoters of these two genes were not responsive to Tip60, Fe65, APP, or various combinations of these constructs. Another recent study suggested that a fusion of the protein citrine to C50/AICD increases the expression of a number of genes, including APP, BACE, Tip60, glycogen synthase kinase-3β, and KAI1 (42). We have been unable to confirm these results by transfection of HEK-293 cells with C50 or C50-Myc, which could reflect a different activity of the reported citrine fusion protein. Overall, we have not been able to confirm any of the proposed C50/AICD target genes that we are aware of at this time. Further work needs to be done to delineate the APP signaling pathway with primary importance being the identification of endogenous target genes that are affected by APP. Progress in this area may have been hindered thus far by the focus on AICD as the transcriptionally active form.
The relevance of APP signaling to Alzheimer disease is unclear. However, it has been shown that many pathogenic mutations in presenilins actually decrease Notch signaling (4, 43–45). Moreover, presenilin-deficient mice exhibit a neurodegenerative phenotype (46). Additionally, a recent study showed that familial Alzheimer disease mutations in APP can inhibit signaling through the APP-Gal4-VP16 fusion (47). Thus, alterations in APP signaling could potentially play a role in the pathogenesis of Alzheimer disease.
Overall, our data provide evidence for APP signaling by a novel mechanism that does not require γ-secretase cleavage by the presenilins and that involves recruitment of signaling factors to the membrane to facilitate phosphorylation and complex formation. This mechanism would allow signaling to be regulated in multiple ways through the activity of CDKs, shuttling of Tip60 between the nucleus and cytoplasm, or possibly through γ-secretase activity, which may modulate activation even though it is not required.
Acknowledgments
We thank Shyan-Yuan Kao for providing the AICD plasmids, Changiz Geula and Menglan Yuan for assistance, Matthew Salanga for help with the confocal microscopy, and Tao Lu and Frank Lee for helpful discussion.
Footnotes
This work was supported by National Institutes of Health Grants AG17974 and NS030352 (to B. A. Y.) and a fellowship from National Institutes of Health Training Grant AG0022 (to M. R. H.).
The abbreviations used are: APP, amyloid precursor protein; BACE, beta-site APP-cleaving enzyme; APPsw, APP containing the Swedish mutation; APPwt, wild-type APP; AICD, APP intracellular domain; Aβ, amyloid β-peptide; HEK-293, human embryonic kidney 293; HA, hemagglutinin; NICD, Notch intracellular domain; ES, embryonic stem; PS-KO, presenilin-1/2 knockout; PS1, presenilin-1; CDK, cyclin-dependent kinase; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine; RIPA-DOC, deoxychelate-containing radioimmune precipitation assay buffer.
References
- 1.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 2.Lichtenthaler SF, Haass C. J Clin Investig. 2004;113:1384–1387. doi: 10.1172/JCI21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selkoe D, Kopan R. Annu Rev Neurosci. 2003;26:565–597. doi: 10.1146/annurev.neuro.26.041002.131334. [DOI] [PubMed] [Google Scholar]
- 4.Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA. Proc Natl Acad Sci U S A. 1999;96:6959–6963. doi: 10.1073/pnas.96.12.6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, Yankner BA. Nat Cell Biol. 2000;2:463–465. doi: 10.1038/35017108. [DOI] [PubMed] [Google Scholar]
- 7.Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, De Strooper B. Nat Cell Biol. 2000;2:461–462. doi: 10.1038/35017105. [DOI] [PubMed] [Google Scholar]
- 8.Tanzi RE, Bertram L. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Sisodia SS, St George-Hyslop PH. Nat Rev Neurosci. 2002;3:281–290. doi: 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- 10.Yankner BA. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 11.Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Nat Med. 1998;4:827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- 12.Breen KC, Bruce M, Anderton BH. J Neurosci Res. 1991;28:90–100. doi: 10.1002/jnr.490280109. [DOI] [PubMed] [Google Scholar]
- 13.Chen M, Yankner BA. Neurosci Lett. 1991;125:223–226. doi: 10.1016/0304-3940(91)90034-q. [DOI] [PubMed] [Google Scholar]
- 14.Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldsteain LS. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 15.LeBlanc AC, Kovacs DM, Chen HY, Villare F, Tykocinski M, Autilio-Gambetti L, Gambetti P. J Neurosci Res. 1992;31:635–645. doi: 10.1002/jnr.490310407. [DOI] [PubMed] [Google Scholar]
- 16.Jin LW, Ninomiya H, Roch JM, Schubert D, Masliah E, Otero DA, Saitoh T. J Neurosci. 1994;14:5461–5470. doi: 10.1523/JNEUROSCI.14-09-05461.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qiu WQ, Ferreira A, Miller C, Koo EH, Selkoe DJ. J Neurosci. 1995;15:2157–2167. doi: 10.1523/JNEUROSCI.15-03-02157.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao X, Sudhof TC. Science. 2001;293:115–201. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 19.Ebinu JO, Yankner BA. Neuron. 2002;34:499–502. doi: 10.1016/s0896-6273(02)00704-3. [DOI] [PubMed] [Google Scholar]
- 20.Koo E, Kopan R. Nat Med. 2004;10:S26–S33. doi: 10.1038/nm1065. [DOI] [PubMed] [Google Scholar]
- 21.Fiore F, Zambrano N, Minopoli G, Donini V, Duilio A, Russo T. J Biol Chem. 1995;270:30853–30856. doi: 10.1074/jbc.270.52.30853. [DOI] [PubMed] [Google Scholar]
- 22.Zambrano N, Bimonte M, Arbucci S, Gianni D, Russo T, Bazzicalupo P. J Cell Sci. 2002;115:1411–1422. doi: 10.1242/jcs.115.7.1411. [DOI] [PubMed] [Google Scholar]
- 23.Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ. J Biol Chem. 2001;276:40288–40292. doi: 10.1074/jbc.C100447200. [DOI] [PubMed] [Google Scholar]
- 24.Kinoshita A, Whelan CM, Smith CJ, Berezovska O, Hyman BT. J Neurochem. 2002;82:839–847. doi: 10.1046/j.1471-4159.2002.01016.x. [DOI] [PubMed] [Google Scholar]
- 25.Kinoshita A, Whelan CM, Berezovska O, Hyman BT. J Biol Chem. 2002;277:28530–28536. doi: 10.1074/jbc.M203372200. [DOI] [PubMed] [Google Scholar]
- 26.Gaughan L, Brady ME, Cook S, Neal DE, Robson CN. J Biol Chem. 2001;276:46841–46848. doi: 10.1074/jbc.M103710200. [DOI] [PubMed] [Google Scholar]
- 27.Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG. Cell. 2002;110:55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- 28.Ikura T, Ogrzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. Cell. 2000;102:463–473. doi: 10.1016/s0092-8674(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 29.McAllister D, Merlo X, Lough J. Gene (Amst) 2002;289:169–176. doi: 10.1016/s0378-1119(02)00546-2. [DOI] [PubMed] [Google Scholar]
- 30.Sabo SL, Lanier LM, Ikin AF, Khorkova O, Sahasrabudhe S, Greengard P, Buxbaum JD. J Biol Chem. 1999;274:7952–7957. doi: 10.1074/jbc.274.12.7952. [DOI] [PubMed] [Google Scholar]
- 31.Kim HS, Kim EM, Lee JP, Park CH, Kim S, Seo JH, Chang KA, Yu E, Jeong SJ, Chong YH, Suh YH. FASEB J. 2003;17:1951–1953. doi: 10.1096/fj.03-0106fje. [DOI] [PubMed] [Google Scholar]
- 32.Zambrano N, Gianni D, Bruni P, Passaro F, Telese F, Russo T. J Biol Chem. 2004;279:16161–16169. doi: 10.1074/jbc.M311027200. [DOI] [PubMed] [Google Scholar]
- 33.Pardossi-Piquard R, Petit A, Kawarai T, Sunyach C, Alves da Costa C, Vincent B, Ring S, D’Adamio L, Shen J, Muller U, St George-Hyslop P, Checler F. Neuron. 2005;46:541–554. doi: 10.1016/j.neuron.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 34.Lemercier C, Legube G, Caron C, Louwagie M, Garin J, Trouche D, Khochbin S. J Biol Chem. 2003;278:4713–4718. doi: 10.1074/jbc.M211811200. [DOI] [PubMed] [Google Scholar]
- 35.la Cour T, Gupta R, Rapack K, Skriver K, Poulsen FM, Brunak S. Nucleic Acids Res. 2003;31:393–396. doi: 10.1093/nar/gkg101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao X, Sudhof TC. J Biol Chem. 2004;279:24601–24611. doi: 10.1074/jbc.M402248200. [DOI] [PubMed] [Google Scholar]
- 37.Ando K, Iijima K, Elliott JI, Kirino Y, Suzuki T. J Biol Chem. 2001;276:40353–40361. doi: 10.1074/jbc.M104059200. [DOI] [PubMed] [Google Scholar]
- 38.Iijima K, Ando K, Takeda S, Satoh Y, Seki T, Itohara S, Greengard P, Kirino Y, Nairn AC, Suzuki T. J Neurochem. 2000;75:1085–1091. doi: 10.1046/j.1471-4159.2000.0751085.x. [DOI] [PubMed] [Google Scholar]
- 39.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 40.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 41.Creaven M, Hans F, Mutskov V, Col E, Caron C, Dimitrov S, Khochbin S. Biochemistry. 1999;38:8826–8830. doi: 10.1021/bi9907274. [DOI] [PubMed] [Google Scholar]
- 42.von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, Konietzko U. J Cell Sci. 2004;117:4435–4448. doi: 10.1242/jcs.01323. [DOI] [PubMed] [Google Scholar]
- 43.Moehlmann E, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, Kaether C, Zheng H, Ghetti B, Haass C, Steiner H. Proc Natl Acad Sci U S A. 2002;99:8025–8030. doi: 10.1073/pnas.112686799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen F, Gu YJ, Hasegawa H, Ruan X, Arawaka S, Fraser P, Westaway D, Mount H, St George-Hyslop P. J Biol Chem. 2002;277:36521–36526. doi: 10.1074/jbc.M205093200. [DOI] [PubMed] [Google Scholar]
- 45.Schroeter EH, Ilagan MX, Brunkan AL, Hecimovic S, Li Y, Xu M, Lewis HD, Saxena MT, De Strooper B, Coonrod A, Tomita T, Iwatsubo T, Moore CL, Goate A, Wolfe MS, Shearman M, Kopan R. Proc Natl Acad Sci U S A. 2003;100:13075–13080. doi: 10.1073/pnas.1735338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saura CA, Choi SY, Beglopoulos V, Malkani S, Zhang D, Shankaranarayana Rao BS, Chattarji S, Kelleher RJ, III, Kandel ER, Duff K, Kirkwood A, Shen J. Neuron. 2004;42:23–36. doi: 10.1016/s0896-6273(04)00182-5. [DOI] [PubMed] [Google Scholar]
- 47.Wiley JC, Hudson M, Kanning KC, Schecterson LC, Bothwell M. J Neurochem. 2005;94:1189–1201. doi: 10.1111/j.1471-4159.2005.03266.x. [DOI] [PubMed] [Google Scholar]
- 48.Passer B, Pellegrini L, Russo C, Siegal RM, Lenardo MJ, Schettini G, Bachmann M, Tabaton M, D’Adamio L. J Alzheimers Dis. 2000;2:289–301. doi: 10.3233/jad-2000-23-408. [DOI] [PubMed] [Google Scholar]