

Abstract
Molecular dynamics simulations have been performed to explore the distribution and translocation of a set of furocoumarins (psoralen derivatives) inside saturated and partially unsaturated lipid membranes. Within the simulations, strong accumulation of the photodynamic drugs is observed near the polar headgroup region, although the populations also extend out into the membrane/water interface as well as to the membrane center. The computed transverse (Dz) diffusion coefficients are in the range 0.01–0.03 × 10−5 cm2 s−1—significantly slower than those reported for small molecules like water, ethane, and ammonia—and are related to the low mobility inside the polar headgroup region. Trimethylpsoralen (TMP) has a very low free energy barrier to transversion, only ∼10 kJ/mol, whereas 5- and 8-methoxy psoralens (5-MOP, 8-MOP) have the largest barriers of the compounds studied—between 25 and 40 kJ/mol. Upper bounds to the permeation coefficients, obtained by integrating the resistance profiles across the bilayers, range from 5.2 × 10−8 cm s−1 for TMP to 4.1 × 10−12 cm s−1 for 5-MOP. The current simulations explain the high level of furocoumarin-lipid membrane complexes found in experimental studies of albino Wistar rats exposed to topical application of 8-MOP, and points to the possibility of membrane photodamage as a viable mechanism in psoralen ultraviolet-A treatment.
INTRODUCTION
Photodynamic therapy has been employed in the treatment of a wide range of diseases over the past 15 years and is generally based on topical application of a photosensitive drug—a polycyclic heteroatomic aromatic compound—followed by irradiation generally in the ultraviolet-A/visible (UV-A/Vis) region of the spectrum (320–400/400–720 nm). The photosensitizer will absorb the radiation and govern the excitation energy into the tissue, thereby inducing a variety of photochemical, redox, and/or radical reactions. Very often, these involve the generation of reactive oxygen species or direct photobinding of the sensitizer by way of its first excited singlet state. The latter are concerted C4-cyclizations between a double bond on the sensitizer and a double bond on the target molecule, yielding a cyclobutane-like cross-linked structure. One of the most illustrative examples thereof is that between the C3=C4 bond of the pyrone ring in psoralen and the C5=C6 bond of thymine in DNA (1), one of the main targets of many of these compounds.
A number of different photosensitizers have been proposed, the most common currently in use being based on the psoralen family or various porphyrin derivatives such as photophrin and foscan. Psoralen compounds (furocoumarins) have been used in photochemical treatment of, e.g., psoriasis, vitiligo, mycosis fungoides, chronic leukemia, or as antibacterial and antiviral agents (2–4). However, other large heterocycles and/or aromatic compounds including anthrapyrazoles, isoquinoline alkaloids, phylloerythrins, and perylenequinones have also been suggested (5).
Despite extensive research in the field, the specific mechanisms of action of many of these compounds are still largely unknown, giving room for theory to assist in the elucidation of their properties as well as possible reaction routes and resulting product distributions. In addition, having more details on the mechanisms involved, computational chemistry can be employed to fine-tune the photosensitizer properties and to explore the chemistries of possible new compounds and their derivatives.
For the drugs to reach their cellular targets, they must first penetrate the lipid membrane of the cell. In the event of UV radiation hitting the cell as the drug resides within the membrane, photodynamic reactions with the lipid molecules may be induced. Such photoinduced cross-links between forucoumarins and lipid membranes are well known to occur (6,7), and small models systems thereof have been investigated both theoretically (8) and experimentally (9,10). For example, in a recent study of 8-methoxy psoralen (8-MOP) reacting with shaved backs of albino Wistar rats, ∼26% of the covalently bound complexes found were to unsaturated lipid membranes, even higher than the observed percentage of covalent complexes to DNA (17%) (11). Hence, photoinduced damage to membranes appears to be an important, albeit hitherto much neglected, mechanism of action of these substances.
In addition, despite the fact that membrane interaction and permeability are key aspects in drug delivery, very little is known on the diffusion of these types of compounds experimentally. Modeling of membrane permeation is also rather limited and has mainly focused on small molecules such as water, ammonia, NO, CO2, ethane, and benzene in saturated dimyristoylphosphatidylcholine (DMPC) or dipalmitoyl-phosphatidylcholine (DPPC) membranes (12–15). It was shown that in these systems the free energy of traversion either increases monotonically as the molecule moves from the water layer toward the center of the lipid bilayer (e.g., water, ammonia, acetamide, methanol) or increases at the interface and then decreases toward a minimum at the bilayer center (e.g., ethane, benzene, methyl acetate, O2). The distributions in membranes and effects on membrane properties of organic pollutants pentachlorophenol and pyrene were recently reported (16,17), showing that in these cases accumulation occurs inside the headgroup region but that very little systematic movements take place after that. In terms of drug-membrane interactions, studies have been reported dealing with the anesthetic haloethane in dioleoylphosphatidylcholine (DOPC) or DPPC bilayers (18,19) and the anticonvulsant drug valproic acid in DPPC (20). The studies of valproic acid revealed a considerable difference in behavior between the neutral and deprotonated form. The neutral species had a local minimum just inside the polar headgroup region and a shallow barrier (∼10 kJ/mol) to translocation across the bilayer middle. The deprotonated form, on the other hand, which is the predominant one in aqueous solution, had no minimum within the bilayer; instead it required ∼100 kJ/mol to permeate across the membrane.
To understand in more detail the aspects of drug delivery involving membrane translocation, as a platform for development of photodynamic drugs with enhanced capabilities, the distribution and diffusion properties of psoralen derivatives are here explored in a detailed molecular dynamics study using both fully saturated DPPC and unsaturated 1-palmitoyl-2-linoleoyl-sn-glycero-3-phosphatidylcholine (PLPC) lipid membrane models. Worth emphasizing in the current context is that in a fully saturated membrane system such as DPPC, direct photobinding is not possible due to the lack of unsaturated C=C double bonds. Modeling of both a saturated and a partly unsaturated membrane model will hence provide insight into possible differences in interactions and reactions inside these systems.
THEORETICAL METHODOLOGY
The GROMACS program (21,22) was employed in the simulations of furocoumarin distribution inside the two lipid bilayer models: i), a saturated membrane consisting of 64 DPPC lipids solvated by 1474 water molecules: potentials and initial bilayer patch by Söderhäll and Laaksonen (23); and ii), an unsaturated membrane containing 128 PLPC solvated by 2453 water molecules: potentials and initial bilayer patch by A. Rouk and co-workers (24). Each bilayer was carefully equilibrated before insertion of the solute molecules.
The GROMACS force field was used throughout. The distribution and permeability of the furocoumarin parent compound psoralen (Pso) and four of its main derivatives (angelicin (Ang), trimethylpsoralen (TMP), 5-methoxy psoralen (5-MOP), and 8-MOP) as depicted in Fig. 1 were simulated inside the two membrane models. In the cases where oxygen interaction parameters in the psoralen heterocycles were lacking, potentials between chemically similar atoms of nitrogen containing heterocycles were employed after initial test calculations. Atomic partial charges and dipole moments were obtained through B3LYP/6-311+G(2df,p) single point calculations after initial optimization at the B3LYP/6-31G(d,p) level, using the Gaussian 03 program (25–28). The simulation parameters (NPT ensemble at T = 300 K, Nosé-Hoover temperature coupling (29,30), semiisotropic Parinello-Rahman pressure coupling (31–33), and particle mesh Ewald summation for the electrostatic interactions) are similar to those employed in previous work (24). A 10-Å cutoff was used for the long-range electrostatic interaction as well as for the short-range Lennard-Jones terms. Bond lengths were constrained using the SHAKE algorithm.
FIGURE 1.

(a) Furocoumarins studied in this work and their computed dipole moments. (b) Snapshots from simulations showing psoralen (encircled) at its minimum and maximum penetration in PLPC during the 20-ns simulation.
The simulated systems were constructed by inserting a furocoumarin molecule into the middle of the bilayer where there is the biggest free volume available; and, by this fact, it is the insert position that results in the least perturbation (according to Marrink et al. (34) a penetrant molecule with a diameter of 0.6 nm can fit on average into almost 1% of the total volume in the middle of the bilayer without disturbing the surrounding lipids; the biggest molecular axis in the Pso molecule is 0.8 nm). Since the molecules are planar, each furocoumarin molecule was inserted exactly into the middle to render the molecule and the bilayer in the same plane. After each insertion a steepest descent minimization was performed to remove close interactions between molecules. The interaction energy between the inserted molecule and the lipids was monitored and found to equilibrate fast, well within the 2 ns of equilibration time. In addition, the insertion of the molecules did not change the average area per lipid of the initial equilibrated bilayers (well within the fluctuations). For the DPPC a value of 0.62 nm2 was obtained, which is very similar to that estimated by Nagle et al. (35) (0.629 ± 0.014 nm2). The area per lipid for the PLPC systems was similar to the area per lipid of the initial equilibrated bilayer system (∼0.67 nm2).
Within the equilibration run, all furocoumarin molecules moved from the middle of the bilayer toward one of the water/phospholipid interfaces, illustrating the amphiphilic character of these drugs. For each system, a 20-ns production run followed in which the system trajectories were collected every 0.2 ps. Since the molecules can move over a fairly large region (∼17.5 Å from the water/bilayer interface toward the center of the bilayer) in a low frequency movement, long simulation times are needed to correctly sample the distributions. For the molecules to move from the bilayer middle to the water/lipid interface during the equilibration process, 1–1.5 ns was required. From the diffusion coefficients of the equilibrium calculations the molecules were found on average to move much less (in 1 ns the molecular centers of mass on average move between 1 and 3 Å). During the simulations, none of the furocoumarins moved out into the water phase or across the bilayer middle to the opposing side of the membrane. For this reason, we display all distributions collected into the same half of the bilayer throughout.
We used a potential of mean force formalism to calculate the furocoumarins free energy profiles across the DPPC lipid bilayer. To calculate the free energy of transfer of a particle across the bilayer normal (the direction of the z axis), we define the reaction coordinate by the z axis and collect the z-component of the force acting on the particle, Fz, at a certain constrained distance between the particle and the bilayer center of mass at different positions z along the reaction coordinate. This gives the free energy for the transfer process between points zi and zf as
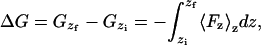 |
(1) |
where the bracket means that we are averaging over the forces collected at a certain constrained point z of the reaction path. In this study, we collected the force acting on the furocoumarin centers of mass every time step during 1200 ps and used a SHAKE algorithm (36) to constrain the distance between the centers of mass of the bilayer and of the furocoumarins (the z-positions of the molecular centers of mass are constrained, allowing the molecules to rotate). The starting systems containing the furocoumarins at different distances from the bilayer center of mass were sampled from the previous partition calculation runs (∼18 different distances were used).
Permeability can be defined as the current density divided by the concentration gradient across the membrane. To calculate the permeability coefficients we followed the procedure developed by Marrink and Berendsen (15). This method is based on the fluctuation dissipation theorem and uses the deviation of the instantaneous force, F(z,t), from the average force acting on the molecule obtained during the constrained dynamics
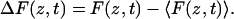 |
(2) |
From this we calculate the local time-dependent friction coefficient, ξ,
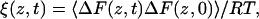 |
(3) |
where T is the absolute temperature and R is the gas constant. The diffusion coefficient, D, can be obtained by integrating the friction coefficient,
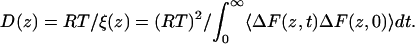 |
(4) |
To integrate the autocorrelation of the force fluctuations, this function was best fitted to a double exponential using a nonlinear fitting procedure (15)
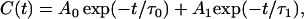 |
(5) |
which illustrates that the molecules move inside the lipid bilayer in two distinct timescales, corresponding to the two decay times τ0 and τ1.
The permeability coefficient, P, can be calculated by integrating over the local resistances across the membrane, R(z), obtained from the previously calculated position-dependent free energies, ΔG(z), and diffusion coefficients, D(z),
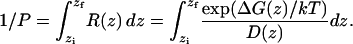 |
(6) |
The simulations provide information on some of the key features of furocoumarin interaction with lipid membranes: in particular the effects of the substrate substituent patterns and the behavior in the two extreme cases of lipid bilayers employed.
RESULTS AND DISCUSSION
Although the molecules already during equilibration move toward the interface, the overall probability to find any of the furocoumarins inside the phospholipid head regions is very low since this is the densest region of the membrane. This is illustrated for Pso in Fig. 2, a and b, and the final density distributions of all derivatives inside the lipid bilayer models are shown in Fig. 2 c (DPPC) and Fig. 2 d (PLPC). The density distributions have Gaussian shapes with maximum probabilities near the polar headgroup regions, very close to the maximum distance of penetration for water molecules into the bilayer. In this context, a recent study of methanol and ethanol in DPPC and POPC bilayers is of interest (37). Starting with all alcohol molecules in the water layer outside the lipid membrane, the molecules were still found to accumulate inside the polar headgroup regions also for these systems, despite being more polar and having less hydrocarbon content than the systems currently under study.
FIGURE 2.

Density profiles for psoralen and (a) the DPPC and (b) the PLPC bilayers. Resulting distributions for the all furocoumarins inside (c) the DPPC and (d) the PLPC bilayers.
The large polarities of the furocoumarins imply that they will not diffuse into the apolar bilayer middle; instead, they are attracted by the polar medium near the interface. On the other hand, the large sizes of the molecules in combination with the high hydrocarbon content also make them avoid the most dense and polar regions; the resulting distribution is a balance between these contributions.
For all molecules studied, the maximum position probability is located at roughly the same distance from the bilayer middle. The trimethylpsoralen (TMP) molecule, with its larger amount of aliphatic substituents, is able to move more deeply inside both bilayers. This is also seen for Pso in the DPPC bilayer and has implications for the permeability of these compounds through the membranes. Within the DPPC bilayer the distribution of Ang is shifted toward the more polar environment near the interface.
More details about the specific movements of the molecules inside the bilayers are obtained from the mean-square displacement (MSD) (38). The MSD is defined by
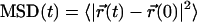 |
(7) |
where  (0) and
(0) and 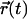 are the positions of a particle at time t = 0 and at a certain time t, respectively. The integral indicates a time average over all similar particles and over different time origins along the simulation. The Einstein relation allows for the calculation of the diffusion coefficient, D, at sufficiently long simulation times (38):
are the positions of a particle at time t = 0 and at a certain time t, respectively. The integral indicates a time average over all similar particles and over different time origins along the simulation. The Einstein relation allows for the calculation of the diffusion coefficient, D, at sufficiently long simulation times (38):
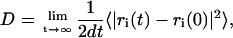 |
(8) |
where d is the dimensionality of the space. This way, one can obtain the MSD for the molecules moving in the bilayer plane (d = 2) and along the bilayer normal (d = 1), respectively. The MSD and the root MSD provide measures of the average distance a molecule travels in the system; and the growth rate of the MSD depends on how often the molecule collides with others, i.e., it is a measure of the ease of diffusion of the substrate.
Using a log-log plot of the MSD time dependence, the Einsteinian limit is reached if the MSD is proportional to tn, where n = 1 (39). Initially, in the short timescale when the particle starts to diffuse, the motion is not perturbed by the surrounding environment (the velocity of the particle is constant) and the diffusion is proportional to t2. Before reaching the Einsteinian regime, anomalous diffusion may occur with 0 < n < 1. The Einsteinian limit corresponds to a random walk (this implies an unbound, randomly oriented particle which does not experience any kind of potential) (40). In Fig. 3, the doubly logarithmic 1D (Dz) MSD plots of all the molecules inside the DPPC and PLPC bilayer are displayed.
FIGURE 3.

Logarithmic plots of the MSD for all the molecules moving along the bilayer normal (1D) inside the DPPC (left) and PLPC (right) bilayers.
In both bilayers and for times <300 ps the MSD is proportional to t0.5 and the molecules display anomalous diffusion. In the current systems, since the molecules move only inside the lipid bilayer (during the 20-ns production phase no psoralen molecule moved across the middle or escaped the lipid bilayer), the molecules can be considered to move inside two flexible walls (bilayer middle and water/lipid interface). The MSD along the bilayer normal will for sufficiently long simulation times (usually larger than 1–2 ns) reach the saturation limit. Although the right parts of these graphs suffer from noise due to lack of statistics (fewer time origins), this limit can be clearly seen in both figures. For these big molecules confined in such a relatively small space (along the z direction), the Einsteinian limit will never be obtained for the Dz diffusion. Similar plots were obtained for the 2D (Dxy) MSD of all the molecules inside the DPPC and PLPC bilayers (Fig. 4).
FIGURE 4.

Logarithmic plots of the MSD for all the molecules moving in the bilayer plane (2D) inside the DPPC (left) and PLPC (right) bilayers.
For the diffusion of the molecules in the PLPC bilayer plane, a change in the slope is visible at values larger than 1–2 ns. In this bilayer, for times <1 ns n ≈ 0.5, and for time origins between 1 ns and 15 ns n ≈ 1 (except for 5-MOP that still has n ≈ 0.5). This change to an Einsteinian regime is not observed in the other bilayer where n is always far from unity, except for Pso (for times >1 ns, n = 0.9). In this bilayer, 5-MOP also presents the lowest value, closer to 0.5.
For diffusion along the bilayer normal, the Einsteinian regime is hence never reached by these large molecules. For diffusion in the bilayer plane, although the Einsteinian regime was not obtained for the diffusion in the DPPC bilayer, the diffusion will eventually become normal, at times larger than the present simulation. This means that with these results, calculation of diffusion coefficients based on the Einstein relation is not accurate and the obtained values are always underestimated. On the other hand, real measurements of diffusion in bilayers operate in length scales ranging from microns to 10 nm. Diffusion coefficients measured by, for instance, quasielastic neutron scattering and by fluorescent recovery after photobleaching can differ by as much as 100-fold (41). It is hence of importance that we compare diffusion of the molecules in similar regimes. For these confined molecules, the linear regime to consider for the calculation of the diffusion coefficient in the direction normal to the interface is located before the MSD gets into saturation. The Dxy MSD is not affected by such constraint, since the topology of the system allows for the molecules to move in an infinite plane. However, since the Einstein limit was not reached in the DPPC bilayer and we are interested in comparing the molecules in similar regimes to get insight about the effect of the substitutions in the psoralen family, we present these values in Table 1.
TABLE 1.
Self-diffusion coefficients inside the DPPC and PLPC bilayers
| DPPC
|
PLPC
|
|||
|---|---|---|---|---|
| 1D | 2D | 1D | 2D | |
| Pso | 0.015 ± 0.002 | 0.043 ± 0.004 | 0.022 ± 0.008 | 0.018 ± 0.005 |
| Ang | 0.022 ± 0.000 | 0.037 ± 0.000 | 0.008 ± 0.006 | 0.029 ± 0.002 |
| 5-MOP | 0.013 ± 0.011 | 0.035 ± 0.002 | 0.029 ± 0.013 | 0.022 ± 0.008 |
| 8-MOP | 0.011 ± 0.002 | 0.033 ± 0.001 | 0.010 ± 0.005 | 0.014 ± 0.001 |
| TMP | 0.014 ± 0.004 | 0.032 ± 0.002 | 0.026 ± 0.003 | 0.019 ± 0.006 |
The error estimates are differences in diffusion coefficients obtained from fits over the two halves of the fit interval.
1D is the self-diffusion coefficient in one dimension along the axis perpendicular to the bilayer plane (z axis). 2D is the self-diffusion coefficient in two dimensions in the bilayer plane (x and y axes). All values are in units of 10−5 cm2 s−1.
Although for some values the error is relatively high, it is clear that the molecules can diffuse more easily in the bilayer plane than along the bilayer normal. This is more striking in the DPPC bilayer where the Dxy diffusion is about twice that for Dz. For the PLPC bilayer, the diffusion coefficient is sometimes larger along the bilayer normal than in the bilayer plane. Although in the DPPC bilayer the largest values are found for Pso and Ang, which are the smallest molecules, this fact does not apply for the PLPC bilayer and no clear trend is found. The time-averaged diffusion coefficients along the bilayer normal are for these compounds ∼0.01–0.03 × 10−5 cm2 s−1, which is even below the lowest local Dz diffusion coefficients found for valproic acid/valproate (lowest values ∼0.1 × 10−5 cm2 s−1) (20) and the small molecules mentioned above (lowest Dz values 0.5–1 × 10−5 cm2 s−1) (12,14).
Several models have been proposed for the passive diffusion of molecules across biological membranes (42). In the free volume model, the bilayer interior is compared to a soft polymer and the molecules make a diffusive step when they jump from one free volume pocket to another. For example, in a crystalline polymer, molecules move between well-defined cavities and gives trajectory projections revealing several well-defined pockets connected by a few lines (43). In Fig. 5, a typical furocoumarin trajectory from this study is projected into three different planes. Since in the bilayer interior, the free volume cavities are not rigid but change their shapes and sizes over time, this produces a more diffuse image. Nevertheless, we can see that the molecule preferentially samples some locations more than others. This corresponds to the molecule being trapped and moving inside a dynamically changing free volume pocket. From time to time, the molecule escapes one pocket and diffuses to another, after which it again moves in a narrow region.
FIGURE 5.

Trajectory projection of the freely moving Ang center of mass in the PLPC bilayer plane (xy, top figure) and in two planes normal to the bilayer (yz, on the left and xz, on the right). The probability to find a molecule in a certain position in the plane increases from dark blue to red.
The collected forces along the bilayer normal were used to calculate the autocorrelation of the deviation of the instantaneous force from the average. The autocorrelation function obtained was then fitted to a double exponential (Eq. 5) from 0 to 20 ps. For the tested molecules, the short decay time was calculated to be between 0.01 and 0.04 ps, whereas the long decay time is located between 4 and 29 ps. The short decay time is related to the very quick response of the molecules to the environment and if we assume a hopping type of diffusion mechanism, the long time decay is related with the time needed for the psoralen molecules to move between different free volume cages.
The Dz diffusion coefficients were calculated by integrating the fitted autocorrelation function and the dependence with the distance to the bilayer center is displayed in Fig. 6 (left plot). The plot on the right-hand side of Fig. 6 shows the diffusion coefficients for the molecules moving in the bilayer plane calculated by the Einstein relation (Eq. 8) and using the constrained dynamics simulations.
FIGURE 6.

Local diffusion coefficients of the different furocoumarins in the PLPC lipid bilayer, as functions of the distances to the bilayer middle: Motion across the membrane bilayer (1D, left) and in the bilayer plane (2D, right), respectively.
Albeit associated with considerable noise, two observations can be made from the data. First of all, there is a significant difference in local diffusion coefficients close to the bilayer middle (0.2–0.6 × 10−5 cm2 s−1) and in the regime near the polar headgroups. The values in the vicinity of the bilayer middle closely resemble the data for other systems listed above, whereas the local diffusion coefficients in the polar headgroup region are highly similar to the time-averaged data in Table 1. The high diffusion rates in the hydrophobic part of the membrane are largely related to the lower density in this region (see Fig. 2). Furthermore, that the overall diffusion coefficients closely resemble those seen in the 10–15-Å region reflects the fact that the compounds spend most of their time in the polar headgroup region (Fig. 2).
Free energy profiles as a function of the distance to the bilayer center (Fig. 7) were calculated for the systems in DPPC using the potential of mean force formalism outlined above (44). The free energy profiles display the occurrence of a local minimum near the polar headgroup region and an increase in free energy as the molecules move from the water/bilayer interface toward the bilayer middle. This is consistent with a surfactant character and, taken together with the low diffusion rates, allows for an accumulation of the substrate inside the lipid bilayer.
FIGURE 7.

Free energy profiles (kJ/mol) for the furocoumarins inside the DPPC bilayer.
The largest barriers to translocation across the bilayer are found for 5- and 8-MOP (37 and 25 kJ/mol, respectively) whereas TMP presents the lowest value, ∼10 kJ/mol. The barrier for 8-MOP is similar to those found for, e.g., water, acetamide, and methanol, albeit these systems do not display local minima inside the membranes which thus disallows accumulation of the substrates (14). The lower value for TMP comes about from the higher hydrocarbon content and lower polarity (lack of methoxy group; see Fig. 1 a) and thus more favorable interactions with the lipid hydrocarbon chains, and is comparable to the barriers calculated for ethane, methylamide, and neutral valporic acid (albeit all three display different free energy profiles as such) (14,20).
With the free energy profile calculated using the potential of mean force formalism and the local diffusion coefficient across the lipid bilayer, the local resistance was calculated using Eq. 6. The resulting resistances to permeation for the different molecules are displayed in Fig. 8. For clarity, the 5-MOP resistance profile was scaled down by a factor of 20. The resistance increases steeply as the molecules move toward the bilayer middle and is clearly dominated by the free energy component. This way, a higher free energy corresponds also to a higher resistance to permeation.
FIGURE 8.

Local resistance profiles of the different furocoumarins in the PLPC lipid bilayer, as functions of the distances to the bilayer middle. For a better comparison, the 5-MOP resistance profile was reduced by a factor of 20.
The permeability coefficients were calculated by integrating the resistance profiles across the bilayer and a value of 2.5 × 10−9 cm s−1 was found for psoralen, 2.2 × 10−10 cm s−1 for Ang, 4.1 × 10−12 cm s−1 for 5-MOP, 6.8 × 10−11 cm s−1 for 8-MOP, and 5.2 × 10−8 cm s−1 for TMP. Again, since the permeability is governed mainly by the free energy component, the permeation decreases in the following sequence, which follows the increase in free energy: TMP > Pso > Ang > 8-MOP > 5-MOP.
The permeation process is usually described by three steps, involving the solvation of the molecule into the bilayer, diffusion through the membrane interior and across the bilayer middle, and finally the return of the molecule to the environment surrounding the bilayer (42). Since in the constrained dynamic simulations our starting point already contained the molecules inside the lipid bilayer (although close to the water/lipid interface), the calculated permeability coefficients do not contain the first and last steps of the process. It should be noted that these molecules are very big and a correct starting point of the molecule in the water layer should account for the existence of bulk water and not just the amount of water required for the lipid bilayer solvation. This means a much increased system size and computation time. Moreover, trying to insert the molecules inside the lipid headgroup region will constitute a major perturbation to the system (one way to fit the molecules in this zone would involve the removal of some lipids with a subsequent long equilibration). On the other hand, since in this zone the major contribution to the increased free energy is entropic in nature, the effect should be similar for all the tested molecules since their volumes are not much different. All these problems and the non-Einsteinian regime found for the diffusion coefficients point to the use of nonequilibrium molecular dynamics techniques in future studies. Nevertheless, although the global permeation coefficients should be lower (the current ones representing an upper bound), the trends found when comparing the relative properties should remain essentially unaltered.
Although chemical reactions can only be correctly described using quantum mechanics, which is very difficult to apply to systems with a large number of molecules, one can use a simpler approach to gain some insight about the addition reaction rate between the psoralen molecules and the double bonds in lipids occurring in lipid bilayers (in our case, we can only consider the PLPC bilayer because it is the unsaturated one). The reaction rate R and the collision rate C can be related through the following equation (45):
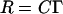 |
(9) |
where Γ is the reaction probability. Assuming that the reaction probability between the psoralens and the lipids is the same (same addition reaction), higher collision rates will hence translate into higher reaction rates.
We define a collision event between a psoralen molecule and a lipid to occur if any psoralen atom that participates in a photoactive double bond (one on the furan side and one on the pyrone side of the molecules) is closer than 4 Å from any lipid atom that also participates in a double bond (two double bonds in each of the PLPC lipid chains). If the collision occurs between the same pair of atoms as in the previous recorded time frame, then a residence time can also be computed. If the same pair of atoms remains for a continuous period within the cutoff radius, only a single collision is recorded and the collision lifetime, τcol, is recorded to account for the residence time.
In the current system, the main question is if the active double bonds of the linoleate and the furocoumarins will be in sufficiently close proximity as these are hit by radiation to enable a photoinduced cyclization. In previous theoretical studies of photochemical cyclization reactions (8,46–48), both the ground state and excited state energy surfaces at distances between 3.5 and 4.5 Å between the reacting centers were found to be very flat. This conclusion was reached for both TMP binding to a lipid model system and for a number of cyclobutane pyrimidine dimer systems in DNA. It is hence reasonable to assume that the mobility within this region will be essentially unhindered from an energetic point of view and that if a system is hit by radiation when at a distance of ∼4 Å, cyclization may readily occur.
Using a 4-Å cutoff we obtained the collision ratios (number of collisions divided by the total number of time frames) for Pso (0.25), Ang (0.26), 5-MOP (0.17), 8-MOP (0.20), and TMP (0.18) inside the PLPC bilayer. If we use a 4.5-Å cutoff we find values that are more than twice the previous ones, except for the 5-MOP molecule, which remains fairly constant. A 3.5-Å cutoff was also tested but a very low number of collisions occur (69 for the psoralen molecule compared to 2547 for a 4-Å cutoff). The fact that the 5-MOP molecule presents a low collision rate is understandable given that the molecule needs to diffuse toward the bilayer middle where the double bonds are located (see Fig. 2 b) and that this molecule presents the highest energy barrier to diffusion. On the other hand, the relatively low result for the TMP molecule that presents the lowest energy barrier and can move more easily to the bilayer middle is rather surprising. For this molecule the bulky substituents appear to hinder close contact between the molecules.
The free energy barrier increases in the following sequence TMP < Pso < Ang < 8-MOP < 5-MOP (Fig. 7) and for the collision ratios, we find that it decreases in the following way: Ang < Pso < 8-MOP < TMP < 5-MOP. If we take into consideration that the collision ratios of Pso and Ang are very similar, we find that except for the TMP molecule the energy barrier is inversely connected with the possibility of a reaction to occur. In Fig. 9, we display the probability distribution of the collision lifetimes, which is seen to follow a power decay with time. This means that for the recorded time lengths, the atoms involved in the reacting bonds come into contact and leave in a short time. The probability of finding a given interaction lifetime follows the same trend as for the collision ratios. One should bear in mind that since the history of the system was written to disk every 0.2 ps, we cannot access the interaction lifetimes between the 2-fs simulation time step and this value.
FIGURE 9.

Probability distributions of collision lifetimes for the furocoumarin molecules inside the PLPC bilayer.
The current data show that once the psoralens get inside the lipid membranes, they tend to remain there and accumulate inside the polar headgroups. The different hydrophobicity character of the substituents gives rise to variations in the barriers for the molecules to traverse the lipid bilayer middle. 8-MOP, which is one of the most utilized furocoumarins for medical applications, has one of the highest barriers to traversion of the compounds investigated and may be expected to have a slower rate of entering into the cell as compared to the more lipophilic TMP. The relatively high barrier for 8-MOP to traverse the membrane may explain the high percentage of covalent lipid-8-MOP bond formation mentioned earlier (11).
For a drug (or a drug-carrier complex) to be optimal it needs to display multiple functionality—it should not only bind efficiently to its target but must also be able to diffuse readily in aqueous as well as apolar environments and avoid degrading side reactions along the way. The efficiency of the drug to penetrate a cell wall without vesicles or facilitated transport implies a delicate balance between water and lipid solubility. We believe that the results presented herein provide information that may assist in enabling a systematic characterization and optimization of novel psoralen derivatives for which membrane permeability is further enhanced. In addition, it provides insight into the design of photoactive drugs where the focus is shifted to membrane interactions and the aim is to accumulate and—upon irradiation—disrupt the membrane structure and function.
CONCLUSIONS
The distribution and diffusion of five different furcoumarin derivatives in DPPC and PLPC lipid bilayer models were investigated using classical molecular dynamics simulations. It is concluded that the compounds reside mainly in the polar headgroup region of the membranes with essentially Gaussian population distributions, extending toward the bilayer middle and the water phase. The time-resolved motions of the molecules reveal that they are able to move between the extreme points (water interface versus bilayer middle) in ∼5 ns.
Local diffusion coefficients display high diffusion rates in the hydrophobic region (∼0.2–0.6 × 10−5 cm2 s−1), whereas in the polar headgroup region the diffusion rates are one order of magnitude lower and close to the overall self-diffusion coefficients. All furocoumarins have a very high number of close contacts between the photochemically active bonds in the furan and pyrone rings and unsaturated carbons in the lipid molecules, indicating that if the membrane is irradiated with the psoralen derivative inside, there is a very high likelihood for photochemical cross-links to be formed between the drug and the lipid molecules.
Of the five molecules investigated, the highest total permeability coefficients are seen for the more hydrophobic compounds, whereas the more polar methoxy-psoralens have the lowest values. This is also reflected in the much higher free energy barriers to traversion of the latter (25–40 kJ/mol) as compared with the TMP molecule that has a free energy barrier of only 10 kJ/mol. This means that the TMP molecules can be expected to translocate across the membranes more readily than the methoxy-substituted species. We can therefore expect more of the 8-MOP and 5-MOP molecules to accumulate within the membranes and hence provide a higher degree of photodamage to these than is the case for species like TMP. This is also in accordance with the experimentally measured high amount of 8-MOP-lipid molecule complexes in treated albino Wistar rats (11).
This study provides a basis for development of more efficient photodynamic compounds—either aiming to penetrate the membranes at higher rates or to accumulate to an even higher degree within the lipid bilayers and degrade these upon photodynamic treatment.
Acknowledgments
The Swedish Science Research Council is gratefully acknowledged for financial support. We also acknowledge the national supercomputing center in Linköping for grants of computing time.
References
- 1.Hearst, J. E., S. T. Isaacs, D. Kanne, H. Rapoport, and K. Straub. 1984. The reactions of psoralen with deoxyribonucleic acid. Q. Rev. Biophys. 17:1–44. [DOI] [PubMed] [Google Scholar]
- 2.Regan, J. D., and J. A. Parrish, editors. 1982. The Science of Photomedicine. Plenum, New York.
- 3.Horspool, W. H., and F. Lenci, editors. 2003. CRC Handbook of Photochemistry and Photobiology, 2nd ed. Taylor and Francis, New York.
- 4.Shima, A., M. Ichahasci, Y. Fujiwara, and H. Takebe, editors. 1993. Frontiers in Photobiology. Excerpta Medica, Amsterdam.
- 5.Ebermann, R., G. Alth, M. Kreitner, and A. Kubin. 1996. Natural products derived from plants as potential drugs for the photodynamic destruction of tumor cells. J. Photochem. Photobiol. B. 36:95–97. [DOI] [PubMed] [Google Scholar]
- 6.Caffieri, S., D. Vedaldi, A. Daga, and F. Dall'Acqua. 1988. Photosensitizing furocoumarins: photocycloaddition to unsaturated fatty acids. In Psoralens in 1988, Past, Present and Future. T. B. Fitzpatrick, P. Forlot, M. A. Pathak, and F. Urbach, editors. John Libbey Eurotext, Montrouge, France. 137–188.
- 7.Zarebska, Z., E. Waszkowska, S. Caffieri, and F. Dall'Acqua. 2000. PUVA (psoralen_UVA) photochemotherapy: processes triggered in the cells. Farmaco. 55:515–520. [DOI] [PubMed] [Google Scholar]
- 8.Li, X. Y., and L. A. Eriksson. 2005. Photoreaction of skin-sensitizing trimethyl psoralen with lipid membrane models. Photochem. Photobiol. 81:1153–1160. [DOI] [PubMed] [Google Scholar]
- 9.Specht, K. G., W. R. Midden, and M. R. Chedekel. 1989. Photocycloaddition of 4,5′,8-trimethylpsoralen and oleic acid methyl ester: product structures and reaction mechanism. J. Org. Chem. 54:4125–4134. [Google Scholar]
- 10.Frank, S., S. Caffieri, A. Raffaelli, D. Vedaldi, and F. Dall'Acqua. 1988. Characterization of psoralen-oleic acid cycloadducts and their possible involvement in membrane photodamage. J. Photochem. Photobiol. B. 44:39–44. [DOI] [PubMed] [Google Scholar]
- 11.Beijersbergen van Henegouwen, G. M. J., E. T. Wihn, S. A. Schononderwoerd, and F. Dall'Acqua. 1989. A method for the determination of PUVA-induced in vivo irreversible binding of 8-methoxypsoralen (8-MOP) to epidermal lipids, proteins and DNA/RNA. J. Photochem Photobiol. B: Biol. 3:631–635. [DOI] [PubMed] [Google Scholar]
- 12.Marrink, S. J., and H. J. C. Berendsen. 1996. Permeation process of small molecules across lipid membranes studied by molecular dynamics simulations. J. Phys. Chem. 100:16729–16738. [Google Scholar]
- 13.Jedlovszky, P., and M. Mezei. 2003. Effect of cholesterol on the properties of phospholipids membranes. 2. Free energy profile of small molecules. J. Phys. Chem. B. 107:5322–5332. [Google Scholar]
- 14.Bemporad, D., J. W. Essex, and C. Luttmann. 2004. Permeation of small molecules through a lipid bilayer: a computer simulation study. J. Phys. Chem. B. 108:4875–4884. [Google Scholar]
- 15.Marrink, S.-J., and H. J. C. Berendsen. 1994. Simulation of water transport through a lipid membrane. J. Phys. Chem. 98:4155–4168. [Google Scholar]
- 16.Mukhopadhyay, P., H. J. Vogel, and D. P. Tieleman. 2004. Distribution of pentachlorophenol in phospholipids bilayers: a molecular dynamics study. Biophys. J. 86:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoff, B., E. Strandberg, A. S. Ulrich, D. P. Tieleman, and C. Posten. 2005. 2H-NMR study and molecular dynamics simulation of the location, alignment, and mobility of pyrene in POPC bilayers. Biophys. J. 88:1818–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koubi, L., M. Tarek, M. L. Klein, and D. Scharf. 2000. Distribution of halothane in a dipalmitoylphosphatidylcholine bilayer from molecular dynamics calculations. Biophys. J. 78:800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacCallum, J. L., P. Mukhopadhyay, H. Luo, and D. P. Tieleman. 2003. Large scale molecular dynamics simulations of lipid-drug interactions. In Proceedings of the 17th Annual International Symposium on High Performance Computing Systems and Applications and the OSCAR Symposium. D. Senechal, editor. NRC Research Press, Ottawa, Canada.
- 20.Ulander, J., and A. D. J. Haymet. 2003. Permeation across hydrated DPPC lipid bilayers: simulation of the titrable amphiphilic drug valproic acid. Biophys. J. 85:3475–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Comm. 91:43–56. [Google Scholar]
- 22.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 7:306–317. [Google Scholar]
- 23.Söderhäll, J. A., and A. Laaksonen. 2001. Molecular dynamics simulations of ubiquinone inside a lipid bilayer. J. Phys. Chem. 105:9308–9315. [Google Scholar]
- 24.Bachar, M., P. Brunelle, D. P. Tieleman, and A. Rauk. 2004. Molecular dynamics simulation of a polyunsaturated lipid bilayer susceptible to lipid peroxidation. J. Phys. Chem. B. 108:7170–7179. [Google Scholar]
- 25.Becke, A. D. 1993. Density-functional thermochemistry, III: the role of exact exchange. J. Chem. Phys. 98:5648–5652. [Google Scholar]
- 26.Lee, C., W. Yang, and R. G. Parr. 1988. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 37:785–789. [DOI] [PubMed] [Google Scholar]
- 27.Stephens, P. J., F. J. Devlin, C. F. Chabalowski, and M. J. Frisch. 1994. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98:11623–11627. [Google Scholar]
- 28.Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, et al. 2004. GAUSSIAN 03, Revision B.02. Gaussian, Inc., Wallingford CT.
- 29.Nosé, S. A. 1984. Molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52:255–268. [Google Scholar]
- 30.Hoover, W. G. 1985. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 31:1695–1697. [DOI] [PubMed] [Google Scholar]
- 31.Parrinello, M., and A. Rahman. 1981. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52:7182–7190. [Google Scholar]
- 32.Parrinello, M., and A. Rahman. 1980. Crystal structure and pair potentials: a molecular-dynamics study. Phys. Rev. Lett. 45:1196–1199. [Google Scholar]
- 33.Parrinello, M., and A. Rahman. 1982. Strain fluctuations and elastic constants. J. Chem. Phys. 76:2662–2666. [Google Scholar]
- 34.Marrink, S. J., R. M. Sok, and H. J. C. Berendsen. 1996. Free volume properties of a simulated lipid membrane. J. Chem. Phys. 104:9090–9099. [Google Scholar]
- 35.Nagle, J. F., R. Zhang, S. Tristram-Nagle, W. Sun, H. I. Petrache, and R. M. Suter. 1996. X-ray structure determination of fully hydrated l alpha phase dipalmitoylphosphatidyl-choline bilayers. Biophys. J. 70:1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryckaert, J. P., G. Ciccoti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- 37.Patra, M., E. Salonen, E. Terama, I. Vattulainen, R. Faller, B. W. Lee, J. Holopainen, and M. Karttunen. 2006. Under the influence of alcohol: the effect of ethanol and methanol on lipid bilayers. Biophys. J. 90:1121–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen, M. P., and D. J. Tildesley. 1990. Computer Simulation of Liquids. Oxford University Press, Oxford.
- 39.Müller-Plathe, F., S. C. Rogers, and W. van Gunsteren. 1992. Computational evidence for anomalous diffusion of small molecules in amorphous polymers. Chem. Phys. Lett. 199:237–243. [Google Scholar]
- 40.Huertas, M. L., V. Cruz, J. J. López Cascales, A. U. Acuña, and J. Garcia de la Torre. 1996. Distribution and diffusivity of a hydrophobic probe molecule in the interior of a membrane: theory and simulation. Biophys. J. 71:1428–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitragotri, S., M. E. Johnson, D. Blankschtein, and R. Langer. 1999. An analysis of the size selectivity of solute partitioning, diffusion, and permeation across lipid bilayers. Biophys. J. 77:1268–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin, B., and A. J. Hopfinger. 1996. Characterization of lipid membrane dynamics by simulation: 3. Probing molecular transport across the phospholipid bilayer. Pharm. Res. 13:1786–1794. [DOI] [PubMed] [Google Scholar]
- 43.Milano, G., G. Guerra, and F. Müller-Plathe. 2002. Anisotropic diffusion of small penetrants in the δ crystalline phase of syndiotactic polystyrene: a molecular dynamics simulation study. Chem. Mater. 14:2977–2982. [Google Scholar]
- 44.Paci, E., G. Ciccotti, M. Ferrario, and R. Kapral. 1991. Activation energies by molecular dynamics with constraints. Chem. Phys. Lett. 176:581–587. [Google Scholar]
- 45.Vieceli, J., O. L. Ma, and D. J. Tobias. 2004. Uptake and collision dynamics of gas phase ozone at unsaturated organic interfaces. J. Phys. Chem. A. 108:5806–5814. [Google Scholar]
- 46.Durbeej, B., and L. A. Eriksson. 2002. Reaction mechanism of thymine dimer formation in DNA induced by UV light. J. Photochem. Photobiol. A: Chemistry. 152:95–101. [Google Scholar]
- 47.Durbeej, B., and L. A. Eriksson. 2003. On the formation of cyclobutane pyrimidine dimers in UV-irradiated DNA: why are thymines more reactive? Photochem. Photobiol. 78:159–167. [DOI] [PubMed] [Google Scholar]
- 48.Li, X. Y., and L. A. Eriksson. 2005. Influence of C5-methylation of cytosine on the formation of cyclobutane pyrimidine dimers. Chem. Phys. Lett. 401:99–103. [Google Scholar]