

Abstract
The human P-glycoprotein (MDR1/P-gp) is an ATP-binding cassette (ABC) transporter involved in cellular response to chemical stress and failures of anticancer chemotherapy. In the absence of a high-resolution structure for P-gp, we were interested in the closest P-gp homolog for which a crystal structure is available: the bacterial ABC transporter MsbA. Here we present the molecular dynamics simulations performed on the transmembrane domain of the open-state MsbA in a bilayer composed of palmitoyl oleoyl phosphatidylethanolamine lipids. The system studied contained more than 90,000 atoms and was simulated for 50 ns. This simulation shows that the open-state structure of MsbA can be stable in a membrane environment and provides invaluable insights into the structural relationships between the protein and its surrounding lipids. This study reveals the formation of a semipore-like structure stabilized by two key phospholipids which interact with the hinge region of the protein during the entire simulation. Multiple sequence alignments of ABC transporters reveal that one of the residues involved in the interaction with these two phospholipids are under a strong selection pressure specifically applied on the bacterial homologs of MsbA. Hence, comparison of molecular dynamics simulation and phylogenetic data appears as a powerful approach to investigate the functional relevance of molecular events occurring during simulations.
INTRODUCTION
The family of ABC (ATP-binding cassette) proteins is composed of an impressive, and still growing, number of proteins of various functions and is expressed at every level of the living kingdom, from bacteria and yeasts to plants and mammals (1). Most of them are membrane proteins, most generally devoted to ATP-dependent transports of substrates of highly various natures, and are thus often referred to as “traffic ATPases”. In bacteria, ABC transporters are involved in either cell import of different metabolites or the efflux of antibiotics and exogenous molecules. In mammals, they are always expelling substrates out of cells, and in man they are involved in different pathophysiological disorders, such as Tangier disease (ABC A1) or cystic fibrosis (C7), and pharmacological processes, such as multidrug resistance (MDR) or absorption-distribution-elimination (ABC B1, C1, or G2) (2). The substrates handled by the human ABC transporters are, as far as they are known, often lipids (ABC A1, A4, B1, B4, D1, G1, G5, G8) (3) or drugs (B1, C1–6, G2) (4–6), and sometimes (B1, C1, G2) both of them (7).
Despite their biomedical importance, very limited insights have been provided into the molecular structures of these membrane proteins (8). Currently, high resolution structures have been obtained for a few isolated nucleotidic binding domains. The entire protein structure has only been obtained for the bacterial transporters BtuCD (9) and (MsbA)2 (10). BtuCD is an importer of vitamin B12, a large and hydrophilic molecule, and is rather atypical for an ABC transporter with its 20 transmembrane helices. In contrast, MsbA presents itself as a homodimer with a moderate sequence identity with the members of the B subfamily of the human ABC proteins, and it mediates the outward transport of the toxic lipid A but also of various lipids (11) and even drugs (12,13). This functional homology makes it an attractive bacterial model for the multidrug transporter P-glycoprotein (P-gp, ABC B1), especially considering that P-gp is suspected to work as a flippase for drugs (14), and this appears indeed the case as it can handle various lipids (15).
The interest in structural data about MsbA is reinforced by the report of crystal structures of three different isoforms from Escherichia coli (10), Vibrio cholera (16), and Salmonella typhimurium (17), which are proposed to be different conformations of the enzyme during its catalytic cycle. However, the E. coli MsbA structure has raised several concerns regarding the insertion of its transmembrane domain in the bilayer (18,19) since the bending of the intramembrane helices with respect to the membrane plane would result in an atypical geometry for the “open” conformation of the dimer. The investigation of the organization of the lipid molecules around this unique protein shape is thus of great interest. In particular, it is desirable to address the dynamic stability of a phospholipid bilayer in its vicinity, which conditions the functional relevance of this protein structure. If such an assembly is stable, the question to address is how phospholipid molecules can accommodate the bending of the transmembrane domain of the protein. In this context, we decided to tackle the question of the membrane perturbation induced at the molecular level by the presence of MsbA in a model lipid bilayer in silico. This study was designed in relation to the lipid translocase function exhibited by MsbA, with the perspective of gaining some insights into its mechanisms of functioning.
Molecular dynamics (MD) simulations (20) offer a powerful way of studying biological macromolecules under approximately physiological conditions. These simulations have been extensively applied to the study of biological membranes (21–23) as well as various membrane-bound proteins (24–28). Recent advances in computer power have allowed the simulation of mixed micelles modeling human bile, with simulation times extending to 150 ns (29). Advances in simulation procedures and computer power mean than timescales exceeding 10 ns and large systems of ∼100,000 atoms are becoming computationally tractable.
In this study, we report extended MD simulations of E. coli MsbA. A previous simulation of this protein has been published (19). However, whereas monomer simulations revealed themselves stable, the MsbA molecule was unstable in its dimeric form and showed large structural drifts over the course of the simulation. Moreover a membrane-mimetic octane slab was used. The interactions between membrane proteins and their lipid environment play important roles in the stability and function of proteins (30). Although an octane slab can represent a correct membrane-mimetic environment for cylindrical proteins, it might not be suited when lipid-protein interactions are critical for protein stability or when the protein does not possess a simple cylindrical shape. In this context, we present a 50-ns simulation of MsbA embedded in a palmitoyl oleoyl phosphatidylethanolamine (POPE) bilayer. For computational tractability reasons and since our main concern is the organization of lipids around the protein, only the transmembrane domain of the protein has been studied. As a result, it is shown that i), protein secondary/tertiary structure is reasonably stable; ii), polar headgroups of close lipids in the cytosolic leaflet partially invaginate, leading to a semipore formation; iii), two individualized lipid molecules can interact with the edge of the protein cavity with a clear specificity with respect to bulk phospholipids; and iv), the amino acid involved in this specificity has been strongly and specifically conserved during the evolution of bacterial MsbA-like ABC transporters, suggesting an important functional role for the observed structure.
METHODS
General strategy and characteristics of the simulation
The simulation of the transmembrane domain of E. coli MsbA inserted in a POPE bilayer is presented in this study (Table 1). Since the main components of the natural membrane in which MsbA is found are phosphatidylethanolamine lipids (31), POPE lipids represent a good approximation for the membrane environment. The MsbA cavity was filled with solvent and the protein was constrained during 16 ns; constraints were then removed and the system was further examined for 34 ns, therefore leading to a total simulation time of 50 ns. System preparation involved three steps: i), protein reconstruction, ii), phospholipid bilayer construction, and iii) protein and bilayer assembly.
TABLE 1.
Simulation summary
| Duration (ns) | Lipids | Waters and ions | Atoms | Restraints |
|---|---|---|---|---|
| 50 | 591 POPE | 18,601 waters and 14 Na+, 34 Cl− | 91,271 | Protein backbone: first 16 ns |
All MD simulations were conducted using the GROMACS v3.2 MD simulation package (32) with parameter set ffgmx. Production simulations were performed in the isobaric-isothermal ensemble (NPT). The temperature of the system was coupled using the Berendsen algorithm (33) at 300 K with coupling constant τT = 0.1 ps. Protein, phospholipids, solvent, and ions were coupled separately. The pressure was coupled using the Berendsen algorithm at 1 bar with coupling constant τP = 2 ps and the compressibility was set to 4.5 × 105 bar−1 in all simulation box directions. Electrostatic interactions were calculated using particle mesh Ewald (34) with a real space cutoff of 0.9 nm. Cutoff for van der Waals interactions was set at 0.9 nm. Time step for integration was 2 fs; coordinates and velocities were saved every 0.5 ps. The LINCS algorithm was used to restrain bond lengths (35).
Protein reconstruction
The crystallographic structure of the MsbA protein from E. coli (Protein Data Bank (PDB) code: 1JSQ) was used as a starting structure for our simulations. The structure only contains an incomplete Cα trace; no side-chain or backbone atoms other than Cα are present. Several residues are missing (residues 1–9 and 208–237 for the first monomer). The transmembrane domain of E. coli MsbA, i.e., residues 1–104, 132–200, and 245–309, was reconstructed (Fig. 1). The nine N-terminal residue Cα atoms were reconstructed to form an α-helix with residues 10–20. The other missing residues (residues 208–237) were not part of the transmembrane domain and were therefore not reconstructed. Missing backbone and side-chain atoms were predicted using MAXSPROUT (36) and SCWRL (37). Residues lying inside the bilayer hydrophobic core were considered neutral. Ionization states of the protein's other side chains were the same as the standard at pH 7, except for buried residues. Buried residues were determined using WHATIF (38) to calculate accessible solvent area and with visual examination.
FIGURE 1.

(A) General presentation of the open-state MsbA structure reconstruction, only considering its transmembrane domain. The ribbons are colored from blue to green from the N- to C-terminus of the M1 monomer and from green to red from the N- to C-terminus of the M2 monomer. (B) Schematic key phospholipids organization on the intracellular side of the protein.
Phospholipid bilayer construction
The bilayer contained 662 POPE lipids; it was generated by extending a preequilibrated bilayer containing 340 lipids (26). The extension was achieved by replicating the 340 lipids box twice in both directions of the membrane plane, therefore leading to a 1360 POPE lipids system. Box dimensions were then reduced by 70% in the plane of the bilayer, yielding a system of 669 lipids. Seven lipids from the top layer were then randomly selected and removed to achieve an equal number of lipids in both layers. The system was then hydrated using a preequilibrated box of simple point charge-extended (SPC/E) water (39). The system was subjected to 1000 steps of steepest descent energy minimization. The coordinates of the lipids were then frozen, allowing the solvent molecules to relax and adapt their coordinates with the membrane during a 500-ps simulation in the canonical ensemble (NVT). The system was then equilibrated in a series of 250-ps simulations in the NPT ensemble with semiisotropic pressure coupling. The compressibility in the plane of the bilayer was fixed to 4.25 × 10−5 bar−1 and was discretely increased from 1.0 × 10−30 bar−1 along the bilayer normal to reach 4.25 × 10−5 bar−1 after 2.25 ns of simulation. The final equilibration run consisted of a 2.5-ns simulation in the NPT ensemble with a compressibility of 4.25 × 10−5 bar−1 in all directions.
Protein and bilayer assembly
The protein was inserted in a preequilibrated POPE bilayer in the fashion outlined by Sansom et al. (40). The protein was oriented such that its principal axis of inertia was aligned along the bilayer normal (z axis) and its other axes of inertia made an angle of 45° with the box axes in the plane of the bilayer. Positioning along the z axis was such that the bands of tryptophan residue side chains were located in the lipid headgroup region. To accommodate the protein, the box dimension along the z-direction was increased to 7.75 nm, and the system was subsequently hydrated using a preequilibrated box of SPC/E water (39). The cavity of the protein was filled with water. Randomly positioned potassium and sodium ions replaced 48 water molecules to balance the charges on the nonneutral protein residues, equivalent to a concentration of 17 mM. The size of the simulation box was 13.33 × 13.21 × 7.75 nm. The resulting system was equilibrated following the procedure summarized in Table 2. After initial steepest descents energy minimization of the system, the protein and lipids coordinates were frozen to allow solvent molecules to relax their coordinates and optimize their interactions with the lipid-protein assembly in a 500-ps simulation in the NVT ensemble. The constraints on lipid molecules were then removed to optimize lipid-protein interactions in a 500-ps simulation in the NVT ensemble. The backbone atoms of the resulting system were then frozen, allowing side-chain atoms to relax their positions and optimize their interactions with lipid and solvent molecules in a 500-ps simulation in the NVT ensemble. The final equilibration step consisted of a 500-ps simulation in the NPT ensemble where backbone atoms of the protein were restrained harmonically using a force constant of 1000 kJ mol−1 nm−2. The resulting system was used in a 50-ns simulation in the NPT ensemble. In the first 16 ns of this simulation, the protein backbone was restrained harmonically using a force constant of 1000 kJ mol−1 nm−2; this restraint was removed for the remainder of the simulation.
TABLE 2.
Equilibration procedure
| Step | Length | Type* | Frozen groups | Restrained groups |
|---|---|---|---|---|
| 1 | 1000 steps | SD | Protein, lipids | – |
| 2 | 500 ps | NVT | Protein, lipids | – |
| 3 | 1000 steps | SD | Protein | – |
| 4 | 500 ps | NVT | Protein | – |
| 5 | 1000 steps | SD | Protein backbone | – |
| 6 | 500 ps | NVT | Protein backbone | – |
| 7 | 500 ps | NPT | – | Protein backbone |
SD refers to steepest descents energy minimization. NVT refers to simulations in the NVT ensemble at 300 K using Berendsen algorithm and a coupling constant τT = 0.1 ps. NPT refers to simulations in the NPT ensemble at 300 K, and 1 bar using a coupling constant τP = 2 ps and a compressibility of 4.5 × 105 bar−1 in all box directions.
Simulations were analyzed using GROMACS routines and locally written scripts. Secondary structure was analyzed using DSSP. All molecular graphics representations were generated using Pymol.
Conservation analysis
Given the sequence of the query protein, close homologous sequences were retrieved from the NR database using Psi-Blast (41). Divergent sequences leading to gaps or insertion in the transmembrane helices were removed from the resulting multiple sequence alignment (MSA) to be consistent with the structure of the protein. After removing any sequence with more than 90% sequence identity to any other as detected using Psi-Blast, a set of 96 sequences was extracted to form the MsbA-like data set.
A phylogenetic tree has been constructed (considering a neighbor joining method using a BLOSUM62 matrix) with the JALVIEW software (42). The 96 sequences were then divided into two major subgroups according to the phylogenetic tree: A1, including E. coli MsbA, which contains the closest homologs of this protein (49 sequences with an average identity of 42%), and A2, which contains remote homologs of E. coli MsbA (47 sequences with an average identity of 20%). Two MSAs were thus considered, MSA1, restricted to Msba functional homologs, and MSA2, gathering the remaining sequences of ABC transporters retrieved from the Blast procedure. For every position of MSA1 and MSA2, an evolutionary conservation score was computed. Conservation scores for each amino acid position in the resulting MSAs were computed using the rate4site program (43). The Bayesian method was applied for the calculation of the conservation scores using the Jones-Taylor-Thornton amino acid substitution model (44). The conservation scores computed by rate4site are a relative measure of evolutionary conservation at each position in the MSA: the lowest score represents the most conserved position in the MSA. Each score S has been rescaled between 0 and 99 using
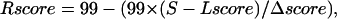 |
where Rscore is the rescaled score, Lscore the lowest score, and Δscore the amplitude of all the scores. The rescaled conservation grades were then mapped onto the protein by replacing the B factors in the PDB file. The most conserved residues correspond to the highest rescaled scores.
The second goal of the phylogenetic analysis was to identify in the MSA the positions subjected to specific evolutionary pressure in the subfamily of MsbA-like bacterial lipid export ABC transporters. To obtain positions which are more specific to the A1 subgroup, conservation scores for each amino acid position in MSA1 were computed using rate4site and compared with those considering MSA2. The rate4site program assigns a confidence interval to each of the inferred evolutionary conservation score. For site specific comparison, nonoverlapping 95% confidence intervals (45) were used to indicate significant rate variations between both the MSA1 and the MSA2. One position was defined as specifically conserved in close homologs of MsbA-like proteins if the estimated confidence intervals between MSA1 and MSA2 did not overlap (the rate variation is defined as positive if the site is more conserved in the truncated MSA1). Only sites with a positive rate variation and a negative upper confidence limit considering MSA1 are defined as specifically conserved in the set of close homologous sequences (negative score indicates strong conservation).
RESULTS
To investigate the MsbA dimer stability as well as the lipid organization around such an assembly, a 50-ns simulation has been performed. Since lipid organization around the MsbA dimer cannot be predicted a priori, the simulation was started from an out of equilibrium structure in which the lipids close to the protein cavity entrance do not possess the equilibrium structure. The simulation has therefore been split up into two parts. In the first part (0–16 ns), the protein backbone was harmonically restrained, allowing lipids to organize themselves around the MsbA dimer and avoiding protein denaturation early in the simulation owing to inappropriate environment. In the second part of the simulation, the restraints were removed. The entire simulation provides information about the lipid-protein interactions, and the second part reports on the stability of these interactions and of the protein. Finally, conservation analysis enables us to examine those results in light of phylogenetic data.
A second ∼20-ns simulation was performed in which the MsbA cavity was filled with POPE lipids (data not shown). The lipids quickly dived inside the cavity (∼1 ns) and remained trapped for the remaining of the simulation. Calculation of the electrostatic potential of the MsbA inner cavity (data not presented) revealed its polar nature. It hence leads us to the conclusion that the cavity was more likely to interact with solvent in its steady state. The arrangement obtained might be an intermediate state or a local energy minimum. In both cases, the simulation time was not sufficient to observe a significant rearrangement of the system. The remainder of this work therefore only presents results referring to simulations where the protein cavity was filled with solvent.
Lipid organization around the protein
Before restraints on the lipid phosphorus along the z axis are removed, the bilayer is flat and the MsbA hinge region is in contact with both the cytosol region and the hydrocarbon chains of lipids in the vicinity. The hinge region corresponds to the interface of the two monomers that is located in the hydrophobic region of the membrane and in contact with the solvent. This region is defined by helix 2 of the first monomer and helix 5 of the second on one side and reciprocally on the other side (Fig. 1). This is a nonphysical arrangement since it involves contact between hydrophobic chains and polar residues of the protein as well as with the solvent (Fig. 2 A). During the last 1.5 ns of equilibration, where restraints on the POPE lipid phosphorus atoms are removed, a migration of the phospholipids headgroups toward the hinge region of MsbA can be observed (Fig. 2 B). This arrangement of POPE lipids around the protein early in the simulation before proper production is started balances out unfavorable interactions between protein, lipids, and solvent. The resulting organization, which can be described as a semipore-like structure, ensures that no hydrophobic groups are in contact with hydrophilic groups. The curvature of the cytosolic monolayer is indeed modified in the proximity of the protein hinge region. However, the membrane perturbation is only very local, not concerning the exoplasmic leaflet and not propagated to long distances: outside a perimeter of 10 Å from the hinge region of MsbA, the bilayer is no longer perturbed and lipid headgroups regain their normal heights similar to that of lipids in the bulk of the membrane (Fig. 3).
FIGURE 2.

Lipid migration in the vicinity of the MsbA hinge region. Structure of the assembly at the beginning of the equilibration (A) (t = −1.5 ns) and at the end of the equilibration (B) (t = 0 ns). Protein is represented in blue, phospholipids chains are represented in gray lines, and headgroup atoms in spheres. For purpose of clarity, solvent and counterions are omitted. The extracellular region is located above the protein and the intracellular region below. The black frame highlights the very local perturbation of the cytosolic leaflet surrounding the protein.
FIGURE 3.

Average height of the phospholipid headgroup atoms in the intracellular layer over the 0–16-ns time range, in nanometers. Headgroup atoms considered in the calculations included those from the phosphatidyl ethanolamine group as well as those from the glycerol part. The grid size was 2.0 Å. The color scale is arbitrary.
Lipid-protein interaction
Two key phospholipids play an important role in the stability of the interface between the hinge region of MsbA and the membrane, POPE79 and POPE217. These phospholipids interact with the protein and remain in the hinge region during the integrality of the simulation. Quantifying lipid-protein interactions, as defined by atoms within 0.35 nm of each other, reveals that these phospholipids interact exclusively with helices 2 and 5 (see Fig. 1 B for a schematic representation). Indeed, interactions with these helices make up the integrality of the interactions with the protein. Both phospholipids have a similar number of interactions with helix 5, which stabilizes between 15 and 20 (Fig. 4). Most of these contacts are van der Waals contacts since helix 5 mostly contains hydrophobic and aromatic residues. On the other hand, POPE79 and POPE217 have a different behavior with respect to helix 2 (Fig. 4). Whereas POPE79 has a limited number of interactions with this helix (<2), POPE217 makes most of its interactions with the protein with helix 2 (∼20–30 interactions). This suggests different conformations for these two POPE molecules.
FIGURE 4.

(A) Overall number of lipid-protein interactions (cutoff 0.35 nm). (B) Number of interactions between lipid POPE79 and helix 2 of monomer 2 (black line) and helix 5 of monomer 1 (gray line). (C) Number of interactions between lipid POPE217 and helix 2 of monomer 1 (black line) and helix 5 of monomer 2 (gray line). (D) Overall number of interactions between protein and lipid POPE79 (black line) and lipid POPE217 (gray line).
The total number of protein-lipid interactions is steadily increasing during the unrestrained part of the simulation. There are ∼1300 interactions after 16 ns and ∼1500 interactions at the end of the simulation. Even though total simulation time reached 50 ns, the protein is still further stabilized by the membrane at the end of the study, and a final steady state is not completely reached.
As a likely consequence for the fact that their locations and conformations are directly dependent upon the initial conformation, and indeed upon the process used to generate the protein-membrane complex, these two phospholipids adopt two different conformations. Whereas lipid POPE79 has its two hydrophobic chains outside of the protein cavity, lipid POPE217 has one chain half inside the cavity (SN2) and the other outside (SN1).
The examination of the average minimum distance of these two phospholipids with residues making helices 2 and 5 reveals the importance of several residues (Fig. 5). In helix 2, arginine Arg-78 and threonine Thr-81 are particularly important for the protein-lipid interface structure (see Fig. 1 A). Both residues are on average <0.2 nm away from POPE217, whereas with POPE79 only residue Thr-81 is at close distance (0.25 nm on average), testifying to the presence of two distinct conformations. In helix 5, most interactions are van der Waals contacts with hydrophobic residues Ile-258, Ser-260, Ala-262, and Phe-265 for POPE79 and Ile-258, Ala-262, and Phe-265 for POPE217.
FIGURE 5.

Average minimum distance of characteristic lipids POPE79 and POPE217 with each residue of the helices making the interface between the two protein monomers (helices 2 and 5). Profiles are calculated in 500-ps windows and averaged over all windows. (A) Minimum distance between lipid POPE79 and residues of helix 2 of monomer 2, (B) minimum distance between lipid POPE79 and residues of helix 5 of monomer 1, (C) minimum distance between lipid POPE217 and residues of helix 2 of monomer 1, and (D) minimum distance between lipid POPE217 and residues of helix 5 of monomer 2.
The number of H-bonds (data not presented) also reveals two distinct behaviors. The total number of H-bonds stabilizes during the constrained part of the simulation and increases after constraints are removed to stabilize at around 80 H-bonds at the end of the simulation. During the unrestrained part of the simulation, local fluctuations of the protein enable new H-bonds to be created, further stabilizing the protein inside the membrane. The two individualized phospholipids have distinct behavior toward H-bonding. POPE217 interacts more with the protein than POPE79 and ∼4 and ∼1 H-bonds during the unrestrained part of the simulation, respectively. POPE79 has between one and two H-Bonds with helix 5 and occasionally one with helix 2. The other phospholipid, POPE217, has a greater number of H-bonds: up to four with helix 5 during the unrestrained part of the simulation. Interactions with helix 2 are more limited, between 1 and 2 H-bonds.
In agreement with these analyses, visual examination of these phospholipids (Fig. 6) shows the presence of one H-bond between POPE79 and threonine Thr-81. The other phospholipid has two H-bonds with arginine Arg-78 and one H-bond with threonine Thr-81.
FIGURE 6.

Snapshot of key protein-lipid interactions seen from the cavity. The extracellular side is located on the bottom and the intracellular side above. The bottom ribbon representation of the MsbA dimer indicates the orientation of the protein. The ribbons are colored from blue to green from the N- to C-terminus of the M1 monomer and from green to red from the N- to C-terminus of the M2 monomer. In both diagrams the protein is shown in “cartoon” format (in green). Selected side chains and lipids are shown in “sticks” format, with nitrogen and oxygen atoms colored blue and red, respectively. (A) Threonine (Thr-81) residue interacting with phospholipid POPE79. The threonine hydroxyl group is H-bonded with the POPE SN2 carbonyl group. (B) Threonine (Thr-81) and arginine (Arg-78) residues interacting with phospholipid POPE217. The threonine hydroxyl group is H-bonded with the POPE SN1 carbonyl group. The arginine amine group is H-bonded with the POPE SN2 carbonyl and ester group.
Lateral motion of lipids
In terms of short scale lateral diffusion, two classes of lipids can be distinguished: bound and free lipids (30). In our analysis, bound lipids were defined as those that had their phosphorus atom within 5 Å of the protein at the beginning of the simulation, whereas free lipids are defined as those not having their phosphorus atom within 5 Å of the protein. The selection was updated every 500 ps. To investigate the influence harmonic restraints might have on the lipid diffusion behavior, lateral diffusion coefficients have also been calculated for time range representative of the restrained and unrestrained part of the simulation, respectively, 1–15 ns and 20–50 ns. Characteristic lipids POPE79 and POPE217 have also been characterized. Values for short scale lateral diffusion coefficients are summarized in Table 3, where POPE44 is representative of a bulk lipid.
TABLE 3.
POPE lipids short scale lateral diffusion coefficients (× 10−5 cm2 s−1)
| Time range (ns) | D*§ < 5 Å | D†‡ > 5 Å | Overall‡ | POPE79§ | POPE217§ | POPE44§ |
|---|---|---|---|---|---|---|
| 0–50 | 0.0217 (±0.0080) | 0.0334 (±0.0051) | 0.0323 (±0.0048) | 0.0034 (±0.0019) | 0.0082 (±0.0090) | 0.0104 (±0.0098) |
| 0–15 | 0.0215 (±0.0086) | 0.0340 (±0.0052) | 0.0334 (±0.0048) | 0.0070 (±0.0030) | 0.0048 (±0.0044) | 0.0094 (±0.0051) |
| 20–50 | 0.0227 (±0.0075) | 0.0334 (±0.0050) | 0.0327 (±0.0047) | 0.0033 (±0.0020) | 0.0114 (±0.0107) | 0.0134 (±0.0125) |
Lipids for which a phosphorus atom is located within 5 Å of the protein at the beginning of the simulation, updated every 500 ps.
Lipids for which a phosphorus atom is located above 5 Å away from the protein at the beginning of the simulation, updated every 500 ps.
Diffusion coefficients calculated in 500-ps frames.
Diffusion coefficients calculated in 5-ns frames.
As frequently seen, free lipids have a larger diffusion coefficient than bound phospholipids but of the same order of magnitude. In contrast, POPE79 and POPE217 have smaller diffusion coefficients, as testified by the values obtained for a lipid located in the bulk, POPE44. The POPE79 diffusion coefficient is smaller than that of POPE217. This can be linked to the different conformations the two phospholipids are adopting, since POPE79 has its two hydrophobic chains outside of the protein cavity whereas POPE 217 has one chain inside the cavity and one chain outside. Lipids transported by MsbA are supposedly recruited inside the membrane before being flipped and expelled in the extracellular side membrane as suggested by Reyes and Chang (17) in the case of lipopolysaccharide (LPS). In our case, POPE217 might be in an intermediate state that would explain a significantly higher diffusion coefficient than POPE79. In light of these considerations, it is interesting to study the influence of restraints on these diffusion coefficients. Restraints have little effect on diffusion coefficients of the free, the bound, and the entire set of phospholipids. They have nonetheless a significant influence on the key phospholipids POPE79 and POPE217. Once restraints are removed, POPE217 diffusion is about two times larger, an observation consistent with the hypothesis of a transition state in which significant protein conformational change is required to allow the lipid to be transported. The POPE79 diffusion coefficient is, on the other hand, reduced by a factor of 2. Local protein movement observed in the unrestrained part of the simulation might allow this phospholipid to reach a stable state by enabling it to reach an energy minimum. These observations can be corroborated by the examinations of root mean square deviations (RMSDs) (see Fig. 8 B). Helix 5 of monomer 1, which is in contact with phospholipid POPE217, is prone to more fluctuations (final Cα RMSD 0.361 nm) than its counterpart of monomer 2 interacting with the other phospholipid (final Cα RMSD 0.316 nm). Helices 2 have similar RMSDs in both monomers.
FIGURE 8.

Time evolution of helices tilt with respect to the bilayer normal (A) and intrahelix Cα RMSDs (B) for monomer 1 (black line) and monomer 2 (gray line). Intrahelix RMSDs are calculated by fitting each helix on itself and measuring RMSDs relative to the fit over time.
Protein stability
Protein stability can be characterized by examining the RMSD of the protein structure as a function of time. RMSDs for the entire protein together with monomer 1 and 2 are presented in Fig. 7. The RMSD of the entire protein continuously increases during the restrained part of the simulation to reach 0.25 nm after 16 ns. After removal of the restraints, RMSD keeps on rising but stabilizes at 0.51 nm after 25 ns without any further significant drift. When plotting the contributions of the two monomers separately, it appears that the contribution from monomer 2 is larger than that of monomer 1, with final values of, respectively, 0.51 and 0.47 nm. These values, although large, are of the same order of magnitude as the one observed by Campbell et al. in their in silico study of MsbA (19). These numbers have to be related to the resolution of the crystal structure used (0.45 nm) and to the fact that it only contains a Cα trace. Furthermore, our study only considers the transmembrane domain of the protein: missing intracellular loops likely have a direct influence on the stability of the assembly.
FIGURE 7.

Time evolution of Cα RMSDs for protein (black line), monomer 1 (black dotted line), and monomer 2 (gray line). Intramonomer RMSDs are calculated by fitting each monomer on itself and measuring RMSDs relative to the fit over time.
The secondary structure reports on the protein stability over the course of the simulation (see Fig. 1 in Supplementary Data). Significant loss of helicity is observed for helix 3 in both monomers, helix 4 in monomer 1, and helix 5 in both monomers. Most of the loss of secondary structure happens on the extracellular side of the protein. This observation can be explained by the fact that intracellular loops joining helices 4 to 5 and helices 2 to 3 have been omitted for computational reasons. Consequently, amino acids located at the cut and in the vicinity of the membrane polar headgroups tend to interact with the latter, causing some helices to slide toward the intracellular side. Helices 3 and 4 are particularly exposed to the membrane headgroups, which could explain the loss of helicity observed on the extracellular side. The loss of helicity observed might be related to its interaction with the elbow helix. Interestingly, the elbow helix, which was model built, is conserved in the integrality of the simulation (especially in monomer 2). This feature was not observed in the study of Campbell et al. (19), which reveals the importance of the membrane choice for the protein stability.
Motions in the protein have been further characterized by examining tilt angles with respect to the membrane normal and the RMSDs of each helix relative to itself as a function of time (Fig. 8). Helices 2, 3, 4, and 6 show similar tilt angle fluctuations in both monomers with a final drift of ∼5°, a value reflecting standard oscillations that might be observed in MD simulations exceeding tens of nanoseconds. There is a significant difference in the final tilt angles of helix 5 in monomers 1 and 2. Whereas the helix in monomer 2, which is in contact with phospholipid POPE79 does not show any significant final drift, helix 5 of monomer 1 shows a final drift of nearly 10°. This helix is in contact with phospholipid POPE217, which undergoes significant conformational change as noted above. We might therefore think that the observed tilt angle drift is part of the mechanism allowing phospholipid POPE217 migration toward the protein cavity and not just standard fluctuation. Concerning helix 1, there is a significant difference between the two monomers. The tilt angle is prone to more fluctuations in the first monomer; the question of whether this observation is significant remains to be elucidated. Intrahelix RMSDs are similar for both monomers except for helices 4 and 5. For the latter, the drift is more important in the first monomer, a logical observation considering the fact that this helix interacts with phospholipid POPE217, which undergoes significant conformational change. Helices 1, 2, and 6 are quite stable considering the overall protein stability, with final drifts of the order of 0.3 nm or less. The remaining helices show larger structural fluctuations with final drifts of the order of 0.4 nm.
Dimer dynamic behavior
Time evolution of the two monomers principal axes of inertia has been investigated. In Fig. 9 we report the angles made by the two monomers with the bilayer normal (z axis), together with the angle between the two monomers' axes of inertia. Both monomers are relatively stable. Monomers are initially oriented with an angle of 35° with respect to the bilayer normal; after restraints are removed, these angles oscillate with an amplitude of <10° to reach final values of 37° and 29°, respectively, for monomers 1 and 2. Considering the length of the simulation, these drifts testify to the stability of the assembly. Observation of the angle between the two monomers' principal axes of inertia reveals a slight closing of the chamber. The angle has an initial value of 70°, which decreases during the simulation to reach a final value of 63°. Our protein model did not include the nucleotide binding domains, leaving helices of the intracellular domain free to interact with the membrane surface. These interactions affect the stability of the assembly and can partly explain the closing of the chamber. Indeed, the concerned amino acids tend to bend the helices to favor interactions with the membrane headgroups. These interactions do not affect the assembly stability on the timescale of this study (final drift of 7°) but might have more significant effects if longer timescales were considered.
FIGURE 9.

Time evolution of the principal axis of inertia of monomers 1 and 2. Angles of the principal axis of inertia of monomers 1 and 2 with the bilayer normal are reported together with the angle between the two monomers' principal axes of inertia.
The dimer stability has been further examined by investigating the time evolution of the number of H-bonds between the two monomers (Fig. 10). Since H-bonds between atoms can form and disappear quite quickly, the total number of H-bonds is subject to significant fluctuations. Hence, only the general trend of the curve is discussed here. The initial number of polar contacts is limited, with one to two H-bonds between the two monomers. This number continuously increases during the restrained part of the simulation (up to 16 ns) to reach a value of ∼6 H-bonds. During the unrestrained part of the simulation (16 ns and beyond), the number of H-bonds initially decreases to about four interactions and finally “stabilizes” at about six interactions from 34 ns to the end of the simulation. This plateau testifies to the stability of the dimer interface.
FIGURE 10.

Time evolution of the number of H-bonds between the two monomers. The cutoffs used to define H-bonds are 0.25 nm for the hydrogen-acceptor distance and 60° for the donor-hydrogen-acceptor angle.
The examination of polar contacts at different times reveals that most H-bonds are between extracellular domain 1 (ECD1) of one monomer and extracellular domain 3 (ECD3) of the other monomer, and reciprocally. Typical H-bonds are illustrated in Fig. 11. ECD1 of the first monomer, which is the loop joining helices 1 and 2, is drawn in blue; and ECD3 of monomer 2, the loop joining helices 5 and 6, is drawn in red. On this snapshot, amino acids Leu-52, Asp-53, Phe-56, Gly-57, Lys-58, and Arg-61 of ECD1 are H-bonded to amino acids Ser-274, Val-275, and Asp-276 of ECD3. Among notable interactions, the H-bond between the carboxyl group of Asp-276's side chain and the NH group of Arg-61's side chain as well as with the NH group of Lys-58's backbone can be highlighted. H-bonds between Asp-53, Ser-274, and Val-275's backbone CO groups and Val-275, Phe-56, and Gly-57's backbone NH groups, respectively, together with H-bonds between Leu-52's backbone CO group and the Ser-274 hydroxyl group are relatively stable and can be observed during most of the second part of the simulation. These observations reveal the existence of a dimer interface which is stabilized by the appearance of numerous H-bonds during the course of the simulation and noticeably interactions involving backbone atoms.
FIGURE 11.

Snapshot of polar contacts between the two monomers at t = 25 ns. The protein is seen from the extracellular side. The protein is shown in “cartoon” format; selected side chains are shown in “sticks” format with nitrogen and oxygen atom colored blue and red, respectively. Helices 1 and 2 of monomer 1 are colored in dark and light blue, respectively, and helices 5 and 6 of monomer 2 are colored in orange and red, respectively. Illustrated H-bonds are those present between extracellular domains 1 and 3.
Conservation analysis
We first analyzed the projection at the protein surface of the evolutionary conservation based on the MSA of 49 MsbA-like ABC transporters. Fig. 12 A shows that conserved positions (colored red) can be found in different regions of the protein. The regions with the highest concentration of conserved positions are located inside the dimer cavity at the level of the cytosolic lipid interface and at the hinge between both monomers (shown in Fig. 12 A as hinge M1 and hinge M2 for monomers 1 and 2, respectively). The residues located at the membrane interface interacting with the lipid headgroups also appear relatively conserved, probably to ensure a proper positioning of the protein in the bilayer. In contrast, the external surface of the protein pointing toward the hydrophobic lipid environment contains more variable positions. Indeed, as long as the hydrophobic character of the residues is kept at the lipid-exposed surface, the structural integrity and function of the protein is likely to be maintained. The structure of the MsbA analyzed in this simulation only contains the transmembrane domain. Hence conservation data shown here only concern this region of the MsbA protein. It is interesting to note that in the entire structure the region containing the ICD region is also conserved to a similar extent as that of the internal cavity. This conservation probably demonstrates the importance of these regions in transmitting a proper signal transduction between the domains involved in the hydrolysis of the ATP and the transmembrane domain involved in the physical transport of substrates.
FIGURE 12.

Conservation analysis calculated from the MSA of MsbA proteins and mapped on the surface of the dimer. The top ribbon representation of the MsbA dimer indicates the orientation of the corresponding surface representations shown below in panels A and B. The ribbons are colored from blue to green from the N- to C-terminus of the M1 monomer and from green to red from the N- to C-terminus of the M2 monomer. (A) The surface of monomer M2 is colored with respect to the conservation scores. Most to least conserved positions are colored from red to white, respectively, yellow corresponding to the average conservation score (left, middle, and right panels show the interior, the lipid-exposed, and the hinge regions of the dimer, respectively). (B) Positions that are specifically and significantly conserved in the MsbA close homologs family but not in the more general LPS ABC transporters family are colored in red at the surface of M2 monomer (left and right panels show the interior and the lipid-exposed regions, respectively).
To further investigate the evolutionary constraints existing in the MsbA protein family, we decided to discriminate between two types of evolutionary pressures: i), those involved in the general transport mechanism of compounds through the membrane likely to be common to the entire ABC transporter superfamily, and ii), the conservation events that specifically occurred in the MsbA close homologs for reasons such as the specificity of the substrate recognition. In that scope we further aligned 47 sequences of ABC transporters more distantly related to MsbA in the evolution. The clustering between the close and remote homologs was based on a rigorous phylogenetic tree analysis of the entire set of 96 sequences. We then analyzed among the conserved positions of MsbA homologs those positions significantly more conserved in the set of close homologs of MsbA than in the set of remote ones. This protocol corresponds to a differential conservation analysis based on the comparison of evolutionary rates and of their associated confidence intervals (see Methods). Interestingly, using a stringent confidence interval (probability above 95%) only two positions stand out, Arg-78 and Trp-165 (Fig. 12 B). Residue Arg-78 stands at the dimer hinge region, whereas residue Trp-165 is located in a loop pointing out toward the extracellular medium. Decreasing the confidence interval to 90%, three other positions are found to exhibit a differential conservation effect: Gln-134, Ala-142, and Ser-186 (see Figs. 2 and 3 in Supplementary Material). These residues are located in helices 3 and 4 close to the inner membrane interface. Although at this stage we cannot further interpret these evolutionary pressures, the conservation pattern suggests that these residues may also play a specific and important role in MsbA close homologs. Among the five positions pinpointed by the differential conservation protocol, Arg-78 remains the most conserved position in E. coli MsbA's close homologs. Arg-78 is precisely the residue found interacting with the two key lipids (POPE79 and POPE217 in our simulation) and stabilizing the semipore-like structure.
DISCUSSION
MsbA is a bacterial ABC transporter first recognized to export lipid A, a toxic metabolic intermediate involved in the biosynthesis of the outer membrane, and now reported to also handle various lipids, LPSs, and even cytotoxic drugs. As a lipid transporter, it is of interest to investigate the relationships between lipids and this protein, both from a general view of membrane biophysics and from the specific perspective of its molecular mechanism of functioning. As a multispecific active exporter of amphiphilic compounds, analyzing its molecular properties is highly desirable to gain some useful knowledge in the molecular enzymology of the mammalian multidrug transporter, P-gp, remarkable for its close structural and functional homologies, which would be valuable in various pharmacological applications. In this context, the evidencing by MD simulations of specific interaction of two phospholipid molecules at the hinge of MsbA dimer structure, in parallel with a very local invagination of the cytosolic leaflet of the membrane surrounding the protein as a consequence of its open-state conformation, can be considered a significant shedding of light on this model of ABC transporter. Moreover, these observations might be of great interest to suggest site-directed mutagenesis experiments.
Assessment of the model quality
MD is an adequate tool for analyzing molecular mechanisms determining protein-lipid interactions. However, the quality of the protein structure modeled is of prime importance for the reliability of the data generated. Actually, since we started from a rather moderate resolution crystal structure, we have chosen to take into account only the transmembrane domain of the protein, without considering the nucleotide binding domains and the other fragments clearly outside the membrane, because they would imply constraints that are too speculative when compared to their expected low repercussion on transmembrane domain dynamics. This has been validated a posteriori as shown by the reasonable stability which is obtained for the protein. In addition, this may be consistent with a report on the related protein P-gp according to which the transmembrane domain alone is sufficient for trafficking to cell surface and drug recognition (46). Missing intracellular domains make difficult a strong assumption of the model quality, and this point will be obviously further tested only after modeling the whole protein. Nevertheless, a PROCHECK (47) analysis performed on the initial structure revealed that all protein-health characteristics were within the acceptable range.
Structural stability of the protein
Even though intrahelical RMSDs denote important structural drifts for some transmembrane segments, the dimer structure is relatively stable. Previous studies performed using an octane slab (19) showed large structural drift of the transmembrane domains over just 1 ns. Our study shows a significant drift that stabilizes at 0.5 nm after 25 ns and remains constant until the end of the simulation. This highlights the critical role played by the nature of the membrane in the stability of the assembly. The structure used in this study was closely based upon the crystallographic MsbA Cα coordinates (PDB code, 1JSQ). The rather poor resolution of this structure partly explains the fluctuations observed in our simulation. The significant rearrangement of transmembrane helices that can be observed is not typical in membrane protein simulations. MsbA is a lipid transporter that supposedly undergoes significant transmembrane domain rearrangement during the transport cycle. As such, the transmembrane domains must consequently be very flexible, which could explain the structural drifts observed in this study. Anyway, this does not preclude the conclusions shown by the simulations about lipid dynamics in the protein environment.
Implication for lipid transport
The presented simulations show that the open-state MsbA structure can be inserted into a lipid membrane and that this structure may be physiologically viable. This (resting) conformation is in agreement with recent data obtained using electron paramagnetic resonance and chemical cross-linking techniques (48). Thanks to the dynamic vision they allow, the molecular simulations also give invaluable insights into the transport mechanism that might be adopted by MsbA to translocate lipids.
MsbA transports lipid A and also phospholipids and LPSs (7). Furthermore, it has been recently shown experimentally that MsbA is responsible for the export of lipid A by mediating its transbilayer translocation (i.e., it displays a “flip-floppase activity”) (49). From the crystal structure obtained for S. typhimurium MsbA, a transport mechanism has been suggested by Reyes and Chang (17) from the sequestration of the polar sugar headgroup of LPS. The first two steps involve i), initial binding to the elbow helix of the apo structure, and ii), lipid headgroup insertion in the chamber of the apo structure. The last two steps involve transporter conformational change related to ATP binding and hydrolysis involving lipid headgroup flip while lipid chains are dragged through the lipid bilayer. In our simulation, analysis of protein-lipid interactions reveals the occurrence of two distinct conformations for phospholipids POPE79 and POPE217. These two singularized phospholipid molecules are actually the result of statistical sampling yielded by MD simulation. These two conformations might be in agreement with the transport mechanism suggested by Reyes and Chang in the sense that they show that the alkyl chains of these lipid molecules can adopt different conformations when interacting with this protein environment. This observation is also in good agreement with the differential conservation analysis that pinpointed the residue Arg-78 as one of the residues highly conserved in close homologs of MsbA although more variable in more distantly related organisms. All along the simulation, this residue is found interacting with the heads of the phospholipids POPE79 and POPE217 and stabilizing the semipore structure. Although lipid A was not included in the simulation, the evolutionary pressure found at position 78 may well reflect the functional specificity of the MsbA for the binding of lipid A.
Otherwise, we observed local and partial invagination in the cytosolic leaflet of lipid headgroups in contact with the protein, this being a direct consequence of the optimal filling of the periproteic space taking into account geometric constraint induced by the open state. This very local membrane structure perturbation might be the preliminary step of the transport mechanism. This would eventually imply translocation of the polar headgroup, coupled with energy-giving ATP hydrolysis, within the chamber of the observed semipore-like assembly. Indeed, it can be assumed that the subsequent energy-requiring translocation step will happen on a far larger timescale and will be accompanied by a significant rearrangement of the protein.
In addition, if we consider, in agreement with Chang's suggestion (16), as a fair possibility that MsbA alternates during its enzymatic cycle between open and closed states, it appears that the most altered part of the protein during such a transconformation is the hinge defined by the dimeric association. The fact that the hinge between both monomers is one of the most conserved regions in the transmembrane domain also supports the functional importance of this region in the transport process. It is then relevant, and even remarkable, that two phospholipids have been individualized on the hinge regions of the protein, located in the contact region of helices 2 and 5. These phospholipids establish specific interactions with the protein, when compared to other phospholipids in the bulk or in the ring surrounding the protein. They adopt two distinct conformations, resulting from the initial setup of the simulation. Interestingly, their location is similar to the one suggested for initial LPS binding in the structure of S. typhimurim MsbA (17), proposed at the level of the hinge of the dimer, involving elbow and helix 1 but also helices 2 and 5 due to the size of the ligand. This constitutes a clear evocation of possible transport site, which would mainly involve six amino acids in each side of the hinge and could be a general characteristic of substrate transport by MsbA. Actually, this would be in full agreement with the fact that the drugs handled by MsbA are amphiphilic and likely come to interact with the protein via the membrane phase after partitioning into the lipids, as described for P-gp.
Connection with P-gp functioning
The mammal multidrug transporter P-gp is responsible for the MDR of some tumor cells against cytotoxic drugs used in anticancer chemotherapy, leading to treatment failures. P-gp presents enough sequence homology with MsbA to raise an interest in analyzing MD data to possibly shed light on its transport function. In particular, P-gp is now described as being rather a translocase, and more precisely a floppase, realizing the active transfer of its hydrophobic substrates, lipids, or drugs, from the cytosolic to the exoplasmic leaflet. As an ABC protein with a tandem structure, it looks like a possible MsbA dimer. Such a “reminiscence” of a dimeric structure is supported by data showing that the coexpression of the two halves of the protein is able to exhibit a drug-stimulated ATPase activity, a fair indication of a full functionality, whereas each separated half is only capable of a basal ATP hydrolysis (50). In the same line, it has been shown by molecular biology techniques that a good flexibility of the linker between the N- and C-terminus is required for P-gp activity (51). From a structural point of view, the low resolution image obtained by tridimensional reconstruction shows an inner chamber with a possible opening/communicating to the near lipid phase (52). This protein structure displays under the action of ATP hydrolysis a large amplitude transconformation (53) involving tertiary structure changes evidenced by various spectroscopic techniques (54–56). Furthermore, projection data from two-dimensional crystals of P-gp are consistent with the MsbA transmembrane domain structure (57), even if more recent structure refinements have shown that P-gp does not have a strictly symmetric transmembrane domain (58). As a whole, since the molecular mechanism of P-gp-mediated drug transport is currently very poorly understood, available data make MsbA a fair structural and functional model for P-gp in the frame of a first approach.
The data generated by molecular simulation of the interactions between MsbA and surrounding lipids have pinpointed few amino acids in the hinge region of the protein, involving helices 2 and 5 from each of the two MsbA monomers, for being able to specifically interact with particular lipid molecules. This is strikingly consistent with experimental data coming from cross-linking assays of the helix-helix neighborhood in P-gp, evidencing a close proximity between helices 2 and 11 as well as between helices 5 and 8 (59). In addition, helix 11 in P-gp has often been involved by directed point mutagenesis experiments in the specificity of drug recognition (60,61). Also, different photolabeling experiments indicated helices 4-5-6, 7-8, and 11-12 as targets for drug binding (62–64). Furthermore, it has been reported that among the 12 helices in P-gp, 7 (1-2-3-5-6-8-11) are rich in aromatic residues conserved among the various isoforms of P-gp, 4 of them (3-5-8-11) being suited for interacting with rhodamine 123, a typical polycyclic drug substrate, even if this is the result of a rather crude molecular modeling process considering each of the P-gp helices separately (65). As a whole, helices 5, 8, and 11 in P-gp thus appears to be consistently involved in the transported substrate recognition. In addition, among the six amino acids of MsbA specifically interacting with POPE lipids, three of them are conserved in the aligned P-gp sequence in each of its half-tandem: Ser-260, Ala-262, and Phe-265 respectively conserved in positions 309-311-314 and 952-954-957. According to available mutagenesis data (60), these residues are located close to some of the various amino acids involved in drug recognition by P-gp.
Finally, the molecular characteristics obtained here for MsbA interaction with membrane lipids, and that can be favorably compared with what is known for P-gp, could also be considered for the other ABC transporters from the B subfamily, which have the closest homology with MsbA. In particular, ABC B4 (also called MDR3) is known as a phosphatidylcholine translocase, mainly expressed in biliary canalicule where it participates to bile secretion (3), and it is conceivable that such a phospholipid transporter shares with MsbA (and P-gp) some similarities at the molecular level for transport mechanism. Also, ABC B2 and B3 (also known as TAP1/2) constitute as a dimer a transporter of hydrophobic peptides, formed by antigenic restriction and loaded into endoplasmic reticulum of macrophages (66), which have flexible structures and could thus be handled in a similar manner to phospholipids. Indeed, in this study, we showed that the coupling between simulation data and phylogenetic analyses could powerfully highlight residues likely to play a role in the transport of a specific class of substrates. The protocol developed here may be applied in the future to other classes of ABC transporters provided enough sequences can be aligned together with the corresponding knowledge on their transport specificities.
As a conclusion, based on the structure of the bacterial ABC transporter MsbA, molecular dynamic simulation of a large atomic system comprising a membrane protein inserted in its lipid membrane environment has allowed us to evidence the functional importance of the hinge region of the dimeric protein and the destabilization of the cytosolic leaflet, which may be proposed as a general mechanism for other ABC transporters of lipids and/or amphiphilic drugs involved in mammalian physiology.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
Our thanks go to the Centre de Calcul Recherche et Technologie (CCRT) of the Commissariat á l'Energie Atomique for access to computing resources. D.Y.H. thanks Massimo Marchi for helpful discussions.
H.M. is supported by a Délégation Générale pour l'Armement (DGA) fellowship.
David Y. Haubertin's present address is AstraZeneca, Centre de Recherche, Z.I. La Pompelle, BP 1050, 51689 Reims cedex 2, France.
References
- 1.Higgins, C. F. 1992. ABC transporters: from microorganisms to man. Annu. Rev. Cell Biol. 8:67–113. [DOI] [PubMed] [Google Scholar]
- 2.Gottesman, M. M., and S. V. Ambudkar. 2001. Overview: ABC transporters and human disease. J. Bioenerg. Biomembr. 33:453–458. [DOI] [PubMed] [Google Scholar]
- 3.Borst, P., N. Zelcer, and A. van Helvoort. 2000. ABC transporters in lipid transport. Biochim. Biophys. Acta. 1486:128–144. [DOI] [PubMed] [Google Scholar]
- 4.Bates, S. E., R. Robey, K. Miyake, K. Rao, D. D. Ross, and T. Litman. 2001. The role of half-transporters in multidrug resistance. J. Bioenerg. Biomembr. 33:503–511. [DOI] [PubMed] [Google Scholar]
- 5.Kruh, G. D., H. Zeng, P. A. Rea, G. Liu, Z. S. Chen, K. Lee, and M. G. Belinsky. 2001. MRP subfamily transporters and resistance to anticancer agents. J. Bioenerg. Biomembr. 33:493–501. [DOI] [PubMed] [Google Scholar]
- 6.Sauna, Z. E., M. M. Smith, M. Muller, K. M. Kerr, and S. V. Ambudkar. 2001. The mechanism of action of multidrug-resistance-linked P-glycoprotein. J. Bioenerg. Biomembr. 33:481–491. [DOI] [PubMed] [Google Scholar]
- 7.Pohl, A., P. F. Devaux, and A. Herrmann. 2005. Function of prokaryotic and eukaryotic ABC proteins in lipid transport. Biochim. Biophys. Acta. 1733:29–52. [DOI] [PubMed] [Google Scholar]
- 8.Schmitt, L., and R. Tampe. 2002. Structure and mechanism of ABC transporters. Curr. Opin. Struct. Biol. 12:754–760. [DOI] [PubMed] [Google Scholar]
- 9.Locher, K. P., A. T. Lee, and D. C. Rees. 2002. The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science. 296:1091–1098. [DOI] [PubMed] [Google Scholar]
- 10.Chang, G., and C. B. Roth. 2001. Structure of MsbA from E-coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science. 293:1793–1800. [DOI] [PubMed] [Google Scholar]
- 11.Doerrler, W. T., M. C. Reedy, and C. R. Raetz. 2001. An Escherichia coli mutant defective in lipid export. J. Biol. Chem. 276:11461–11464. [DOI] [PubMed] [Google Scholar]
- 12.Woebking, B., G. Reuter, R. A. Shilling, S. Velamakanni, S. Shahi, H. Venter, L. Balakrishnan, and H. W. van Veen. 2005. Drug-lipid A interactions on the Escherichia coli ABC transporter MsbA. J. Bacteriol. 187:6363–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reuter, G., T. Janvilisri, H. Venter, S. Shahi, L. Balakrishnan, and H. W. van Veen. 2003. The ATP binding cassette multidrug transporter LmrA and lipid transporter MsbA have overlapping substrate specificities. J. Biol. Chem. 278:35193–35198. [DOI] [PubMed] [Google Scholar]
- 14.Higgins, C. F., and M. M. Gottesman. 1992. Is the multidrug transporter a flippase? Trends Biochem. Sci. 17:18–21. [DOI] [PubMed] [Google Scholar]
- 15.Romsicki, Y., and F. J. Sharom. 2001. Phospholipid flippase activity of the reconstituted P-glycoprotein multidrug transporter. Biochemistry. 40:6937–6947. [DOI] [PubMed] [Google Scholar]
- 16.Chang, G. 2003. Structure of MsbA from Vibrio cholera: a multidrug resistance ABC transporter homolog in a closed conformation. J. Mol. Biol. 330:419–430. [DOI] [PubMed] [Google Scholar]
- 17.Reyes, C. L., and G. Chang. 2005. Structure of the ABC transporter MsbA in complex with ADP.vanadate and lipopolysaccharide. Science. 308:1028–1031. [DOI] [PubMed] [Google Scholar]
- 18.Stenham, D. R., J. D. Campbell, M. S. Sansom, C. F. Higgins, I. D. Kerr, and K. J. Linton. 2003. An atomic detail model for the human ATP binding cassette transporter P-glycoprotein derived from disulfide cross-linking and homology modeling. FASEB J. 17:2287–2289. [DOI] [PubMed] [Google Scholar]
- 19.Campbell, J. D., P. C. Biggin, M. Baaden, and M. S. Sansom. 2003. Extending the structure of an ABC transporter to atomic resolution: modeling and simulation studies of MsbA. Biochemistry. 42:3666–3673. [DOI] [PubMed] [Google Scholar]
- 20.Karplus, M., and J. A. McCammon. 2002. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 9:646–652. [DOI] [PubMed] [Google Scholar]
- 21.Essex, J. W., M. M. Hann, and W. G. Richards. 1994. Molecular dynamics simulation of a hydrated phospholipid bilayer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 344:239–260. [DOI] [PubMed] [Google Scholar]
- 22.Tieleman, D. P., S. J. Marrink, and H. J. Berendsen. 1997. A computer perspective of membranes: molecular dynamics studies of lipid bilayer systems. Biochim. Biophys. Acta. 1331:235–270. [DOI] [PubMed] [Google Scholar]
- 23.Tobias, D. J., K. C. Tu, and M. L. Klein. 1997. Atomic-scale molecular dynamics simulations of lipid membranes. Curr. Opin. Colloid Interface Sci. 2:15–26. [Google Scholar]
- 24.Sansom, M. S. 1998. Models and simulations of ion channels and related membrane proteins. Curr. Opin. Struct. Biol. 8:237–244. [DOI] [PubMed] [Google Scholar]
- 25.Tieleman, D. P., I. H. Shrivastava, M. R. Ulmschneider, and M. S. Sansom. 2001. Proline-induced hinges in transmembrane helices: possible roles in ion channel gating. Proteins. 44:63–72. [DOI] [PubMed] [Google Scholar]
- 26.Tieleman, D. P., and H. J. Berendsen. 1998. A molecular dynamics study of the pores formed by Escherichia coli OmpF porin in a fully hydrated palmitoyloleoylphosphatidylcholine bilayer. Biophys. J. 74:2786–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valadie, H., J. J. Lacapère, Y. H. Sanejouand, and C. Etchebest. 2003. Dynamical properties of the MscL of Escherichia coli: a normal mode analysis. J. Mol. Biol. 332:657–674. [DOI] [PubMed] [Google Scholar]
- 28.Roux, B. 2005. Ion conduction and selectivity in K(+) channels. Annu. Rev. Biophys. Biomol. Struct. 34:153–171. [DOI] [PubMed] [Google Scholar]
- 29.Marrink, S. J., and A. E. Mark. 2002. Molecular dynamics simulations of mixed micelles modeling human bile. Biochemistry. 41:5375–5382. [DOI] [PubMed] [Google Scholar]
- 30.Deol, S. S., P. J. Bond, C. Domene, and M. S. Sansom. 2004. Lipid-protein interactions of integral membrane proteins: a comparative simulation study. Biophys. J. 87:3737–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burnell, E., L. van Alphen, A. Verkleij, and B. de Kruijff. 1980. 31P nuclear magnetic resonance and freeze-fracture electron microscopy studies on Escherichia coli. I. Cytoplasmic membrane and total phospholipids. Biochim. Biophys. Acta. 597:492–501. [DOI] [PubMed] [Google Scholar]
- 32.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. [Online]. 7:306–317. [Google Scholar]
- 33.Berendsen, H. J., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 34.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an N.log(N) method for Ewald sum in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 35.Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- 36.Holm, L., and C. Sander. 1991. Database algorithm for generating protein backbone and side-chain co-ordinates from a C alpha trace application to model building and detection of co-ordinate errors. J. Mol. Biol. 218:183–194. [DOI] [PubMed] [Google Scholar]
- 37.Canutescu, A. A., A. A. Shelenkov, and R. L. Dunbrack. 2003. A graph-theory algorithm for rapid protein side-chain prediction. Protein Sci. 12:2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vriend, G. 1990. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8:52–56. [DOI] [PubMed] [Google Scholar]
- 39.Berendsen, H. J. C., J. R. Grigera, and T. P. Straatsma. 1987. The missing term in effective pair potentials. J. Phys. Chem. 91:6269–6271. [Google Scholar]
- 40.Bond, P. J., J. D. Faraldo-Gomez, and M. S. Sansom. 2002. OmpA: a pore or not a pore? Simulation and modeling studies. Biophys. J. 83:763–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clamp, M., J. Cuff, S. M. Searle, and G. J. Barton. 2004. The Jalview Java alignment editor. Bioinformatics. 20:426–427. [DOI] [PubMed] [Google Scholar]
- 43.Mayrose, I., D. Graur, N. Ben-Tal, and T. Pupko. 2004. Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol. Biol. Evol. 21:1781–1791. [DOI] [PubMed] [Google Scholar]
- 44.Jones, D. T., W. R. Taylor, and J. M. Thornton. 1992. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8:275–282. [DOI] [PubMed] [Google Scholar]
- 45.Park, T., J. B. Loomis, and M. Creel. 1991. Confidence intervals for evaluating benefits estimates from dichotomous choice contingent valuation studies. Land Econ. 67:64–73. [Google Scholar]
- 46.Loo, T. W., and D. M. Clarke. 1999. The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J. Biol. Chem. 274:24759–24765. [DOI] [PubMed] [Google Scholar]
- 47.Laskowski, R. A., M. W. MacArthur, D. S. Moss, and J. M. Thornton. 1993. Procheck-a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291. [Google Scholar]
- 48.Buchaklian, A. H., A. L. Funk, and C. S. Klug. 2004. Resting state conformation of the MsbA homodimer as studied by site-directed spin labeling. Biochemistry. 43:8600–8606. [DOI] [PubMed] [Google Scholar]
- 49.Doerrler, W. T., H. S. Gibbons, and C. R. Raetz. 2004. MsbA-dependent translocation of lipids across the inner membrane of Escherichia coli. J. Biol. Chem. 279:45102–45109. [DOI] [PubMed] [Google Scholar]
- 50.Loo, T. W., and D. M. Clarke. 1994. Reconstitution of drug-stimulated ATPase activity following co-expression of each half of human P-glycoprotein as separate polypeptides. J. Biol. Chem. 269:7750–7755. [PubMed] [Google Scholar]
- 51.Hrycyna, C. A., L. E. Airan, U. A. Germann, S. V. Ambudkar, I. Pastan, and M. M. Gottesman. 1998. Structural flexibility of the linker region of human P-glycoprotein permits ATP hydrolysis and drug transport. Biochemistry. 37:13660–13673. [DOI] [PubMed] [Google Scholar]
- 52.Rosenberg, M. F., R. Callaghan, R. C. Ford, and C. F. Higgins. 1997. Structure of the multidrug resistance P-glycoprotein to 2.5 nm resolution determined by electron microscopy and image analysis. J. Biol. Chem. 272:10685–10694. [DOI] [PubMed] [Google Scholar]
- 53.Rosenberg, M. F., G. Velarde, R. C. Ford, C. Martin, G. Berridge, I. D. Kerr, R. Callaghan, A. Schmidlin, C. Wooding, K. J. Linton, and C. F. Higgins. 2001. Repacking of the transmembrane domains of P-glycoprotein during the transport ATPase cycle. EMBO J. 20:5615–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sonveaux, N., A. B. Shapiro, E. Goormaghtigh, V. Ling, and J. M. Ruysschaert. 1996. Secondary and tertiary structure changes of reconstituted P-glycoprotein. A Fourier transform attenuated total reflection infrared spectroscopy analysis. J. Biol. Chem. 271:24617–24624. [DOI] [PubMed] [Google Scholar]
- 55.Sonveaux, N., C. Vigano, A. B. Shapiro, V. Ling, and J. M. Ruysschaert. 1999. Ligand-mediated tertiary structure changes of reconstituted P-glycoprotein. A tryptophan fluorescence quenching analysis. J. Biol. Chem. 274:17649–17654. [DOI] [PubMed] [Google Scholar]
- 56.Liu, R., A. Siemiarczuk, and F. J. Sharom. 2000. Intrinsic fluorescence of the P-glycoprotein multidrug transporter: sensitivity of tryptophan residues to binding of drugs and nucleotides. Biochemistry. 39:14927–14938. [DOI] [PubMed] [Google Scholar]
- 57.Lee, J. Y., I. L. Urbatsch, A. E. Senior, and S. Wilkens. 2002. Projection structure of P-glycoprotein by electron microscopy. Evidence for a closed conformation of the nucleotide binding domains. J. Biol. Chem. 277:40125–40131. [DOI] [PubMed] [Google Scholar]
- 58.Rosenberg, M. F., R. Callaghan, S. Modok, C. F. Higgins, and R. C. Ford. 2005. Three-dimensional structure of P-glycoprotein: the transmembrane regions adopt an asymmetric configuration in the nucleotide-bound state. J. Biol. Chem. 280:2857–2862. [DOI] [PubMed] [Google Scholar]
- 59.Loo, T. W., M. C. Bartlett, and D. M. Clarke. 2004. Disulfide cross-linking analysis shows that transmembrane segments 5 and 8 of human P-glycoprotein are close together on the cytoplasmic side of the membrane. J. Biol. Chem. 279:7692–7697. [DOI] [PubMed] [Google Scholar]
- 60.Ambudkar, S. V., S. Dey, C. A. Hrycyna, M. Ramachandra, I. Pastan, and M. M. Gottesman. 1999. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 39:361–398. [DOI] [PubMed] [Google Scholar]
- 61.Ambudkar, S. V., C. Kimchi-Sarfaty, Z. E. Sauna, and M. M. Gottesman. 2003. P-glycoprotein: from genomics to mechanism. Oncogene. 22:7468–7485. [DOI] [PubMed] [Google Scholar]
- 62.Wu, Q., P. Y. Bounaud, S. D. Kuduk, C. P. Yang, I. Ojima, S. B. Horwitz, and G. A. Orr. 1998. Identification of the domains of photoincorporation of the 3′- and 7-benzophenone analogues of taxol in the carboxyl-terminal half of murine mdr1b P-glycoprotein. Biochemistry. 37:11272–11279. [DOI] [PubMed] [Google Scholar]
- 63.Demeule, M., A. Laplante, G. F. Murphy, R. M. Wenger, and R. Beliveau. 1998. Identification of the cyclosporin-binding site in P-glycoprotein. Biochemistry. 37:18110–18118. [DOI] [PubMed] [Google Scholar]
- 64.Safa, A. R. 2004. Identification and characterization of the binding sites of P-glycoprotein for multidrug resistance-related drugs and modulators. Curr. Med. Chem. Anticancer Agents. 4:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pawagi, A. B., J. Wang, M. Silverman, R. A. Reithmeier, and C. M. Deber. 1994. Transmembrane aromatic amino acid distribution in P-glycoprotein. A functional role in broad substrate specificity. J. Mol. Biol. 235:554–564. [DOI] [PubMed] [Google Scholar]
- 66.Abele, R., and R. Tampe. 1999. Function of the transport complex TAP in cellular immune recognition. Biochim. Biophys. Acta. 1461:405–419. [DOI] [PubMed] [Google Scholar]