

Abstract
We report a computational study of the small peptide Met-enkephalin based on the ECEPP/2 and ECEPP/3 force fields using the basin paving method. We have located a new global minimum when using the ECEPP/3 force field with peptide angles ω fixed at 180°. With this new result, we can conclude that the lowest energy configurations of Met-enkephalin predicted based on all four versions of ECEPP have a classic γ-turn centered at residue Gly3 and a β-turn at residues Gly3-Phe4. However, minor differences between the structures also exist.
INTRODUCTION
In computer simulations of protein folding, small peptides are often used as model systems for uncovering the physical properties of larger systems. From a computational perspective, small peptides are also ideal benchmarks for evaluating the efficiency of newly developed computational algorithms for protein folding. It is possible to thoroughly search the configuration space of a short peptide within a reasonable amount of computational time—such thoroughness is required to reveal the general physical properties of protein folding.
Met-enkephalin, first identified from the enkephalin mixture from brains (1), is one of the most used model peptides. It has a short residue sequence of Tyr1-Gly2-Gly3-Phe4-Met5. Met-enkephalin is an endogenous opioid pentapeptide found in brains, pituitary and peripheral tissues, and is involved in a variety of physiological processes. Experimental studies showed that this peptide exhibits several different structures in aqueous solutions (2).
In computer simulations of this peptide, especially in those adopting the ECEPP/2 (3,4) or ECEPP/3 (5) force fields, NH2 and COOH are often chosen as the N-terminus and C-terminus neutral groups, respectively. The pentapeptide consists of a total of 75 atoms described by 24 independent backbone and side-chain dihedral angles. Even such a small peptide gives rise to a complex conformational space, and the total number of local minima was estimated to be no less than 1011 (6). Because of the complexity in its configuration space and the short sequence allowing significant computational studies, Met-enkephalin has been extensively studied computationally (6–34), and has been regarded as a benchmark model used frequently for testing simulation methods (14,16–19,29–33) in recent years.
The lowest energies of Met-enkephalin without explicit solvation effects were previously determined based on the ECEPP/2 potential (6,10–12,14,15) and the ECEPP/3 potential (11,13). Local energy minima not much higher than global minima were sampled and classified by Freyberg and Braun (10) based on the ECEPP/2 potential, and by Eisenmenger and Hansmann (11) based on both ECEPP/2 and ECEPP/3 potentials (with the peptide dihedral angles ω fixed at 180°). Using the mutually orthogonal Latin-squares technique, Vengadesan and Gautham (21) performed the conformational studies of this peptide based on the ECEPP/3 potential, and compared the structures with those of Leu-enkephalin. Hansmann and co-workers studied the energy landscape (8), characteristic temperatures (35), and thermodynamics and kinetics (7) of folding of the peptide using the ECEPP/2 potential. Employing the reference interaction site model and various simulation methods, Kinoshita et al. (25,26) and Mitsutake et al. (27,28) studied Met-enkephalin in aqueous solutions. Berg and Hsu (29), using this peptide as a model protein, discussed various solvent-accessible area methods. Shen and Freed (30) performed a comparison study of explicit and implicit solvent effects by adopting this peptide as the model. Carlacci (20) analyzed the conformations of the peptide with the consideration of solvation and ionization. Using Met-enkephalin as a testbed, Zaman et al. (31) compared the peptide's dynamical properties implied by different force fields and suggested that a significant difference can be generated between the explicit-atom and united-atom potentials. Montcalm et al. (32) compared the differences between the global minimum structures of the peptide, obtained based on ECEPP/2, AMBER, and CHARMM force fields. Using the parallel tempering simulation method (36,37), Evans and Wales (9) analyzed the lowest energy conformations, the free energy landscape, and the dynamics of this peptide using the CHARMM potential. Simulations were also performed by Rathore et al. (19), by extending the Wang-Landau algorithm (38,39) to continuous systems, to determine the density of states. Berg (33) used this model protein to test the newly proposed funnel transformation method, which is suitable for global optimization. From another point of view, i.e., the helical contents of its structures, Sanbonmatsu and García (34) performed a structural study of Met-enkephalin in explicit aqueous solution.
The lowest energies previously found for Met-enkephalin, without the inclusion of solvation effects, are summarized in Table 1 for four versions of the ECEPP force field: ECEPP/2 and ECEPP/3, each with the peptide angles ω fixed or relaxed. Also included in the table are some new lowest energies obtained in this work. Their conformations and comparisons with previous studies will be discussed in Results and Discussions.
TABLE 1.
The lowest energies (in kcal/mol) obtained in previous studies of Met-enkephalin and in this work based on the ECEPP/2 and ECEPP/3 force fields
COMPUTATIONAL METHOD
In our study of Met-enkephalin, a recently proposed stochastic optimization algorithm, the basin paving (BP) method (16,40), was employed to search for the lowest energy configurations for all four cases of the force fields. This Monte Carlo (MC) method adopts the histogram accumulation strategy (38,39,41–45) of the energy-landscape paving (41) method as one of the components, and includes a critical revision to the acceptance criterion of lower energy movements. This revision ensures that during the computational search, a new low energy configuration is always examined seriously; in the meantime, the histogram-based method promotes the probability of surpassing higher energy barriers to escape local entrapments.
As another component in BP, to precisely locate the minima of a complex energy landscape in our MC simulation, the energy itself is replaced by the reduced energy landscape (14,46–49) as the target function. Within this strategy, the original energy function, E(r), where r is a 3N-dimensional variable with N the number of representative atoms in the system, is first transformed into a reduced form, 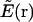 , by adopting the value of a “nearby” local energy minimum,
, by adopting the value of a “nearby” local energy minimum, 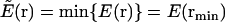 , where min {…} stands for a deterministic minimization procedure that pins down the “nearby” minimum with a high precision. In our simulations we used the quasi-Newton optimization approach (50).
, where min {…} stands for a deterministic minimization procedure that pins down the “nearby” minimum with a high precision. In our simulations we used the quasi-Newton optimization approach (50).
Computationally, at MC step t in BP, the MC transition probability between the existing configuration with a reduced energy 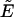 and an attempted configuration with
and an attempted configuration with 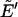 can be expressed as
can be expressed as
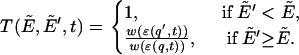 |
(1) |
In this transition probability, a Boltzmann weight
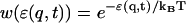 |
(2) |
is used, where kBT is the thermal energy that needs to be externally specified in a typical MC procedure. A deformed energy,
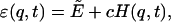 |
(3) |
is considered in the above weight, where q is a physical quantity for which the histogram H(q, t) is collected, and c is an adjustable free parameter used to optimize the search algorithm.
The method used in this article, basin paving, is an extension of the Monte Carlo with minimization (MCM) method (14), or the equivalent basin hopping (BH) method (46,47). The Boltzmann weight is used in MCM or BH, which would cause entrapment of the simulation procedure at a local minimum within limited computational time. Instead, in BP, we have introduced a generalized weighting scheme that overcomes this problem, in the same spirit as the multicanonical basin hopping method that we recently introduced (48,49). Mathematically, setting c = 0 in BP gives the same Boltzmann weight as used in MCM or BH. In comparison with multicanonical basin hopping, BP further ensures the acceptance of low energy conformations by the unconditional allowance of each low energy movement.
In these simulations, we took parameters  , c = 1 kcal/mol, and T = 50 K in Eqs. 2 and 3. To compute the potential energy E based on different versions of ECEPP, we used the program package SMMP (51). For the discussion of structures presented in the next section, we have used the definition of β- and γ-turn types as listed in Table 2. Further details of the definition can be found in Richardson (52), Rose et al. (53), Hutchinson and Thomton (54), Venkatachalam (55), Némethy and Printz (56), Némethy and Scheraga (57), and Milner-White et al. (58). To determine the peptide turns for conformational analysis, the program package PROMOTIF (54) has been employed. As implemented in PROMOTIF by default (54), when determining a β-turn, these φ- and ψ-angles are allowed to vary by 30°, with a more flexible tolerance of allowing one angle to deviate by 40°. However, for a γ-turn determination, the torsional angles are all allowed to vary by 40°.
, c = 1 kcal/mol, and T = 50 K in Eqs. 2 and 3. To compute the potential energy E based on different versions of ECEPP, we used the program package SMMP (51). For the discussion of structures presented in the next section, we have used the definition of β- and γ-turn types as listed in Table 2. Further details of the definition can be found in Richardson (52), Rose et al. (53), Hutchinson and Thomton (54), Venkatachalam (55), Némethy and Printz (56), Némethy and Scheraga (57), and Milner-White et al. (58). To determine the peptide turns for conformational analysis, the program package PROMOTIF (54) has been employed. As implemented in PROMOTIF by default (54), when determining a β-turn, these φ- and ψ-angles are allowed to vary by 30°, with a more flexible tolerance of allowing one angle to deviate by 40°. However, for a γ-turn determination, the torsional angles are all allowed to vary by 40°.
TABLE 2.
The classification of peptide turns
| Turns | φi+1 | ψi+1 | φi+2 | ψi+2 |
|---|---|---|---|---|
| β-turns | ||||
| Type I | −60 | −30 | −90 | 0 |
| Type I′ | 60 | 30 | 90 | 0 |
| Type II | −60 | 120 | 80 | 0 |
| Type II′ | 60 | −120 | −80 | 0 |
| Type III | −60 | −30 | −60 | −30 |
| Type III′ | 60 | 30 | 60 | 30 |
| Type VIa | −60 | 120 | −90 | 0 |
| Type VIb | −120 | 120 | −60 | 0 |
| Type IV | Turns excluded from all the above | |||
| γ-turns | ||||
| Classic | 75 | −64 | ||
| Inverse | −79 | 69 | ||
An ideal β-turn is formed by four residues labeled from i to (i + 3) with the two central residues having the dihedral angles listed in the table. An ideal γ-turn is formed by three residues labeled from i to (i + 2) with the dihedral angles of the (i + 1)st residue shown in the table. Details can be found in Richardson (52), Rose et al. (53), Hutchinson and Thomton (54), Venkatachalam (55), Némethy and Printz (56), Némethy and Scheraga (57), and Milner-White et al. (58).
RESULTS AND DISCUSSIONS
For all four versions of the force field, i.e., ECEPP/2 and ECEPP/3 with the peptide dihedral angles ω either fixed or relaxed, we list the lowest energies and their corresponding configurations obtained from BP search, in Table 3. In the table, the labels E/2 and E/3 denote the configurations obtained using ECEPP/2 and ECEPP/3, respectively. If the peptide dihedral angles ω are fixed at 180°, a subscript π is added to the labels. The graphical representation of the structures obtained is displayed in Fig. 1 from plots a–d, corresponding to the cases E/2π, E/2, E/3π, and E/3, respectively. Furthermore, we have explicitly drawn the hydrogen bonds by dashed lines, using the cutoff length 2.2 Å as the upper bound to identify the existence of a hydrogen bond.
TABLE 3.
Internal coordinates corresponding to lowest-energy minimum for Met-enkephalin, found in this work (columns 3–6) and previous work (last two columns)
| Torsion | E/2π | E/2 | E/3π | E/3 | 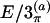 |
E/3(b) | |
|---|---|---|---|---|---|---|---|
| Tyr1 | χ1 | −179.8 | −172.6 | 59.9 | −173.2 | −174.2 | −173.2 |
| χ2 | 68.6 | −101.3 | 94.1 | −100.7 | −85.2 | −100.5 | |
| χ6 | −34.7 | 14.1 | −21.3 | 13.7 | 2.8 | 13.6 | |
| φ | −86.3 | −85.8 | 168.1 | −83.1 | −162.7 | −83.5 | |
| ψ | 153.7 | 156.2 | 0.9 | 155.8 | −41.7 | 155.8 | |
| ω | 180.0 | −176.9 | 180.0 | −177.1 | 180.0 | 177.2 | |
| Gly2 | φ | −161.5 | −154.5 | 126.8 | −154.2 | 65.8 | −154.3 |
| ψ | 71.1 | 83.7 | −21.2 | 85.8 | −87.0 | 86.0 | |
| ω | 180.0 | 168.6 | 180.0 | 168.5 | 180.0 | 168.5 | |
| Gly3 | φ | 64.1 | 83.7 | 83.7 | 83.0 | −157.3 | 83.0 |
| ψ | −93.5 | −73.9 | −61.6 | −75.0 | 34.9 | −75.1 | |
| ω | 180.0 | −170.1 | 180.0 | −170.0 | 180.0 | −169.9 | |
| Phe4 | χ1 | 179.8 | 58.8 | 58.6 | 58.9 | 52.4 | 58.8 |
| χ2 | −100.0 | −85.4 | 92.9 | −85.5 | −96.0 | −85.5 | |
| φ | −81.7 | −137.0 | −128.2 | −136.8 | −158.8 | −136.9 | |
| ψ | −29.2 | 19.3 | 18.8 | 19.1 | 159.5 | 19.1 | |
| ω | 180.0 | −174.1 | 180.0 | −174.1 | 180.0 | −174.1 | |
| Met5 | χ1 | −65.1 | 52.8 | 55.7 | 52.9 | −66.1 | 52.9 |
| χ2 | −179.2 | 175.3 | −178.6 | 175.3 | −179.6 | 175.3 | |
| χ3 | −179.3 | −179.8 | 177.0 | −179.9 | −179.9 | −179.9 | |
| χ4 | −179.9 | 61.4 | −179.3 | −178.6 | 60.1 | 61.4 | |
| φ | −80.7 | −163.6 | −162.1 | −163.4 | −82.4 | −163.5 | |
| ψ | 143.5 | 160.4 | 7.5 | 160.8 | 134.1 | 161.0 | |
| ω | 180.0 | −179.7 | 180.0 | −179.8 | 180.0 | −179.8 | |
| E (kcal/mol) | −10.72 | −12.91 | −10.90 | −12.43 | −10.85 | −11.71 |
In the columns labeled E/2 and E/3, the coordinates were obtained based on the ECEPP/2 and ECEPP/3 potentials, respectively. A subscript π indicates the fact that the peptide angles ω of the structure were fixed at 180° in the minimization process. The last two columns, labeled 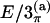 and E/3(b), contain the internal coordinates that can be found in Eisenmenger and Hansmann (11) and Androulakis et al. (13), respectively, for comparison.
and E/3(b), contain the internal coordinates that can be found in Eisenmenger and Hansmann (11) and Androulakis et al. (13), respectively, for comparison.
FIGURE 1.

The lowest energy structures of Met-enkephalin for different versions of ECEPP force fields: (a) ECEPP/2 with ω ≡ π; (b) ECEPP/2 with ω relaxed; (c) ECEPP/3 with ω ≡ π; (d) ECEPP/3 with ω relaxed; and (e) ECEPP/3 with ω ≡ π (found in (11)). The hydrogen bonds (dashed lines) are displayed in units of Å. The figures were generated using PyMol (59).
As can be seen in Tables 1 and 3, the lowest energies that we obtained for cases E/2 and E/2π matches previous results (6,10–12,14) exactly. The reproduced internal coordinates are listed in Table 3. According to the β-turn nomenclature defined in Table 2, the listed internal coordinates contains a Type-II′ β-turn for case E/2π, and a Type-IV β-turn for case E/2, both at Gly3-Phe4. For convenience, we denote them as Gly3-Phe4 Type-II′ and Gly3-Phe4 Type-IV β-turns, respectively. Both structures also contain a classic γ-turn centered at Gly2 according to the classification shown in Table 2.
The global energy minimum structures we have determined for the two cases E/2 and E/2π contain various hydrogen bonds. In both cases, a relatively strong hydrogen bond formed by NH of Gly2 and CO of Met5 is common. However, a hydrogen bond between CO of Gly2 and NH of Gly3 appears in the global energy minimum configuration of E/2, but not of E/2π. On the contrary, a hydrogen bond between CO of Gly2 and NH of Phe4 can be seen in case of E/2π, but not in E/2.
For E/2π, the folding landscape of the peptide, together with a determination of the energy minimum, was examined in Hansmann et al. (8); the lowest-energy structure has a similar configuration to that displayed in Fig. 1 a. That configuration contains two hydrogen bonds with bond length <2.2 Å. One is formed by the amino group (NH) of residue Gly2 and the carboxyl group (CO) of Met5, and the other is formed by CO of Gly2 and NH of Met5. Note that the structural determination described in Hansmann et al. (8) is based on KONF90, which contains a different convention for the ECEPP force field. As a direct result, the energy minimum found in this reference cannot be directly compared with those listed in Table 1. Furthermore, according to the classification shown in Table 2, the internal coordinates in Table 3 for E/2π contain a Gly3-Phe4 Type-IV β-turn. This can be contrasted with the conclusion in Hansmann et al. (8) where a Type-II β-turn was identified.
The internal coordinates of the lowest energy ECEPP/2 structure listed in Montcalm et al. (32) (which were obtained from (15) and further used for comparison with those determined from the AMBER and CHARMM force fields) are very close to those listed in Table 3. The internal coordinates in that reference suggested a Gly3-Phe4 Type-II′ β-turn, rather than a Gly3-Phe4 Type-IV β-turn obtained here based on Table 2.
When the ECEPP/3 potential with ω = 180° and with ω relaxed was used, we obtained energy minima with internal coordinates listed in the columns under E/3π and E/3 of Table 1, respectively. The conformations of the energy minima for the two cases are plotted in Fig. 1, c and d, respectively. Both structures contain a Gly3-Phe4 Type-IV β-turn and a Gly3-centered classic γ-turn. The later is also a common feature in the energy-minimum structures of E/2π and E/2.
From the torsional angles listed in Table 3, and the structures shown in Fig. 1, b and d, we conclude that the configurations of both force fields, the ECEPP/2 and ECEPP/3 potentials with ω relaxed, are very close to one another. Even the hydrogen bonds formed by these two structures are the same. However, a minor structural difference can be noticed between the energy minimum of E/3π (Fig. 1 c) and of the other three cases. The orientation of residues Tyr1, Gly2, and the C-terminus carboxyl group COOH are pointing to different directions. As a result, the hydrogen-bond formations are different. A hydrogen bond between the CO group of Gly2 and the NH group of Phe4 can be seen in structures Fig. 1, b and d; however, a different hydrogen bond between the CO group of Tyr1 and the added terminus OH group, appears in the structure in Fig. 1 c.
The energy minimum for E/3π, E = −10.90 kcal/mol, is ∼0.05 kcal/mol lower than the value previously reported in Eisenmenger and Hansmann (11). While the energy difference is not large, our new result exhibits distinctive differences in the minimum energy conformation. For convenience, we have also reproduced the internal coordinates of the later case, denoted as  , in Table 3, and the corresponding structure in Fig. 1 e. Visually, we can already see the difference between plots in Fig. 1, c and e. A more careful analysis of the internal coordinates reveals that structure Fig. 1 e contains a Gly2-Gly3 Type-IV β-turn and a Gly2-centered classic γ-turn, which can be contrasted with the Gly3-Phe4 Type-IV β-turn and the Gly3-centered classic γ-turn of the new structure found in this work. It is also interesting to note that the plot in Fig. 1 e contains hydrogen bonds between the CO group of Tyr1 and both the NH group of Gly3 and Phe4, which is very different from the hydrogen bonds in plots Fig. 1, a–d.
, in Table 3, and the corresponding structure in Fig. 1 e. Visually, we can already see the difference between plots in Fig. 1, c and e. A more careful analysis of the internal coordinates reveals that structure Fig. 1 e contains a Gly2-Gly3 Type-IV β-turn and a Gly2-centered classic γ-turn, which can be contrasted with the Gly3-Phe4 Type-IV β-turn and the Gly3-centered classic γ-turn of the new structure found in this work. It is also interesting to note that the plot in Fig. 1 e contains hydrogen bonds between the CO group of Tyr1 and both the NH group of Gly3 and Phe4, which is very different from the hydrogen bonds in plots Fig. 1, a–d.
In another comparison, the lowest-energy configuration obtained in this work for E/3 is similar to the one found in Androulakis et al. (13). We have listed their coordinates in the last column, labeled E/3(b), in Table 3. There is a difference between the minimal energies obtained from Androulakis et al. (13), E = −11.71 kcal/mol, compared to the present value of E = −12.43 kcal/mol. Our new value, however, is not the indication of a new energy minimum, but a reflection of minor difference in the versions of the force field used. In Androulakis et al. (13), two extra terms, the cysteine loop-closing term and the cysteine torsional term, were included in the potential energy. The conformation also contains a Gly3-Phe4 Type-IV β-turn and a Gly3-centered classic γ-turn.
SUMMARY
Using the new basin hopping global optimization method, we have determined the lowest energy minima of Met-enkephalin from four versions of the ECEPP force field, ECEPP/2 and ECEPP/3 with ω relaxed or fixed at 180°. We conclude that the lowest energy minimum configurations of ECEPP/2 and ECEPP/3, with ω relaxed, are virtually identical. With our newly discovered lowest energy minimum for ECEPP/3 with ω fixed at 180°, we also conclude that the minimum configuration of this case shares many common features with those of the other three versions of the force fields; this can be contrasted with an earlier conclusion that the global minimum configuration of this particular case, based on the exactly same version of the parameterization of the force field, is structurally different.
Acknowledgments
The authors are grateful to the financial support provided by the National Science and Engineering Research Council of Canada (NSERC). This work was made possible by the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET: www.sharcnet.ca) and the Hyper Dense Research Automat (HYDRA: hydra.uwaterloo.ca) cluster.
References
- 1.Hughes, J., T. W. Smith, H. W. Kosterlitz, L. A. Fothergill, B. A. Morgan, and H. R. Morris. 1975. Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature. 258:577–579. [DOI] [PubMed] [Google Scholar]
- 2.Graham, W. H., E. S. Carter II, and R. P. Hicks. 1992. Conformational analysis of Met-enkephalin in both aqueous solution and in the presence of sodium dodecyl sulfate micelles using multidimensional NMR and molecular modeling. Biopolymers. 32:1755–1764. [DOI] [PubMed] [Google Scholar]
- 3.Nemethy, G., M. S. Pottle, and H. A. Scheraga. 1983. Energy parameters in polypeptides. 9. Updating of geometrical parameters, nonbonded interactions, and hydrogen bond interactions for the naturally occurring amino acids. J. Phys. Chem. 87:1883–1887. [Google Scholar]
- 4.Sippl, M. J., G. Nemethy, and H. A. Scheraga. 1984. Intermolecular potentials from crystal data. 6. Determination of empirical potentials for O-H…O = C hydrogen bonds from packing configurations. J. Phys. Chem. 88:6231–6233. [Google Scholar]
- 5.Nemethy, G., K. D. Gibson, K. A. Palmer, C. N. Yoon, G. Paterlini, A. Zagari, S. Rumsey, and H. A. Scheraga. 1992. Energy parameters in polypeptides. 10. Improved geometrical parameters and nonbonded interactions for use in the ECEPP/3 algorithm, with application to proline-containing peptides. J. Phys. Chem. 96:6472–6484. [Google Scholar]
- 6.Li, Z., and H. A. Scheraga. 1988. Structure and free energy of complex thermodynamic systems. J. Mol. Struct. THEOCHEM. 179:333–352. [Google Scholar]
- 7.Hansmann, U. H. E., and J. N. Onuchic. 2001. Thermodynamics and kinetics of folding of a small protein. J. Chem. Phys. 115:1601–1606. [Google Scholar]
- 8.Hansmann, U. H. E., Y. Okamoto, and J. N. Onuchic. 1999. The folding funnel landscape for the peptide Met-enkephalin. Proteins Struct. Funct. Genet. 34:472–483. [DOI] [PubMed] [Google Scholar]
- 9.Evans, D. A., and D. J. Wales. 2003. The free energy landscape and dynamics of Met-enkephalin. J. Chem. Phys. 119:9947–9955. [Google Scholar]
- 10.von Freyberg, B., and W. Braun. 1991. Efficient search for all low energy conformations of polypeptides by Monte Carlo methods. J. Comput. Chem. 12:1065–1076. [Google Scholar]
- 11.Eisenmenger, F., and U. H. E. Hansmann. 1997. Variation of the energy landscape of a small peptide under a change from the ECEPP/2 force field to ECEPP/3. J. Phys. Chem. B. 101:3304–3310. [Google Scholar]
- 12.Meirovitch, H., E. Meirovitch, A. G. Michel, and M. Vásquez. 1994. A simple and effective procedure for conformational search of macromolecules: application to Met- and Leu-enkephalin. J. Phys. Chem. 98:6241–6243. [Google Scholar]
- 13.Androulakis, I. P., C. D. Maranas, and C. A. Floudas. 1997. Prediction of oligopeptide conformations via deterministic global optimization. J. Global Opt. 11:1–34. [Google Scholar]
- 14.Li, Z., and H. A. Scheraga. 1987. Monte Carlo-minimization approach to the multiple-minima problem in protein folding. Proc. Natl. Acad. Sci. USA. 84:6611–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michel, A. G., C. Ameziane-Hassani, and N. Bredin. 1992. Low-energy conformational domains of polypeptides, characterized by a random-search and minimization procedure. Can. J. Chem. 70:596–603. [Google Scholar]
- 16.Zhan, L., J. Z. Y. Chen, and W.-K. Liu. 2006. Monte Carlo basin paving: an improved global optimization method. Phys. Rev. E. 73:015701 (R). [DOI] [PubMed] [Google Scholar]
- 17.Hansmann, U. H. E. 1997. Parallel tempering algorithm for confirmational studies of biological molecules. Chem. Phys. Lett. 281:140–150. [Google Scholar]
- 18.Sugita, Y., and Y. Okamoto. 1999. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314:141–151. [Google Scholar]
- 19.Rathore, N., T. A. Knotts IV, and J. J. de Pablo. 2003. Density of states simulations of proteins. J. Chem. Phys. 118:4285–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlacci, L. 1998. Conformational analysis of Met-enkephalin: solvation and ionization considerations. J. Comput. Aided Mol. Des. 12:195–213. [DOI] [PubMed] [Google Scholar]
- 21.Vengadesan, K., and N. Gautham. 2004. Conformational studies on enkephalins using the MOLS technique. Biopolymers. 74:476–494. [DOI] [PubMed] [Google Scholar]
- 22.Mitsutake, A., M. Irisa, Y. Okamoto, and F. Hirata. 1999. Classification of low-energy confirmations of Met-enkephalin in the gas phase and in a model solvent based on the extended scaled particle theory. Bull. Chem. Soc. Jpn. 72:1717–1729. [Google Scholar]
- 23.Koča, J., and P. H. J. Carlsen. 1995. Conformational behavior and flexibility of Met-enkephalin. J. Mol. Struct. THEOCHEM. 337:17–24. [Google Scholar]
- 24.Kříž, Z., P. H. J. Carlsen, and J. Koča. 2001. Confirmational features of linear and cyclic enkephalins. A computational study. J. Mol. Struct. THEOCHEM. 540:231–250. [Google Scholar]
- 25.Kinoshita, M., Y. Okamoto, and F. Hirata. 1997. Solvation structure and stability of peptides in aqueous solutions analyzed by the reference interaction site model theory. J. Chem. Phys. 107:1586–1599. [Google Scholar]
- 26.Kinoshita, M., Y. Okamoto, and F. Hirata. 2001. Solvent effects on conformational stability of peptides: RISM analyses. J. Mol. Liq. 90:195–204. [Google Scholar]
- 27.Mitsutake, A., M. Kinoshita, Y. Okamoto, and F. Hirata. 2000. Multicanonical algorithm combined with the RISM theory for simulating peptides in aqueous solution. Chem. Phys. Lett. 329:295–303. [Google Scholar]
- 28.Mitsutake, A., M. Kinoshita, Y. Okamoto, and F. Hirata. 2004. Combination of the replica-exchange Monte Carlo method and the reference interaction site model theory for simulating a peptide molecule in aqueous solution. J. Phys. Chem. B. 108:19002–19012. [Google Scholar]
- 29.Berg, B. A., and H.-P. Hsu. 2004. Metropolis simulations of Met-enkephalin with solvent-accessible area parametrizations. Phys. Rev. E. 69:026703. [DOI] [PubMed] [Google Scholar]
- 30.Shen, M.-Y., and K. F. Freed. 2002. Long time dynamics of Met-enkephalin: comparison of explicit and implicit solvent models. Biophys. J. 82:1791–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaman, M. H., M.-Y. Shen, R. S. Berry, and K. F. Freed. 2003. Computer simulation of Met-enkephalin using explicit atom and united atom potentials: similarities, differences, and suggestions for improvement. J. Phys. Chem. B. 107:1685–1691. [Google Scholar]
- 32.Montcalm, T., W. Cui, H. Zhao, F. Guarnieri, and S. R. Wilson. 1994. Simulated annealing of Met-enkephalin: low energy states and their relevance to membrane-bound, solution and solid-state conformations. J. Mol. Struct. THEOCHEM. 308:37–51. [Google Scholar]
- 33.Berg, B. A. 2003. Metropolis importance sampling for rugged dynamical variables. Phys. Rev. Lett. 90:180601. [DOI] [PubMed] [Google Scholar]
- 34.Sanbonmatsu, K. Y., and A. E. García. 2002. Structure of Met-enkephalin in explicit aqueous solution using replica exchange molecular dynamics. Proteins Struct. Funct. Genet. 46:225–234. [DOI] [PubMed] [Google Scholar]
- 35.Hansmann, U. H. E., M. Masuya, and Y. Okamoto. 1997. Characteristic temperatures of folding of a small peptide. Proc. Natl. Acad. Sci. USA. 94:10652–10656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marinari, E., G. Parisi, and J. J. Ruiz-Lorenzo. 1998. Spin Glasses and Random Fields. A. P. Young, editor. World Scientific, Singapore.
- 37.Hukushima, K., and K. Nemoto. 1996. Exchange Monte Carlo method and application to spin glass simulations. J. Phys. Soc. Jpn. 65:1604–1608. [Google Scholar]
- 38.Wang, F., and D. P. Landau. 2001. Efficient, multiple-range random walk algorithm to calculate the density of states. Phys. Rev. Lett. 86:2050–2053. [DOI] [PubMed] [Google Scholar]
- 39.Wang, F., and D. P. Landau. 2001. Determining the density of states for classical statistical models: a random walk algorithm to produce a flat histogram. Phys. Rev. E. 64:056101. [DOI] [PubMed] [Google Scholar]
- 40.Zhan, L. 2005. Fast stochastic global optimization methods and their applications to cluster crystallization and protein folding. Ph.D. thesis, University of Waterloo, Waterloo, Ontario.
- 41.Hansmann, U. H. E., and L. T. Wille. 2002. Global optimization by energy landscape paving. Phys. Rev. Lett. 88:068105.. [DOI] [PubMed] [Google Scholar]
- 42.Berg, B. A., and T. Neuhaus. 1991. Multicanonical algorithms for first-order phase transitions. Phys. Lett. B. 267:249–253. [DOI] [PubMed] [Google Scholar]
- 43.Berg, B. A., and T. Neuhaus. 1992. Multicanonical ensemble: a new approach to simulate first-order phase transitions. Phys. Rev. Lett. 68:9–12. [DOI] [PubMed] [Google Scholar]
- 44.Lee, J. 1993. New Monte Carlo algorithm: entropic sampling. Phys. Rev. Lett. 71:211–214. [DOI] [PubMed] [Google Scholar]
- 45.Hao, M.-H., and H. A. Scheraga. 1994. Monte Carlo simulation of a first-order transition for protein folding. J. Phys. Chem. 98:4940–4948. [Google Scholar]
- 46.Wales, D. J., and H. A. Scheraga. 1999. Global optimization of clusters, crystals, and biomolecules. Science. 285:1368–1372. [DOI] [PubMed] [Google Scholar]
- 47.Wales, D. J., and J. P. K. Doye. 1997. Global optimization by basin-hopping and the lowest energy structures of Lennard-Jones clusters containing up to 110 atoms. J. Phys. Chem. A. 101:5111–5116. [Google Scholar]
- 48.Zhan, L., B. Piwowar, W.-K. Liu, P. J. Hsu, S. K. Lai, and J. Z. Y. Chen. 2004. Multicanonical basin hopping: a new global optimization method for complex systems. J. Chem. Phys. 120:5536–5542. [DOI] [PubMed] [Google Scholar]
- 49.Zhan, L., J. Z. Y. Chen, W.-K. Liu, and S. K. Lai. 2005. Asynchronous multicanonical basin hopping method and its application to cobalt nanoclusters. J. Chem. Phys. 122:244707. [DOI] [PubMed] [Google Scholar]
- 50.Gill, P. E., W. Murray, and M. H. Wright. 1981. Practical Optimization. Academic Press, London.
- 51.Eisenmenger, F., U. H. E. Hansmann, S. Hayryan, and C.-K. Hu. 2001. [SMMP], a modern package for simulation of proteins. Comput. Phys. Commun. 138:192–212. [Google Scholar]
- 52.Richardson, J. S. 1981. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 34:167–339. [DOI] [PubMed] [Google Scholar]
- 53.Rose, G. D., L. M. Gierasch, and J. A. Smith. 1985. Turns in peptides and proteins. Adv. Protein Chem. 37:1–109. [DOI] [PubMed] [Google Scholar]
- 54.Hutchinson, E. G., and J. M. Thornton. 1996. PROMOTIF—a program to identify and analyze structural motifs in proteins. Protein Sci. 5:212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venkatachalam, C. M. 1968. Stereochemical criteria for polypeptides and proteins. V. Conformation of a system of three linked peptide units. Biopolymers. 6:1425–1436. [DOI] [PubMed] [Google Scholar]
- 56.Némethy, G., and M. P. Printz. 1972. The γ-turn, a possible folded conformation of the polypeptide chain. Comparison with the β turn. Macromolecules. 5:755–758. [Google Scholar]
- 57.Némethy, G., and H. A. Scheraga. 1980. Stereochemical requirements for the existence of hydrogen bonds in β-turn. Biochem. Biophys. Res. Commun. 95:320–327. [DOI] [PubMed] [Google Scholar]
- 58.Milner-White, E. J., B. M. Ross, R. Ismail, K. Belhadj-Mostefa, and R. Poet. 1988. One type of γ-turn, rather than the other, gives rise to chain reversal in proteins. J. Mol. Biol. 204:777–782. [DOI] [PubMed] [Google Scholar]
- 59.DeLano, W. L. http://www.pymol.org.