

Abstract
Impairment of tissue cohesion and the reorganization of the extracellular matrix are crucial events during the progression toward invasive cell phenotype. We studied the in vitro invasion patterns of nine brain tumor cell lines in three-dimensional collagen gels. Cell-cell and cell-matrix interactions were quantified and correlated with the expression level of specific molecules: N-cadherin, matrix metalloproteinases, and their inhibitor. Pattern evolution was studied as a function of time and collagen concentration. Cells with low metalloproteinase expression or high tissue cohesion showed limited invasive potential. Higher metalloproteinase expression and intermediate tissue cohesion resulted in configurations with hypercellular zones surrounding regions mostly devoid of cells and with digested collagen, akin to pseudopalisades in surgically removed malignant astrocytoma specimens. In physical terms, these configurations arise as the result of competition between cell-cell and cell-matrix interactions. Our findings suggest specific ways to characterize, control, or engineer cell migratory patterns and hint at the importance of the interplay between biophysical and biomolecular factors in the characterization of invasive cell behavior and, more generally, in epithelial-mesenchymal transitions.
INTRODUCTION
Brain tumors result in fatality mostly because of their aggressive invasive character. For a cell to be able to detach from a primary tumor and migrate to a distant location, significant changes in its adhesive properties to other cells and to the surrounding matrix must take place. Both of these factors have been studied extensively and shown to be altered in malignant transformations (1). Migrating cells in general exert forces and reorganize their three-dimensional (3D) extracellular matrices (ECM) (2–4). The details of local matrix reorganization in collagen I by invading brain tumor cells have recently been reported (5,6). Degradation of ECM, a hallmark of malignant invasion, takes place through the complex activity of metalloproteinases (MMP) (7–9) and their inhibitors (TIMP) (9,10). The predominant adhesion molecule in brain tissue is a member of the cadherin family of cell adhesion molecules (11,12), N-cadherin (13,14), although the presence of other types, such as T-cadherin (15,16) and E-cadherin (17), has also been reported. The role of N-cadherin in the acquisition of invasive phenotype is still controversial. In contrast to the well-established tumor suppressor role of E-cadherin, N-cadherin might influence the process in a positive or negative manner. In the prostate, during malignant transformation, cells switch from E-cadherin to N-cadherin expression (18). N-cadherin expression in breast cancer cell lines correlates with greater invasive potential (19). Higher N-cadherin level was observed with increasing tumor grade in astrocytic tumors (20), whereas another study found no correlation between N-cadherin expression and the in vitro wound-filling capacity of glioblastoma cell lines (21). The invasive potential of certain glioblastoma cell lines was found to decrease with increasing overall tissue cohesivity but not with decreasing N-cadherin expression (22). However, N-cadherin was downregulated in recurrent malignant astrocytic tumors as compared to the primary lesions and also correlated with the dissemination of tumor cells into the cerebrospinal fluid (23). A further complication stems from the fact that invasion assays have not typically been performed in true three-dimensional matrices, whereas it has been shown that such environment is crucial to the understanding of tumor progression (24).
Motivated by the limited information on the simultaneous effect of cell-cell and cell-matrix interaction in malignant cell invasion (25), we performed a comprehensive study on the migration pattern of nine brain tumor cell lines (along with normal astrocytes) in 3D collagen I matrices. Multicellular aggregates (a widely used model in invasion studies (6,26–29), as representation of the corresponding tissues, were used to quantify cell-cell and cell-matrix interactions. Because tissues composed of adhesive and motile cells are known to mimic the behavior of liquids (30,31), cell-cell adhesion was parameterized in terms of apparent tissue surface tension, a measure of overall tissue cohesivity (32). Measurements were correlated with aggregation assays, N-cadherin expression, and cell morphology. Cell-matrix affinity was assessed by the extent of invasion as a function of time and collagen concentration as well as by quantification of MMP and TIMP expression.
MATERIALS AND METHODS
Cell cultures
Six glioblastoma (GBM1–4, U87, U373), two ependymoma (EM1–2), and one esthesioneuroblastoma (ENB) cell lines were investigated. Histopathological and experimental data on these lines are summarized in Table 1. Seven human brain tumor cell lines were established by mechanical dispersion and further cultivation at the National Institute of Neurosurgery in Budapest, Hungary as described by Hegedüs et al. (33). The cell line GBM1 is identical to HB, a cell line described previously (33,34). All cell cultures used in this study had passage numbers higher than 10. U87 and U373 are standard cell lines from American Type Culture Collection (ATCC, Manassas, VA). Cells were grown on 75 cm2 tissue culture dishes in medium consisting of DMEM (GIBCO/BRL, Gaithersburg, MD) supplemented with 10% FBS (U.S. Bio-Technologies, Pottstown, PA), 10 μg/ml of penicillin-streptomycin, gentamicin, kanamycin sulfate (Sigma-Aldrich, St Louis, MO). Control rat primary astrocytes were obtained as described in Xu et al. (35).
TABLE 1.
Data for the analyzed cell lines
| Code | Diagnosis | Age/sex | WHO grade | Aggr | Cadherin level | Surface tension | Invasion pattern |
|---|---|---|---|---|---|---|---|
| U87 | Glioblastoma | NA | 4 | — | + | — | Dispersed |
| GBM3 | Glioblastoma | 48/f | 4 | — | + | — | Dispersed |
| GBM4 | Glioblastoma | 66/m | 4 | — | + | — | Dispersed |
| GBM2 | Gliosarcoma | 72/m | 4 | + | ++ | 9 ± 3 | Ring |
| ENB | Esthesioneuroblastoma | 21/f | 3 | + | ++ | 17 ± 5 | Ring |
| GBM1 | Glioblastoma | 51/m | 4 | + | ++ | 26 ± 5 | Ring |
| EM2 | Ependymoma | 1/m | 3 | ++ | +++ | 46 ± 3 | Compact |
| U373 | Glioblastoma | 44/f | 4 | ++ | +++ | 46 ± 9 | Compact |
| EM1 | Ependymoma | 2/m | 3 | ++ | +++ | 56 ± 6 | Compact |
| Rat primary astrocytes | NA | NA | ++ | +++ | 41 ± 3 | None | |
Surface tension values (dyne/cm) are shown with standard errors. The invasion pattern of cylindrical aggregates in 1 mg/ml collagen gel is observed after 96 h. The abbreviation “Aggr” stands for aggregation ability. For the meaning of “+” and “—”, signs see text. The grade of malignancy was estimated in accordance with the WHO grading criteria.
Aggregation assay
Confluent cultures were washed twice with Hanks' balanced salt solution (HBSS) containing 2 mM CaCl2, then treated for 10 min with 0.1% trypsin (GIBCO/BRL). The cells were resuspended in DMEM/FBS medium. To follow aggregation, single-cell suspensions were incubated in a gyratory shaker at 120 rpm with 5% CO2 at 37°C for 10 h. Suspension droplets were photographed after 4 and 10 h. The calcium dependency of aggregation was proved by addition of 10 mM EDTA to the medium.
Western blot
Confluent cell cultures were lysed in NP40 lysis buffer for 20 min on ice. Cell lysates were centrifuged at 14,000 rpm for 10 min. Protein measurement was performed using the BCA protein assay kit (Pierce, Rockford, IL). An aliquot with 30 μg of total protein was loaded for each lane on an 8% SDS-PAGE gel. Blots were blocked in 5% dry milk in Tris-buffered saline (TBS) and incubated with anti-N-cadherin primary antibody (C-2542, Sigma-Aldrich; 1:1000) and anti-mouse peroxidase-conjugated secondary antibody (A 5906, Sigma-Aldrich; 1:3000).
Flow cytometry
For flow cytometry cells were harvested from confluent tissue culture dishes with trypsin and washed in phosphate-buffered saline (PBS). After fixation in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) in PBS, cells were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich). Following extensive washing with ice-cold HBSS containing 1% FBS, cells were incubated with the primary anti-pancadherin antibody (Ab 6529, Ab Cam, Cambridge, MA) for 30 min on ice. The primary antibody was coupled to phycoerythrin-conjugated antirabbit antibody (Ab 7007). After each step, cells were washed at least twice with ice-cold PBS containing 2% FBS. Flow cytometry was performed on a FACScan cell analyzer (Becton Dickinson, Franklin Lakes, NJ). For each cell line, three measurements were recorded from three independent experiments.
Cell aggregate preparation
Confluent cultures grown on 75 cm2 TC dishes were washed twice with Hanks' balanced salt solution (HBSS) containing 2 mM CaCl2, then treated for 10 min with 0.1% trypsin (GIBCO/BRL). Depleted cells were centrifuged at 905 × g for 3 min. The resulting pellet was transferred into capillary micropipettes of 500 μm diameter. Following a 15-min incubation in medium containing 2 mM CaCl2 at 37°C with 5% CO2, the firm cylinders of cells extruded from the pipettes were cut into 500-μm fragments. For invasion assays, the cylindrical aggregates were immediately embedded into collagen gel. For scanning electron microscopy and cohesivity studies, spheroidal aggregates were used. These were obtained from the cylindrical aggregates, which, on further incubation in 10-ml tissue culture flasks (Bellco Glass, Vineland, NJ) with 3 ml DMEM on a gyratory shaker at 120 rpm with 5% CO2 at 37°C, in most cases reproducibly rounded into spheroids in 24–36 h.
Scanning electron microscopy of aggregates' surface
Cell morphology on the surface of aggregates was analyzed by field emission scanning electron microscopy (FESEM). Spherical aggregates were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) in PBS on a low-speed shaker. Subsequently, samples were rinsed three times in PBS. Dehydration was performed by an increasing concentration series of ethanol as follows: 10%, 25%, 50%, 75%, and 95% for 30 min each and finally in 100% ethanol overnight. After critical point drying with CO2 in a Samdri-PVT-3B (Tousimis, Rockville, MD), aggregates were mounted on a holder with double-sided carbon tape and sputter coated with platinum to a nominal thickness of 2 nm. Aggregates were examined with a Hitachi S4700 cold-cathode field-emission scanning electron microscope at 5 kV accelerating voltage.
Measurement of tissue surface tension
The compression apparatus used in this work to measure liquid tissue properties is shown in Fig. 1. Modified from previously used similar devices (36), it is monitored by Labview software (National Instruments, Austin, TX) to record the entire force relaxation following uniaxial compression of a spherical aggregate between the compression plates. To minimize adhesion to the plates, these were coated with poly (2-hydroxyethylmethacrylate) (polyHEMA) (37). A typical measurement was performed as follows. The initially spheroidal aggregate was placed on the lower plate of the apparatus in CO2-independent medium (GIBCO/BRL) with antibiotics (as described above) and rapidly compressed with the help of a stepping motor to produce a deformation of a definite magnitude. To avoid irreversible damage to cells, aggregates were compressed to a maximum of 30% of their original diameter. The relaxation process was followed until the compressive force reached a constant equilibrium value (45–60 min), at which point the plates were separated, and the aggregate left to regain its original shape. Measurements in the rare cases when the aggregate did not regain its precompressed shape were discarded. The shape of the aggregate before, during, and after compression was recorded by a Spot Insight CCD camera (Diagnostic Instruments, Sterling Heights, MI) fitted to a horizontally positioned dissecting microscope (SZ60, Olympus). The surface tension of the model tissue was evaluated using the Laplace equation, 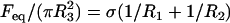 . Here,
. Here, 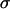 is the tissue's apparent surface tension (i.e., interfacial tension with the surrounding tissue culture medium),
is the tissue's apparent surface tension (i.e., interfacial tension with the surrounding tissue culture medium), 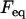 is the equilibrium value of the compressive force,
is the equilibrium value of the compressive force, 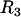 is the radius of the circular contact area of the compressed aggregate with the plates.
is the radius of the circular contact area of the compressed aggregate with the plates. 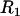 and
and 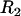 are the radii of curvature of the aggregate's surface, respectively along its equatorial plane, and its peripheral contour, which is assumed to be circular (see inset in Fig. 1). The geometric parameters were determined by an in-house built tracking program with a precision of 3 μm. The program evaluates the aggregate's recorded contour on the basis of variation in gray-scale values in its vicinity. To check tissue liquidity (i.e., the independence of
are the radii of curvature of the aggregate's surface, respectively along its equatorial plane, and its peripheral contour, which is assumed to be circular (see inset in Fig. 1). The geometric parameters were determined by an in-house built tracking program with a precision of 3 μm. The program evaluates the aggregate's recorded contour on the basis of variation in gray-scale values in its vicinity. To check tissue liquidity (i.e., the independence of 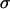 on the compressive force), aggregates were compressed two or three times with varying force with 60- min recovery in the uncompressed state. Average
on the compressive force), aggregates were compressed two or three times with varying force with 60- min recovery in the uncompressed state. Average 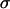 values were calculated from at least six independent measurements.
values were calculated from at least six independent measurements.
FIGURE 1.

Compression apparatus for surface tension measurement. The aggregate (A) is positioned on the lower compression plate (LCP) in the inner chamber (IC) filled with CO2-independent medium containing 2% FBS and the antibiotics mentioned above. The outer chamber is connected to a circulating water pump, which maintains the temperature of the system at 37°C. The upper compression plate (UCP) is suspended from the arm of a Cahn/Ventron (M2000, Cerritos, CA) recording electrobalance (B) through a nickel-chromium wire (NCW). A stepper motor (M) mounted on the lower assembly (LA) raises or lowers the LCP as needed. The balance records the apparent weight of the UCP and works on the null principle: when the LCP is raised and compresses the aggregate against the UCP, the latter is maintained at its initial position by the balance's electromagnetic feedback mechanism. The inset shows the geometric parameters of a compressed aggregate used for the evaluation of the surface tension (see text).
Invasion assay
Rat-tail collagen type I (Sigma-Aldrich) was dissolved in 1 M acetic acid, and the pH was adjusted to 7.0 by 2 M NaOH and HEPES buffer solution (GIBCO/BRL). The solution was further diluted with regular culture medium. An invasion assay was performed as follows. A first layer of collagen gel was prepared by spreading 150 μl of the collagen solution into a small square layer at the bottom of a 10 cm2 Petri dish. To initiate gelation, the dish was kept 10 min at room temperature and then in the incubator at 37°C for 10–15 min. Subsequently, the dish was stored in a cold room until use. Before an experiment, 125 μl of collagen solution was spread on the top of the first layer, and four to six aggregates were placed into this second layer. After 10 min at room temperature, the dish was transferred to the incubator. On completion of gelation in the second layer, 2.7 ml of medium was added to the preparation. Samples were subsequently kept at 37°C and 5% CO2. The medium was changed every other day. The invasion and pattern formation in time were followed by phase-contrast and dark-field microscopy for up to 7 days. Images were taken at 24-h intervals on an Olympus IX70 microscope by a Nikon Coolpix 5000 digital camera. All experiments were repeated independently.
Quantitative real-time PCR
Total RNA was isolated from confluent cell cultures with TriZOL (Invitrogen, Carlsbad, CA), following manufacturer's instructions. Purity and final concentrations of mRNA were determined by spectrophotometric analysis at A280 and A260. Complementary DNAs were constructed from 1 μg of total RNA from each sample using the Roche Diagnostics First Strand Synthesis Kit (Indianapolis, IN) per manufacturer's instructions. Q-RT-PCR was performed with an ABI 7700 system (Applied Biosystems, Foster City, CA). Gene-specific TaqMan Gene Expression Assays (ABI) with manufacturer designed and validated probe and primer sets and universal cycling conditions were used for quantitative gene expression studies of human MMPs, TIMP, and the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase. The PCR reaction mixtures included TaqMan Universal PCR Master Mix, 0.1 mM of each gene-specific primer and 1.25 units of Taq DNA polymerase. The primer sets did not work with normal rat astrocytes; thus, these cells were not included in the PCR study.
RESULTS
Cadherin expression correlates with the ability to aggregate
The N-cadherin expression level of the various cell lines was assessed by Western blotting (Fig. 2 A). This analysis revealed that the nine lines formed three well-separated groups with low (GBM3, GBM4, U87), intermediate (GBM1, ENB, GBM2), and high (EM1, EM2, U373) N-cadherin expression. Fig. 2 A shows two cell lines from each group. Because the type of cadherins might be altered in tumor cells, the expression level was also estimated by flow cytometry following immunolabeling with a pancadherin antibody targeting the conservative, intracellular part of the protein (Fig. 2 B). Comparison of Fig. 2 A and B shows that the predominant cadherin in the investigated cell lines is N-cadherin. The appropriate cell membrane localization of cadherins was verified by selective immunofluorescence microscopy using anti-N-cadherin antibody (data not shown). The highest cadherin expressers (+++, column 6 in Table 1) formed large clumps (>100 cells; ++, column 5) after 4 h of incubation in a gyratory shaker. Intermediate expressers (++, column 6) formed smaller clumps (+, column 5), whereas cell lines with the lowest expression (+, column 6) showed limited aggregation in the shaker assay after 4 or even 10 h of incubation (dash, column 5). These were not capable of reproducibly forming stable aggregates suitable for further investigations.
FIGURE 2.

(A) N-cadherin expression of the brain tumor cell lines was assessed by Western blotting. (A very long exposure of the gel led to a light band at the U87 and GBM4 column, an indication of very low expression.) (B) Overall cadherin expression was also estimated by flow cytometry using pancadherin immunolabeling. The horizontal axis corresponds to the measured intensity of phycoerythrin (FL2-H), which is the detector of the used fluorochrome (emitting at 575 nm). (C) Dependence of apparent surface tension on cadherin expression in six brain tumor cell lines and rat primary astrocytes. Note that the surface tension of the normal (rat) astrocyte aggregate is smaller than some of the tumor aggregates (we had no access to normal human astrocytes). However, in light of the different origin of these cells (i.e., human versus rat), these surface tensions should not be directly compared (see also Fig. 4). Cadherin expression levels assessed by flow cytometry were normalized by the cell surface. Error bars represent standard errors.
Tissue cohesion correlates with cadherin expression
Spherical aggregates of the seven cell types with relative intermediate or high cadherin levels were compressed between parallel plates using the apparatus shown in Fig. 1. The surface tension of these aggregates was determined as explained in the section Materials and Methods. Results in Table 1 (column 7) indicate that, for the tested cell lines, higher cadherin expression implies stronger aggregate cohesion. Note that aggregates composed of the high N-cadherin expressers are nearly as cohesive as aggregates of rat primary astrocytes. This suggests that the adhesion apparatus of these cells has not yet been substantially transformed. In the tested range, linear correlation (R2 = 0.95) was found between average surface tension and effective cadherin surface density (Fig. 2 C). (Effective cadherin surface density was calculated by normalizing the intensity of the measured flow cytometry fluorescence by the average cell surface area, which greatly varied among the cell lines studied.) Similar linearity was found by Foty and Steinberg (32).
Cell morphology is affected by cadherin expression
No apparent correlation between the morphology of 2D cell cultures and cadherin expression has been reported. We observed a dramatic variation in the surface morphology of 3D multicellular spheroids with cadherin expression. The three cell lines with the highest cadherin expression formed tight aggregates with smooth surfaces, similar to primary astrocytes. Cells on the surface were flat, displaying stretched cell membrane, akin to morphology in 2D cultures (Fig. 3, EM1). In many instances junctions between the cells were visible at higher magnifications (not shown). In contrast, aggregates of cells with intermediate cadherin expression showed a rougher surface with both flat and round cells (Fig. 3, GBM1). The surface of aggregates composed of the lower cadherin expressers (Fig. 3, ENB) was berry-like. Cells were mostly round and displayed a large number of membrane blebs and processes; adhesion foci were only occasionally visible.
FIGURE 3.

Surface morphology of spheroids. Scanning electron microscopy shows a great number of small membrane extensions on the round and spindle-shaped cells forming aggregates with low cohesion (ENB). A number of flat cells can be observed on the surface of aggregates with intermediate cohesion (GBM1) among many other less-stretched cells. The aggregates with high cohesion (EM1) have tightly connected, well-spread cells on their surface, which is thus smooth. Scale bars 3 μm.
Tissue cohesion influences the invasion of brain tumor cells in vitro
In order to biophysically characterize the invasive capacity of the various cell lines, cylindrical aggregates were prepared and embedded in type I collagen gel. The evolving patterns were monitored through an inverted microscope with the axis of the cylinder parallel to that of the microscope. Thus, the 2D patterns shown in Figs. 4 and 5 are representative of cell invasion along the entire height of the cylinder.
FIGURE 4.

Pattern of invasion into collagen is determined by tissue cohesion. Cylindrical aggregates (∼500 μm diameter) were placed into 1 mg/ml collagen and imaged at regular time intervals. Note that normal astrocytes practically do not invade the collagen matrix.
FIGURE 5.

Collagen concentration influences the invasion pattern. All images were taken after 96 h of incubation of cylindrical aggregates in the gel. (Migration of normal astrocytes into the gel, shown in Fig. 4, is insensitive to collagen concentration and thus is not displayed here.)
Control primary astrocytes showed very limited invasion of collagen (Fig. 4). Many of the cells in aggregates with high 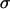 , even after 96 h, remained in relatively compact clumps (Fig. 4, EM1; Fig. 5, EM2 for collagen concentration ≥1 mg/ml; the figures contain at least one cell line from each of the low, intermediate, and high cadherin expresser groups). Those that invaded the matrix displayed characteristic spindle shape. Interestingly, the three cell lines not capable of reproducibly forming spherical aggregates and thus having the lowest
, even after 96 h, remained in relatively compact clumps (Fig. 4, EM1; Fig. 5, EM2 for collagen concentration ≥1 mg/ml; the figures contain at least one cell line from each of the low, intermediate, and high cadherin expresser groups). Those that invaded the matrix displayed characteristic spindle shape. Interestingly, the three cell lines not capable of reproducibly forming spherical aggregates and thus having the lowest 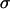 (Figs. 4 and 5, U87) showed global invasion patterns not much different from aggregates with high
(Figs. 4 and 5, U87) showed global invasion patterns not much different from aggregates with high 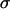 (Fig. 4, EM1; Fig. 5, EM2), although locally the latter appear more dispersed. Tumor cells in aggregates with intermediate
(Fig. 4, EM1; Fig. 5, EM2), although locally the latter appear more dispersed. Tumor cells in aggregates with intermediate 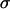 , showed peculiar organization. In every experiment they quickly migrated into the gel, forming ring-like structures with digested collagen and small compact clumps within the ring (Fig. 4, GBM1; Fig. 5, GMB1, GMB2). More cohesive aggregates occasionally showed similar behavior but only at low collagen concentration (Fig. 5, EM2). The migration rate of cells in the intermediate-cohesivity group was considerably higher than that of those in the low- and high-cohesivity groups (Fig. 6 A). The number and displacement of migrating cells typically decreased with increasing collagen concentration. The thicker gel also slowed ring formation (Figs. 5 and 6 B). Collagen concentration had little effect on the dispersion of cells with the lowest cadherin expression (U87). These results imply that no strict correlation exists between the magnitude of
, showed peculiar organization. In every experiment they quickly migrated into the gel, forming ring-like structures with digested collagen and small compact clumps within the ring (Fig. 4, GBM1; Fig. 5, GMB1, GMB2). More cohesive aggregates occasionally showed similar behavior but only at low collagen concentration (Fig. 5, EM2). The migration rate of cells in the intermediate-cohesivity group was considerably higher than that of those in the low- and high-cohesivity groups (Fig. 6 A). The number and displacement of migrating cells typically decreased with increasing collagen concentration. The thicker gel also slowed ring formation (Figs. 5 and 6 B). Collagen concentration had little effect on the dispersion of cells with the lowest cadherin expression (U87). These results imply that no strict correlation exists between the magnitude of 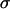 and the invasion pattern even though the latter does depend on the former.
and the invasion pattern even though the latter does depend on the former.
FIGURE 6.

(A) Time dependence of invasion for aggregates of different cohesivity values in 1 mg/ml collagen gel. The effective radius,  was determined at distinct time points as
was determined at distinct time points as 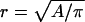 , where
, where 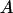 is the measured contiguous area covered by the cells (see Fig. 4). Migration rates, given in parentheses, were obtained from the slopes of linear fits. Data points represent averages over three to six independent experiments, and error bars represent standard deviations. (Where no error bars are seen, they were smaller than the linear size of the symbols denoting data points.) (B) The dependence of the migration rate on collagen concentration for the GBM1 cell line. Rates were determined as explained above. For all linear fits,
is the measured contiguous area covered by the cells (see Fig. 4). Migration rates, given in parentheses, were obtained from the slopes of linear fits. Data points represent averages over three to six independent experiments, and error bars represent standard deviations. (Where no error bars are seen, they were smaller than the linear size of the symbols denoting data points.) (B) The dependence of the migration rate on collagen concentration for the GBM1 cell line. Rates were determined as explained above. For all linear fits, 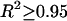 .
.
Cell-matrix interaction influences the invasion pattern of brain tumor cells in vitro
In order to quantitatively characterize matrix remodeling in the course of invasion by brain tumor cells, we first considered their measured expression level of MMP-1, MMP-2, and MMP-9 together with TIMP-1 using quantitative real time (Q-R)-PCR. These three metalloproteinases (of the 24 so far reported (9)) were chosen because they all are needed for the complete degradation of collagen I (8). Collagenase (MMP-1) cleaves fibrillar collagen, whereas gelatinases (MMP-2 and MMP-9) digest the denatured collagen fragments. We considered here only TIMP-1 for the following reason. It is the most multifunctional of the four known TIMPs with diverse functions (10). It is the direct inhibitor of MMP-1 and most probably of MMP-9. Because it controls TIMP-2 expression, it also acts as inhibitor of MMP-2 (10). (Note the almost perfect correlation between MMP-2 and TIMP-1 expression in Fig. 7.)
FIGURE 7.

Collagen invasion is dependent on MMP expression. The bar graphs show the relative copy numbers of MMP and TIMP mRNAs, as quantified by Q-RT-PCR (relative to the mRNA of a standard house-keeping gene). Patterns on the top correspond to 96 h of migration in 1 mg/ml collagen.
As results in Fig. 7 show, MMPs influence the invasive capacity of all the cell lines studied. However, similarly to tissue cohesivity, no strict correlation between invasion pattern and MMP expression can be established. For example, even though the weakly adhesive U87 cells have significantly lower MMP-1 levels than the strongly adhesive EM2 cells, their invasion patterns are globally rather similar. (Note that their TIMP-1 expression is also quite similar.) High MMP expression seems to be typical for ring-forming cells (in particular GBM1 and GBM2 in Fig. 7), which form aggregates with intermediate cohesion. Curiously, their ability to digest collagen is not affected by their relatively high TIMP-1 level. As discussed below, a consistent interpretation of the observed invasion patterns can be given by simultaneously considering both tissue cohesivity and cell-matrix interactions.
DISCUSSION
The spreading of tumors implies that invading cells go through epithelial-mesenchymal transition. Such transitions are driven by the competition between tissue cohesion and affinity to the extracellular matrix. These factors and their modifications in the course of the disease are under genetic control. In particular, it was found that the mRNA expression profiles of glioblastoma cells residing at the tumor core (and thus exposed to predominantly cell adhesive forces) and invasive rim (exposed also to the surrounding matrix) are different (38). However, migration involves movement, which is controlled by global cellular physical quantities (e.g., velocity). The interplay between molecular and physical mechanisms in the spreading of tumors is vital to the understanding of how cancer progresses.
To gain insight into this interplay, we employed a large number of human brain tumor cell lines to quantitatively correlate their biophysical and molecular properties with their invasive characteristics in 3D collagen I matrices. Tissue cohesion was assessed through the measurement of apparent tissue surface tension (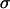 ), using multicellular spherical aggregates (the analog of liquid drops; Fig. 2 C). Cell-matrix interaction was studied by quantifying the expression level of the relevant matrix metalloproteinases and their inhibitor (Fig. 7).
), using multicellular spherical aggregates (the analog of liquid drops; Fig. 2 C). Cell-matrix interaction was studied by quantifying the expression level of the relevant matrix metalloproteinases and their inhibitor (Fig. 7).
The cohesivity of a tissue is determined by all the adhesive mechanisms acting between its constituent cells (not only that related to cadherins). Nevertheless, we found linearity between 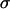 and effective N-cadherin surface density (
and effective N-cadherin surface density (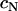 ) over an almost order-of-magnitude variation in the former quantity. (Winters et al. (29) measured
) over an almost order-of-magnitude variation in the former quantity. (Winters et al. (29) measured 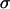 for three glioblastoma cell lines including U87, for which they reported a value of 7 dyne/cm, lower than any of our values listed in Table 1. Together with our flow cytometry results, U87 would still fall on the curve in Fig. 2 C.) The observed linearity, however, must eventually cease with decreasing
for three glioblastoma cell lines including U87, for which they reported a value of 7 dyne/cm, lower than any of our values listed in Table 1. Together with our flow cytometry results, U87 would still fall on the curve in Fig. 2 C.) The observed linearity, however, must eventually cease with decreasing 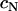 because the true function
because the true function 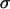 (
(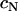 ) must obey the condition
) must obey the condition 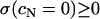 (the curve in Fig. 2 C would cross the vertical axis at negative values). Linearity, as shown in Fig. 2 for larger
(the curve in Fig. 2 C would cross the vertical axis at negative values). Linearity, as shown in Fig. 2 for larger 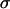 values, suggests that at higher N-cadherin expression it is this molecule that dominates the cohesion of the studied brain tissues, whereas at low
values, suggests that at higher N-cadherin expression it is this molecule that dominates the cohesion of the studied brain tissues, whereas at low 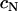 , other adhesive mechanisms take over or become as important. When results on tissue cohesion alone were compared with the outcome of invasion assays (Figs. 4 and 5), no strict correlation between N-cadherin expression and the spreading patterns could be established.
, other adhesive mechanisms take over or become as important. When results on tissue cohesion alone were compared with the outcome of invasion assays (Figs. 4 and 5), no strict correlation between N-cadherin expression and the spreading patterns could be established.
Spreading tumor cells are able to degrade their surrounding matrix. Thus, to characterize cell-matrix interactions, we concentrated on those metalloproteinases in the studied cell lines that are most relevant for the degradation of collagen I (i.e., MMP-1, MMP-2, and MMP-9) as well as on the most ubiquitous inhibitor of these enzymes (TIMP-1). Our results (Fig. 7) indicate that similarly to tissue cohesion, MMP expression alone does not explain the observed invasion patterns. However, a consistent picture occurs when tissue cohesion (Fig. 2 C) and cell-matrix interaction (Fig. 7) are simultaneously considered.
Fig. 7 reveals a striking interplay of these two decisive factors of invasive potential. Cells with either a low MMP pool and relatively low cohesivity (and thus small 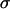 ; the three leftmost cell lines, not shown in Fig. 2 C) or strongest cohesivity (the three rightmost cell lines) have similarly limited migratory capacity. (The effect of relatively high MMP-2 expression of the U87 cells seems to be abolished by their equally high TIMP-1 expression.) Cell lines with intermediate cohesion and relatively high MMP expression (the middle three) exhibit complex migratory patterns as a consequence of competitive interactions. (Note that the initial cylindrical aggregates used in the invasion assays were all of same size.) Cells of these tissues simultaneously try to maximize their contact to each other (mostly through their N-cadherins) and to degrade the matrix (through their MMPs). As a consequence of this competition (i.e., between cell-cell adhesion and matrix proteolytic activity), as a compromise, they display ringlike patterns. Within the ring, cells benefit from their adhesion. At the same time, because the interior of the ring is devoid of collagen, they also benefit from their MMPs. (Note that these ring patterns cannot be explained by the differential gene expression of rim and core cells mentioned above (38) because the latter do not exist.) The appearance of these rings also depends on the concentration of matrix molecules (Fig. 5) and thus on the gel's mechanical properties (39). Similar structures, termed pseudopalisades (40,41), have been reported earlier in surgically excised tumors but, to our knowledge, not in vitro.
; the three leftmost cell lines, not shown in Fig. 2 C) or strongest cohesivity (the three rightmost cell lines) have similarly limited migratory capacity. (The effect of relatively high MMP-2 expression of the U87 cells seems to be abolished by their equally high TIMP-1 expression.) Cell lines with intermediate cohesion and relatively high MMP expression (the middle three) exhibit complex migratory patterns as a consequence of competitive interactions. (Note that the initial cylindrical aggregates used in the invasion assays were all of same size.) Cells of these tissues simultaneously try to maximize their contact to each other (mostly through their N-cadherins) and to degrade the matrix (through their MMPs). As a consequence of this competition (i.e., between cell-cell adhesion and matrix proteolytic activity), as a compromise, they display ringlike patterns. Within the ring, cells benefit from their adhesion. At the same time, because the interior of the ring is devoid of collagen, they also benefit from their MMPs. (Note that these ring patterns cannot be explained by the differential gene expression of rim and core cells mentioned above (38) because the latter do not exist.) The appearance of these rings also depends on the concentration of matrix molecules (Fig. 5) and thus on the gel's mechanical properties (39). Similar structures, termed pseudopalisades (40,41), have been reported earlier in surgically excised tumors but, to our knowledge, not in vitro.
How a migratory cell moves through an extracellular matrix (ECM) typically depends on integrin-ECM ligand (e.g., collagen, fibronectin) interactions (42). In particular, for nonmalignant cells it was shown that, on two-dimensional ECM substrates, the rate of migration or speed displays a biphasic dependence on ligand concentration (43): at low ligand concentration speed increases, whereas at high ligand concentration speed decreases. Intermediate ligand concentration results in the greatest speed. These findings are consistent with the mechanism of normal cell migration, which is driven by the traction forces such cells exert on the matrix. The magnitude of the traction force increases with the amount of available ligand. At high ligand concentration, however, movement through the dense network of ECM macromolecules hinders motion.
In the case of malignant invasion, besides integrin-ECM ligand binding, cell-matrix interaction also involves matrix degradation through MMPs. Which of these two effects dominates the invasion pattern depends on cell type. Although we have not considered the role of integrins, we believe our data are consistent with the MMP activity being the more dominant in glioblastomas. We found that the higher the expression level of MMPs, the faster is invasion (Fig. 6 A), whereas the denser the matrix, the slower is invasion (because there is more material to degrade), and no biphasic dependence of the speed of migration on ligand concentration is observed (Fig. 6 B). (In transformed NIH3T3 cells, it was found that overexpression of the β4 integrin subunit increases invasive capacity (44)).
In summary, our results strongly suggest that despite the complex molecular nature of brain tumor spreading, the emerging global cellular patterns can consistently be characterized in terms of two competing effects: cell-cell and cell-matrix interaction. Our findings also suggest specific ways to characterize, control, or engineer cell migratory patterns by the tuning of these interactions. Competitive interactions are known to drive phase transitions in physical systems (e.g., liquid-gas transition). Here they seem to be responsible for the characteristic cell configurations that accompany the epithelial-mesenchymal-like transition during which brain tumor cells change their phenotype from the more adhesive to the more migratory.
Acknowledgments
The authors are grateful to Lynn Matrisian for critical remarks. They acknowledge the help of Ilona Fazekas and Edit Kerekes in establishing the various tumor cell cultures.
This study was partially supported by grants from the National Aeronautics and Space Administration and the National Science Foundation.
Balázs Hegedüs' present address is Dept. of Neurology, Washington University School of Medicine, St. Louis, MO 63110.
References
- 1.Cavallaro, U., and G. Christofori. 2001. Cell adhesion in tumor invasion and metastasis: loss of the glue is not enough. Biochim. Biophys. Acta. 1552:39–45. [DOI] [PubMed] [Google Scholar]
- 2.Korff, T., and H. G. Augustin. 1999. Tensional forces in fibrillar extracellular matrices control directional capillary sprouting. J. Cell Sci. 112:3249–3258. [DOI] [PubMed] [Google Scholar]
- 3.Meshel, A. S., Q. Wei, R. S. Adelstein, and M. P. Sheetz. 2005. Basic mechanism of three-dimensional collagen fibre transport by fibroblasts. Nat. Cell Biol. 7:157–164. [DOI] [PubMed] [Google Scholar]
- 4.Sawhney, R. K., and J. Howard. 2002. Slow local movements of collagen fibers by fibroblasts drive the rapid global self-organization of collagen gels. J. Cell Biol. 157:1083–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon, V. D., M. T. Valentine, M. L. Gardel, D. Andor-Ardo, S. Dennison, A. A. Bogdanov, D. A. Weitz, and T. S. Deisboeck. 2003. Measuring the mechanical stress induced by an expanding multicellular tumor system: a case study. Exp. Cell Res. 289:58–66. [DOI] [PubMed] [Google Scholar]
- 6.Kaufman, L. J., C. P. Brangwynne, K. E. Kasza, E. Filippidi, V. D. Gordon, T. S. Deisboeck, and D. A. Weitz. 2005. Glioma expansion in collagen I matrices: analyzing collagen concentration-dependent growth and motility patterns. Biophys. J. 89:635–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakraborti, S., M. Mandal, S. Das, A. Mandal, and T. Chakraborti. 2003. Regulation of matrix metalloproteinases: an overview. Mol. Cell. Biochem. 253:269–285. [DOI] [PubMed] [Google Scholar]
- 8.Curry, T. E., and K. G. Osteen. 2004. The matrix metalloproteinase system: changes, regulation, and impact throughout the ovarian and uterine reproductive cycle. Endocr. Rev. 24:428–465. [DOI] [PubMed] [Google Scholar]
- 9.Visse, R., and H. Nagase. 2003. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 92:827–839. [DOI] [PubMed] [Google Scholar]
- 10.Gardner, J., and A. Ghorpade. 2003. Tissue inhibitor of metalloproteinase (TIMP)-1: the TIMPed balance of matrix metalloproteinases in the central nervous system. J. Neurosci. Res. 74:801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gumbiner, B. M. 2000. Regulation of cadherin adhesive activity. J. Cell Biol. 148:399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeichi, M., S. Nakagawa, S. Aono, T. Usui, and T. Uemura. 2000. Patterning of cell assemblies regulated by adhesion receptors of the cadherin superfamily. Philos. Trans. R. Soc. Lond. B Biol. Sci. 355:885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Derycke, L. D., and M. E. Bracke. 2004. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int. J. Dev. Biol. 48:463–476. [DOI] [PubMed] [Google Scholar]
- 14.Puch, S., S. Armeanu, C. Kibler, K. R. Johnson, C. A. Muller, M. J. Wheelock, and G. Klein. 2001. N-cadherin is developmentally regulated and functionally involved in early hematopoietic cell differentiation. J. Cell Sci. 114:1567–1577. [DOI] [PubMed] [Google Scholar]
- 15.Huang, Z. Y., Y. Wu, N. Hedrick, and D. H. Gutmann. 2003. T-cadherin-mediated cell growth regulation involves G2 phase arrest and requires p21(CIP1/WAF1) expression. Mol. Cell. Biol. 23:566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeuchi, T., A. Misaki, S. B. Liang, A. Tachibana, N. Hayashi, H. Sonobe, and Y. Ohtsuki. 2000. Expression of T-cadherin (CDH13, H-cadherin) in human brain and its characteristics as a negative growth regulator of epidermal growth factor in neuroblastoma cells. J. Neurochem. 74:1489–1497. [DOI] [PubMed] [Google Scholar]
- 17.Schwechheimer, K., L. Zhou, and W. Birchmeier. 1998. E-cadherin in human brain tumours: loss of immunoreactivity in malignant meningiomas. Virchows Arch. 432:163–167. [DOI] [PubMed] [Google Scholar]
- 18.Tomita, K., A. van Bokhoven, G. J. van Leenders, E. T. Ruijter, C. F. Jansen, M. J. Bussemakers, and J. A. Schalken. 2000. Cadherin switching in human prostate cancer progression. Cancer Res. 60:3650–3654. [PubMed] [Google Scholar]
- 19.Hazan, R. B., L. Kang, B. P. Whooley, and P. I. Borgen. 1997. N-cadherin promotes adhesion between invasive breast cancer cells and the stroma. Cell Adhes. Commun. 4:399–411. [DOI] [PubMed] [Google Scholar]
- 20.Utsuki, S., Y. Sato, H. Oka, B. Tsuchiya, S. Suzuki, and K. Fujii. 2002. Relationship between the expression of E-, N-cadherins and beta-catenin and tumor grade in astrocytomas. J. Neurooncol. 57:187–192. [DOI] [PubMed] [Google Scholar]
- 21.Shinoura, N., N. E. Paradies, R. E. Warnick, H. Chen, J. J. Larson, J. J. Tew, M. Simon, R. A. Lynch, Y. Kanai, S. Hirohashi, et al. 1995. Expression of N-cadherin and alpha-catenin in astrocytomas and glioblastomas. Br. J. Cancer. 72:627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asano, K., C. D. Duntsch, Q. Zhou, J. D. Weimar, D. Bordelon, J. H. Robertson, and T. Pourmotabbed. 2004. Correlation of N-cadherin expression in high grade gliomas with tissue invasion. J. Neurooncol. 70:3–15. [DOI] [PubMed] [Google Scholar]
- 23.Asano, K., O. Kubo, Y. Tajika, K. Takakura, and S. Suzuki. 2000. Expression of cadherin and CSF dissemination in malignant astrocytic tumors. Neurosurg. Rev. 23:39–44. [DOI] [PubMed] [Google Scholar]
- 24.Bissell, M. J., and D. Radisky. 2001. Putting tumours in context. Nat. Rev. Cancer. 1:46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kokenyesi, R., K. P. Murray, A. Benshushan, E. D. Huntley, and M. S. Kao. 2003. Invasion of interstitial matrix by a novel cell line from primary peritoneal carcinosarcoma, and by established ovarian carcinoma cell lines: role of cell-matrix adhesion molecules, proteinases, and E-cadherin expression. Gynecol. Oncol. 89:60–72. [DOI] [PubMed] [Google Scholar]
- 26.Bauman, G. S., B. J. Fisher, W. McDonald, V. R. Amberger, E. Moore, and R. F. Del Maestro. 1999. Effects of radiation on a three-dimensional model of malignant glioma invasion. Int. J. Dev. Neurosci. 17:643–651. [DOI] [PubMed] [Google Scholar]
- 27.Del Duca, D., T. Werbowetski, and R. F. Del Maestro. 2004. Spheroid preparation from hanging drops: characterization of a model of brain tumor invasion. J. Neurooncol. 67:295–303. [DOI] [PubMed] [Google Scholar]
- 28.Nygaard, S. J., P. H. Pedersen, T. Mikkelsen, A. J. Terzis, O. B. Tysnes, and R. Bjerkvig. 1995. Glioma cell invasion visualized by scanning confocal laser microscopy in an in vitro co-culture system. Invasion Metastasis. 15:179–188. [PubMed] [Google Scholar]
- 29.Winters, B. S., S. R. Shepard, and R. A. Foty. 2005. Biophysical measurement of brain tumor cohesion. Int. J. Cancer. 114:371–379. [DOI] [PubMed] [Google Scholar]
- 30.Steinberg, M. S. 1963. Reconstruction of tissues by dissociated cells. Some morphogenetic tissue movements and the sorting out of embryonic cells may have a common explanation. Science. 141:401–408. [DOI] [PubMed] [Google Scholar]
- 31.Steinberg, M. S., and T. J. Poole. 1982. Liquid behavior of embryonic tissues. In Cell Behaviour. R. Bellains, A. S. G. Curtis, and G. Dunn, editors. Cambridge University Press, Cambridge. 583–697.
- 32.Foty, R. A., and M. S. Steinberg. 2005. The differential adhesion hypothesis: a direct evaluation. Dev. Biol. 278:255–263. [DOI] [PubMed] [Google Scholar]
- 33.Hegedüs, B., A. Czirok, I. Fazekas, T. B'Abel, E. Madarász, and T. Vicsek. 2000. Locomotion and proliferation of glioblastoma cells in vitro: statistical evaluation of videomicroscopic observations. J. Neurosurg. 92:428–434. [DOI] [PubMed] [Google Scholar]
- 34.Hegedüs, B., J. Zach, A. Czirok, J. Lovey, and T. Vicsek. 2004. Irradiation and Taxol treatment result in non-monotonous, dose-dependent changes in the motility of glioblastoma cells. J. Neurooncol. 67:147–157. [DOI] [PubMed] [Google Scholar]
- 35.Xu, J., Y. I. Weng, A. Simonyi, B. W. Krugh, Z. Liao, G. A. Weisman, and G. Y. Sun. 2002. Role of PKC and MAPK in cytosolic PLA2 phosphorylation and arachadonic acid release in primary murine astrocytes. J. Neurochem. 83:259–270. [DOI] [PubMed] [Google Scholar]
- 36.Foty, R. A., C. M. Pfleger, G. Forgacs, and M. S. Steinberg. 1996. Surface tensions of embryonic tissues predict their mutual envelopment behavior. Development. 122:1611–1620. [DOI] [PubMed] [Google Scholar]
- 37.Folkman, J., and A. Moscona. 1978. Role of cell shape in growth control. Nature. 273:345–349. [DOI] [PubMed] [Google Scholar]
- 38.Mariani, L., W. S. McDonough, D. B. Hoelzinger, C. Beaudry, E. Kaczmarek, S. W. Coons, A. Giese, M. Moghaddam, R. W. Seiler, and M. E. Berens. 2001. Identification and validation of P311 as a glioblastoma invasion gene using laser capture microdissection. Cancer Res. 61:4190–4196. [PubMed] [Google Scholar]
- 39.Roeder, B. A., K. Kokini, J. E. Sturgis, J. P. Robinson, and S. L. Voytik-Harbin. 2002. Tensile mechanical properties of three-dimensional type I collagen extracellular matrices with varied microstructure. Trans. ASME. 124:214–222. [DOI] [PubMed] [Google Scholar]
- 40.Brat, D. J., A. A. Castellano-Sanchez, S. B. Hunter, M. Pecot, C. Cohen, E. H. Hammond, S. N. Devi, B. Kaur, and E .G. Van Meir. 2004. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 64:920–927. [DOI] [PubMed] [Google Scholar]
- 41.Brat, D. J., and E. G. Van Meir. 2004. Vaso-occlusive and prothrombotic mechanisms associated with tumor hypoxia, necrosis, and accelerated growth in glioblastoma. Lab. Invest. 84:397–405. [DOI] [PubMed] [Google Scholar]
- 42.Hynes, R. O. 2004. The emergence of integrins: a personal and historical perspective. Matrix Biol. 23:333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palecek, S. P., J. C. Loftus, M. H. Ginsberg, D. A. Lauffenburger, and A. F. Horwitz. 1997. Integrin-ligand binding properties govern cell migration speed thorugh cell-substratum adhesivness. Nature. 385:537–540. [DOI] [PubMed] [Google Scholar]
- 44.Bon, G., V. Folgiero, G. Bossi, L. Felicioni, A. Marchetti, A. Sacchi, and R. Falcioni. 2006. Loss of beta4 integrin subunit reduces the tumorigenicity of MCF7 mammary cells and causes apoptosis upon hormone deprivation. Clin. Cancer Res. 12:3280–3287. [DOI] [PubMed] [Google Scholar]