

Abstract
The availability of the structures of the cytochrome b6f complex (cyt b6f), plastocyanin (PC), and cytochrome c6 (cyt c6) from Chlamydomonas reinhardtii allowed us, for the first time, to model electron transfer interactions between the luminal domains of this complex (including cyt f and the Rieske FeS protein) and its redox partners in the same species. We also generated a model structure in which the FeS center of the Rieske protein was positioned closer to the heme of cyt f than observed in the crystal structure and studied its interactions with both PC and cyt c6. Our data showed that the Rieske protein in both the original crystal structure and in our modeled structure of the cyt b6f complex did not physically interfere with binding position or orientation of PC or cyt c6 on cyt f. PC docked on cyt f with the same orientation in the presence or the absence of the Rieske protein, which matched well with the previously reported NMR structures of complexes between cyt f and PC. When the FeS center of the Rieske protein was moved close to the heme of cyt f, it even enhanced the interaction rates. Studies using a cyt f modified in the 184–191 loop showed that the cyt f structure is a more important factor in determining the rate of complex formations than is the presence or the absence of the Rieske protein or its position with respect to cyt f.
INTRODUCTION
The cytochrome b6f (cyt b6f) complex is an oligomeric membrane protein complex that is one of the three major redox enzyme complexes residing in the thylakoid membrane of higher plants and algae (1,2). This complex is analogous to the cytochrome bc1 (cyt bc1) complex in mitochondria and photosynthetic bacteria (3). There are two crystal structures available for the cyt b6f complex, one from the green alga Chlamydomonas reinhardtii (2) and the other from the cyanobacterium Mastigocladus laminosus (1). The functional, physiological, cyt b6f complex is a dimer. In the C. reinhardtii cyt b6f complex, the extramembrane domains of both cyt f and the Rieske proteins lie on the luminal side of the thylakoid membrane (2). In the cyt b6f complex, cyt f accepts an electron from the Rieske subunit and transfers it to the mobile proteins PC or cyt c6, which then transfer the electron to Photosystem I (4).
The Rieske FeS protein is a transmembrane molecule consisting of a helical membrane anchor and a globular protein located on the luminal side of the membrane. The extramembrane segment of the Rieske protein consists of a 12-kDa β-sheet protein (residues 80–206 in C. reinhardtii), connected via a linker (residues 72–79 to its transmembrane domain (residues: 33–78). The electron density is not well defined for part of the extramembrane domain of the Rieske protein in the C. reinhardtii cyt b6f structure. Also, the linker region is not visible, suggesting high mobility of the extramembrane domain of the Rieske protein (2). In both cyt b6f complexes the FeS cluster of the Rieske protein lies too far away from the heme of cyt f for efficient electron transfer, suggesting that the Rieske FeS protein must move to transfer an electron to the cyt f heme (Fig. 1 (1,2)).
FIGURE 1.

Distances between the Rieske FeS cluster and the cyt f heme in the crystal structure (cyt f+Rieske subcomplex-far) and the close model structure (cyt f+Rieske subcomplex-close) in the C. reinhardtii cyt b6f complex. All of the distances are in Angstroms.
Movement of the extramembrane domain of the Rieske protein has previously been reported in the cytochrome bc1 (cyt bc1) complex. The position of the Rieske FeS protein is nearly identical (to within ∼4 Å) in the two available crystal structures of the cyt b6f complex and most closely resembles the “Rieske down” position in the cyt bc1 complexes close to the Q0 site (3,5–8). Some experimental evidence exists suggesting the movement of the luminal domain of the Rieske protein in the cyt b6f complex. Heimann et al. (9) reported the dependence of cyt b6f complex redox reactions on the luminal viscosity and suggested that the movement of the Rieske protein was part of the rate-limiting step for charge transfer through the cyt b6f complex. On the other hand, mutations in the linker region of the Rieske protein in the cyt b6f complex had less of an effect than those in the cyt bc1 complex. This implied a more limited movement of the Rieske protein in the cyt b6f complex (10,11). Soriano et al. (12) studied the interactions between the Rieske protein and cyt f in vitro and observed that the interactions were independent of pH and ionic strength, implying no significant involvement of electrostatic forces. They proposed that efficient electron transfer between the Rieske protein and cyt f in vivo could be facilitated by guided trajectories of the Rieske extramembrane domain.
Cyt f is functionally analogous to the cytochrome c1 subunit of mitochondrial and bacterial cyt bc1 complexes, although their structures are completely different. The cyt f protein is a transmembrane molecule consisting of a helical membrane anchor and a globular protein on the luminal side of the membrane. The extramembrane segment of cyt f in the lumen of the thylakoid membrane is a 28-kDa β-sheet protein consisting of two domains, the larger of which binds the heme (13–15). Five important lysine residues on cyt f produce a prominent basic patch that contributes to a positive electrostatic field, which attracts negative charges on PC or cyt c6 (13,16). Mutations of these highly conserved residues to neutral residues resulted in a large decrease in the interaction rate with PC (17,18).
PC is an 11-kDa “blue” copper, β-sheet protein with two clusters of negatively charged residues called the upper and lower patches or clusters (4,19–22). The upper cluster consists of residues 59–61. However, PCs from all algae including C. reinhardtii and some species of higher plants have a two-residue deletion in this region. In these species, the negative charge at position 60 is replaced by one at position 85. The lower cluster consists of residues 42–44 and either 45 or 79, which are conserved in all higher plant and green algal PCs. There is another negatively charged residue in this region, namely, D53. These eight anionic residues produce a large negative electrostatic field in PC (23).
Cyt c6 is a high-potential cytochrome related to mammalian cyt c. Cyt c6 is present in only some algae and cyanobacteria (24), although a cyt c6-like protein has recently been reported in a higher plant, Arabidopsis (25). Many species of algae and cyanobacteria produce cyt c6 in response to copper deficiency. Cyt c6 from C. reinhardtii is a 10-kDa α-helical protein with no sequence homology to PC, although it is interchangeable with PC (26,27). Both of these molecules are roughly spherical and have similar size, pI, and Em (24). Also, both proteins have a similar pattern of negatively charged residues on their surfaces, resulting in very similar electrostatic potentials (28).
Both the experimental in vitro data (for a detailed discussion of this see Haddadian and Gross (29)) and the computational modeling studies support the electrostatic nature of the interactions of cyt f with its redox partners. The computational studies include manual docking (16), a combination of Monte Carlo and molecular dynamics simulations (30), Brownian Dynamics (BD) simulations (31–34), and modeling of the interactions of cyt f and PC from spinach (35). However, experimental work by Soriano et al. (36) and Zhou et al. (37) showed much smaller electrostatic interactions between cyt f and PC in vivo in C. reinhardtii.
We now have expanded these studies to include, for the first time, the effect of the Rieske FeS protein on the interactions of both PC and cyt c6 with the cyt b6f complex from Chlamydomonas, using BD simulations. In particular, we have addressed questions such as whether the Rieske protein affects the number of complexes formed, the interaction rates, and the orientation of PC and cyt c6 within the complexes.
For the BD simulations, we built a cyt f plus Rieske subcomplex containing the luminal portions of both proteins using data from the crystal structure of the cyt b6f complex, called cyt f+Rieske subcomplex-far (because the FeS center of the Rieske FeS protein is far from the heme of cyt f). We also built a second cyt f plus Rieske subcomplex (named cyt f+Rieske subcomplex-close) by moving and rotating the Rieske protein to bring its FeS center closer to the heme of cyt f, similar to the model suggested by Kurisu et al. (1) (see Fig. 1).
In addition, we examined the effects of varying the cyt f structure on the interaction of both PC and cyt c6 with both cyt f+Rieske subcomplexes. This is important because, in our previous work, we concluded that varying the structure of the 184–191 loop on the small domain of cyt f caused large changes in the interaction of cyt f with both PC and cyt c6 (38). The question arises as to whether the structure of this loop also affects the interaction of the cyt f+Rieske subcomplexes with both PC and cyt c6. To address this, we made the same loop modification on the cyt f in the cyt f+Rieske subcomplexes and studied the effects on the interactions with both PC and cyt c6.
Our data showed that the Rieske protein in both the close and the far cyt f+Rieske subcomplexes did not physically interfere with binding position or orientation of PC or cyt c6 on cyt f. PC docked on cyt f with the same orientation in the presence or the absence of the Rieske protein, which matched well with the previously reported NMR structures of complexes between cyt f and PC. When the FeS center of the Rieske protein was moved close to the heme of cyt f, it even enhanced the interaction rates. The structural studies on cyt f showed that the conformation of the 184–189 loop on cyt f is a more important factor in determining the rate of complex formations than is the presence or the absence of the Rieske protein or its position with respect to cyt f.
METHODS
Molecular structures
The 3D structures for the cyt b6f complex, PC, and cyt c6 were obtained from the Protein Data Bank (PDB) (http://www.rcsb.org/pdb/ (39)). The C. reinhardtii cyt b6f complex used was that of PDB code 1Q90 (2). We used only the extramembrane domains of the cyt f and Rieske subunits of this structure to model the interactions with PC and cyt c6. The C. reinhardtii PC structure used was that of PDB code 2PLT (19); and the cyt c6 structure used was that of PDB code 1CYJ (27).
Building a model of a cyt f+Rieske subcomplex with the FeS center of the Rieske protein close to the heme of cyt f
As discussed earlier, the position of the Rieske FeS clusters in the cyt b6f complexes is too far away from the heme of cyt f for efficient electron transfer between them. Based on the cyt b6f complex crystal structures, the Rieske protein occupies a cleft between the cyt f large and small domains and faces the hydrophobic surface of cyt f and its heme ligand, His-25. We built our close model subcomplex based on that suggested by Kurisu et al. (1) for Mastigocladus laminosus cyt b6f complex with the goal of bringing the FeS center of the Rieske protein as close as possible to the heme of cyt f. As was described by these authors, the extramembrane domain of Rieske protein requires a 25° rotation toward cytochrome f for an efficient electron transfer between these proteins. In contrast to the Mastigocladus laminosus cyt b6f complex, the electron density is not well defined for part of the extramembrane domain of the Rieske protein in the crystal structure of the C. reinhardtii cyt b6f complex, and also, the linker region connecting this domain to the transmembrane domain is not visible. Therefore, we both translated and rotated the Rieske protein (∼25°) to arrive at our close model. We also checked and prevented the steric clashes of the Rieske protein with the rest of the cyt b6f complex in the close model structure. We do not claim that our model structure is the final close structure between cyt f and Rieske proteins, but it is much closer than that observed in the crystal structure of C. reinhardtii cyt b6f complex. Distances between the Rieske FeS cluster and the cyt f heme in the crystal structure and the close model structure are shown in Fig. 1. This model was built using program Deep View (Swiss-Pdb Viewer; 40).
Modifying the 184–191 loop on the small domain of cyt f
Using Deep View (40), we also replaced the loop of residues 184–191 of the small domain of cyt f subunit from C. reinhardtii cyt b6f complex with its corresponding loop of structure B in C. reinhardtii cyt f in the PDB code 1CFM (15) to produce the modified cyt f structure (herein called: cyt f-modified). No energy minimization was performed, keeping the orientation of the side chains unchanged. For a detailed description, see Haddadian and Gross (38).
Molecular representations
All molecular representations were made using the program GRASP (41). The electrostatic fields used for the figures only were also calculated using GRASP. The internal and external dielectric constants of the proteins were 4 and 78, respectively. The ionic strength was 10 mM, and the pH was 7.0. (See Figs. 1 and 4 as generated by the program Deep View (40).)
FIGURE 4.

Overlay of the cyt f structure B from the PDB code 1CFM on the extramembrane domain of the cyt f subunit from the cyt b6f complex (PDB code 1Q90). The 1CFM-B cyt f backbone and all of its basic residues were colored by their temperature factors (β-factors), in which the molecule is colored from dark blue for low β-factors to red for high β-factors (the heme is colored red for visualization purpose). The backbone and all of the basic residues of the 1Q90 cyt f are shown in gray, and its heme is in black. As can be seen, the loop of residues A-184–G-191 of the 1CFM-B cyt f has some of the highest β-factors compared to the rest of the molecule (indicating its high flexibility) and is oriented very differently than the 1Q90 loop. The key basic residues in both cyt fs are labeled. This figure was taken from Haddadian and Gross (38) and generated by the program Deep View (40).
Electrostatic calculations for BD simulations
The BD program that we used for our simulations applies a modified Tanford-Kirkwood pK algorithm (42) to assign charges on the molecules. In addition, the charge on H-37 and H-87 on C. reinhardtii PC and one of the histidine residues on C. reinhardtii cyt f (H-25) was set to zero because these residues are ligated to the metal centers (the other histidine residue on cyt f lies far from the metal center). The N-terminal residue on cyt f (Y-1) is a heme ligand; therefore, it was also assigned a charge of zero. The charge on the single histidine of C. reinhardtii cyt c6 was also set to zero because it is a heme ligand. C-84 is a ligand to the Cu atom on PC, and its sulfur atom was assigned a net charge of −1 (23); the Cu atom was given a charge of +2. The heme charges for both cyt f and cyt c6 were Fe (+2), two ring nitrogen atoms (−1 each), and the two propionic acid side chains (−1 each).
The charge assignments on the Rieske iron-sulfur cluster and the residues coordinating it were those of Izrailev et al. (8), in which the FeS cluster ligands H-136 and H-155 were assigned a charge of 0.279 on their NE2 atoms. The sulfur atoms on the other two ligands C-134 and C-152 were assigned a charge of −0.408 each. The FE1 atom was assigned a charge of 0.8740; the FE2 atom a charge of 0.6380; the S1 atom a charge of −0.6270; and the S2 atom a charge of −0.6270.
The electrostatic potentials were calculated using the Warwicker/Watson finite difference method to solve a linearized Poisson-Boltzmann equation (43). MacroDox uses a 61 × 61 × 61 cubic grid with its center positioned at the center of the mass of the protein to solve for the electrostatic potential. We used a grid spacing of 3.6 Å, followed by a smaller spacing of 1.2 Å for the electrostatic potential calculations (see Gross and Pearson (21) for a discussion of the effect of grid size on the results obtained).
MacroDox simulations
The simulations were carried out using program MacroDox v. 3.2.1 (S. H. Northrup, Tennessee Technological University, http://pirn.chem.tntech.edu/macrodox.html) exactly as described in detail by Gross and Pearson (21) and Haddadian and Gross (29,38). Typically, five sets of 10,000 trajectories at 10 mM ionic strength and pH 7.0 were carried out (to minimize the error values in the simulations).
MacroDox determines the closest approach of the two molecules based on a set of preselected reaction criteria. In our simulations, these reaction criteria were chosen as the Cu-Fe distance for PC and heme-heme distance for cyt c6 to select for the electron transfer-active complexes. The shorter the distance between metal centers, the higher the chance of electron transfer (44,45). For the cyt c6 interactions, because the heme ring is a possible route for electron transport to the Fe atom, and the orientation of the hemes with respect to each other is important in the electron transfer, we decided to use the heme-heme distance as the criterion (this criterion has been used previously for BD simulations in heme proteins; 29,46). Particularly, because the cyt c6 heme is exposed to the surface at two locations (one more than the other), this would allow us to consider all of the possible orientations of cyt c6 on cyt f. Based on this criterion, at each step of the trajectory the distances between the carbon atoms at four corners of the heme pyrrol ring (CH groups) and the same four atoms on the other protein are measured, and the one with the shortest distance is recorded as the heme-heme distance.
At each step of a trajectory MacroDox measures the Fe-Cu distance in PC interactions and heme-heme distance in cyt c6 interactions. At the end of the trajectory, the complex formed between the proteins with the smallest distance value is considered the successful one. For each successful complex formed, the program records the distance between the metal centers, the structure of the complex formed in the form of a PDB file, the 15 closest electrostatic contacts in the complex, and the electrostatic interaction energy for the complex. After all of the trajectories have been concluded, the number of successes at any distance is determined and plotted as a function of Cu-Fe or heme-heme distance.
Interaction rates were calculated using equations derived by Northrup et al. (46–50). These equations calculate the rate constant for association of the two molecules, ka, from the fraction of trajectories that meet the preset reaction criteria. In this study, a cutoff value of 17 Å for the Fe-Cu distance for PC and a cutoff value of 14.5 Å for heme-heme distance for cyt c6 were used. (This is discussed further in the Results section.)
All of the simulations were carried out on a Silicon Graphics O2 workstation (IRIX 6.5).
RESULTS
In this work, we modeled the interaction between a cyt f+Rieske subcomplex consisting of the luminal, extramembrane domains of the cyt f and Rieske subunits from C. reinhardtii cyt b6f complex and its redox partners from the same species. The goal of these simulations was to study the extent to which Rieske FeS protein affects the binding of PC and cyt c6 on cyt f with the Rieske protein in both its close and far positions. All of the simulations were carried out using the BD simulation program MacroDox at 10 mM ionic strength and pH 7.0.
The interaction of the Cyt f+Rieske subcomplex-far with PC
Fig. 2 A compares the results obtained for PC interacting with cyt f alone with those for PC interacting with the cyt f+Rieske subcomplex-far. The reaction criterion used was the Cu-Fe distance. All of the complexes observed under the peaks are caused by electrostatic interactions because only a very small number of complexes are formed at small Cu-Fe distances (i.e., ≤17 Å in our studies) in the absence of an electrostatic field (29). The complexes within the peaks, also because of their small metal-metal distances, have a higher chance of being involved in electron transfer and are considered electron transfer-active (44,45). Fewer complexes were formed for the Rieske subcomplex-far compared to cyt f alone, also taken from the crystal structure of the cyt b6f complex (PDB code 1Q90; Fig. 3 A and Table 1). The decrease in the number of complexes formed could be due to either the electrostatic field or to the bulk structure of the Rieske protein attached to cyt f. To test the first possibility, we set all of the charges on the Rieske protein to zero in the charge file used for the MacroDox simulations while leaving the structure unchanged. Note that the charges were not removed from either cyt f or PC. This resulted in an increase in the number of complexes formed (Fig. 2 A).
FIGURE 2.

Interactions between the cyt f+Rieske subcomplex-far with PC (A) and cyt c6 (B) at 10 mM ionic strength and pH 7.0. Five sets of 10,000 trajectories each were carried out, after which the average of the number of the complexes formed at the closest approach of each trajectory was plotted as a function of the Fe-Cu distance for PC interactions and heme-heme distance for cyt c6 interactions. The number of complexes with closest metal-to-metal distances between 15.25 and 15.5 Å is shown on the abscissa and plotted at 15.5 Å. See the Methods sections for a description of the cyt f-modified complexes.
FIGURE 3.

Interactions between the cyt f+Rieske subcomplex-close with PC (A) and cyt c6 (B) at 10 mM ionic strength and pH 7.0. The conditions are the same as for Fig. 2. For a description of the close model and the cyt f-modified complexes, see Methods section.
TABLE 1.
Number of complexes formed with Cu-Fe distances ≤17 Å and the calculated rates of association (ka) for the interactions of PC with the cyt f+Rieske subcomplexes from C. reinhardtii
| Number of complexes/10,000 trajectories* | Corrected value† | ka* (×108) M−1 s−1 | Corrected value† | |
|---|---|---|---|---|
| Cyt f-unmodified alone | 137 ± 4 | – | 4.5 ± 0.3 | – |
| Cyt f+Rieske subcomplex-far‡ | 46 ± 1 | 108 ± 5 | 1.2 ± 0.2 | 3.7 ± 0.5 |
| Cyt f+Rieske subcomplex-far-no charge§ | 75 ± 3 | – | 2.0 ± 0.3 | – |
| Cyt f-modified¶ alone | 449 ± 8 | – | 14.3 ± 0.7 | – |
| Cyt f-modified+Rieske subcomplex-far | 528 ± 10 | 566 ± 14 | 13.8 ± 0.6 | 17.2 ± 1.0 |
| Cyt f-modified+Rieske subcomplex-far no charge | 411 ± 6 | – | 10.9 ± 0.5 | – |
| Cyt f+Rieske subcomplex-close‡ | 181 ± 9 | 243 ± 12 | 5.0 ± 0.4 | 7.4 ± 0.5 |
| Cyt f+Rieske subcomplex-close no charge | 75 ± 6 | – | 2.1 ± 0.2 | – |
| Cyt f-modified+Rieske subcomplex-close | 554 ± 11 | 645 ± 14 | 14.9 ± 0.5 | 19.5 ± 1.0 |
| Cyt f-modified+Rieske subcomplex-close no charge | 358 ± 5 | – | 9.7 ± 0.6 | – |
The complexes included were those with the Cu-Fe distances ≤17 Å, which are considered electron transfer-active. The second-order association rate constants, ka, were calculated for the formation of these complexes, as described in the Methods section. Five sets of 10,000 trajectories each were carried out to obtain the error values.
The corrected values for the number of complexes formed for all of the cyt f+Rieske subcomplexes were obtained as follows. First, the number of complexes formed for cyt f+Rieske subcomplexes with all of the Rieske charges set to zero were subtracted from that obtained for the cyt f-unmodified alone. Second, the difference obtained was added to the number of complexes formed for the cyt f+Rieske subcomplexes with all of the Rieske charges in place. In the case of modified cyt f, the subtractions were made from the number of complexes formed by the cyt f-modified alone structure. The values of the interaction rates were corrected in the same way. For an explanation of the corrected values, see the Discussion section.
The cyt f+Rieske subcomplex-far is the structure from the crystal structure of the C. reinhardtii cyt b6f complex. The model of the cyt f+Rieske subcomplex-close was built by moving the extramembrane domain of the Rieske protein closer to the extramembrane domain of cyt f (refer to the Methods section and Fig. 1).
For the cyt f+Rieske-no charge subcomplexes, only the charges on the Rieske protein were set to zero in the charge file used for the MacroDox simulations. Note that the charges were not removed from either cyt f or PC.
In the cyt f-modified structure, the loop of residues 184–191 on the cyt f molecule (PDB code 1Q90) was replaced by that from the cyt f structure B from PDB code 1CFM (refer to the Methods section).
The total number of complexes formed with Fe-Cu distances ≤17 Å and the corresponding association rates, ka, are presented in Table 1. The 17 Å cutoff distance was chosen to include a maximum number of complexes formed as a result of the electrostatic field while eliminating any formed by random diffusion alone. However, selection of the cutoff distances affects only the magnitude of the association rates but not their relative effectiveness (see Gross and colleagues (21,29,33) for a discussion of this point). It should be noted that we are not reporting absolute rate values; only their relative order is considered in this study.
The changes in the interaction rates were similar to the changes in the number of complexes formed. There was an ∼4-fold decrease in the interaction rate for the cyt f+Rieske subcomplex-far compared to cyt f alone (from 4.5 ± 0.3 to 1.2 ± 0.2 × 108 M−1s−1). The removal of the charges on the Rieske increased the ka value slightly to 2.0 ± 0.3 × 108 M−1s−1), which was still smaller than that observed for cyt f alone.
The interaction of cyt f+Rieske subcomplex-far with cyt c6
For the interactions with cyt c6, a heme-heme cutoff value of 14.5 Å was chosen to include almost the entire peaks. As was the case for PC, fewer complexes were formed for cyt f+Rieske subcomplex-far interacting with cyt c6 than for cyt f alone (Fig. 2 B, Table 2). However, in contrast to the results for PC, there was no increase in the number of complexes formed when the charges were removed from the Rieske protein. Note that, as was the case for the PC experiments, the charges were not removed from either cyt f or cyt c6. The changes in the interaction rates were similar to the changes in the number of complexes formed (Table 2).
TABLE 2.
Number of complexes formed with heme-heme distances ≤14.5 Å and the corresponding association rate constants (ka) for the interactions of cyt c6 with cyt f+Rieske subcomplexes from C. reinhardtii
| Number of complexes/10,000 trajectories* | Corrected value† | ka* (×108) M−1 s−1 | Corrected value† | |
|---|---|---|---|---|
| Cyt f-unmodified alone | 91 ± 3 | – | 3.0 ± 0.3 | – |
| Cyt f+Rieske subcomplex-far‡ | 36 ± 2 | 89 ± 4 | 1.0 ± 0.15 | 2.9 ± 0.4 |
| Cyt f+Rieske subcomplex-far no charge§ | 38 ± 2 | – | 1.1 ± 0.2 | – |
| Cyt f-modified¶ alone | 493 ± 6 | – | 16.0 ± 0.7 | – |
| Cyt f-modified+Rieske subcomplex-far | 551 ± 8 | 617 ± 11 | 14.7 ± 0.7 | 19.2 ± 1.1 |
| Cyt f-modified+Rieske subcomplex-far no charge | 427 ± 3 | – | 11.5 ± 0.4 | – |
| Cyt f+Rieske subcomplex-close‡ | 56 ± 3 | 123 ± 3 | 1.6 ± 0.2 | 3.9 ± 0.2 |
| Cyt f+Rieske subcomplex-close no charge | 24 ± 1 | – | 0.68 ± 0.15 | – |
| Cyt f-modified+Rieske subcomplex-close | 408 ± 8 | 653 ± 9 | 11.3 ± 0.5 | 20.4 ± 1.0 |
| Cyt f-modified+Rieske subcomplex-close no charge | 248 ± 7 | – | 6.9 ± 0.4 | – |
The complexes included were those with the heme-heme distances ≤14.5 Å, which are considered electron transfer-active. The second-order association rate constants, ka, were calculated for the formation of these complexes, as described in the Methods section. Five sets of 10,000 trajectories each were carried out to obtain the error values.
The corrected values for the number of complexes formed for all of the cyt f+Rieske subcomplexes were obtained as follows. First, the number of complexes formed for cyt f+Rieske subcomplexes with all of the Rieske charges set to zero were subtracted from that obtained for the cyt f-unmodified alone. Second, the difference obtained was added to the number of complexes formed for the cyt f+Rieske subcomplexes with all of the Rieske charges in place. In the case of modified cyt f, the subtractions were made from the number of complexes formed by the cyt f-modified alone structure. The values of the interaction rates were corrected in the same way. For an explanation of the corrected values, see the Discussion section.
The cyt f+Rieske subcomplex-far is the structure from the crystal structure of the C. reinhardtii cyt b6f complex. The model of the cyt f+Rieske subcomplex-close was built by moving the extramembrane domain of the Rieske protein closer to the extramembrane domain of cyt f (refer to the Methods section and Fig. 1).
For the cyt f+Rieske-no charge subcomplexes, only the charges on the Rieske protein were set to zero in the charge file used for the MacroDox simulations. Note that the charges were not removed from either cyt f or cyt c6.
In the cyt f-modified structure, the loop of residues 184–191 on the cyt f molecule was replaced by that from the cyt f structure B from PDB code 1CFM (refer to the Methods section).
Note that the peak for complex formation occurred at a heme-heme distance of 13.5 Å compared to 15.5 Å for Cu-Fe distance in PC. The difference results from the fact that the Cu atom is buried ∼2 Å beneath the surface of the PC molecule, but two of the heme pyrrole rings of cyt c6 are exposed on the surface.
The interactions of cyt f+Rieske subcomplex-close with PC
As was mentioned earlier, the position of the FeS cluster in the extramembrane domain of the Rieske protein in both of the algal and cyanobacterial cyt b6f complexes is too far from the cyt f heme for efficient electron transfer between them (1,2). The important question is: what is the effect on complex formations with PC and cyt c6 of bringing the FeS center of the Rieske protein closer to cyt f? To check for this, we built a close model of the cyt f+Rieske subcomplex for the C. reinhardtii cyt b6f complex, similar to the structure suggested by Kurisu et al. (1) for the Mastigocladus laminosus cyt b6f complex (herein called the cyt f+Rieske subcomplex-close). In this model, the extramembrane domain of the Rieske protein was both translated and rotated to bring its FeS cluster closer to the cyt f heme (Fig. 1).
The interactions of cyt f+Rieske subcomplex-close with PC are shown in Fig. 3 A and summarized in Table 1. Unlike the case of cyt f+Rieske subcomplex-far, a greater number of complexes were formed for cyt f+Rieske subcomplex-close than for cyt f alone. When the charges on the Rieske protein were set to zero, the number of complexes decreased compared to both cyt f+Rieske subcomplex-far and cyt f alone (Table 1).
The rate of association for cyt f+Rieske subcomplex-close was the same as that for cyt f alone to within the limit of error (5.0 ± 0.4 and 4.5 ± 0.3 × 108 M−1s−1, respectively, Table 1). These results contrast with those for cyt f+Rieske subcomplex-far, which showed a lower interaction rate than for cyt f alone (Table 1). Removal of the charges from cyt f+Rieske subcomplex-close reduced the interaction rate to 2.1 ± 0.2 × 108 M−1s−1, which is similar to that obtained for cyt f+Rieske subcomplex-far under the same conditions. In other words, the position of the Rieske protein did not affect complex formation when the electrostatic field of this protein was absent.
The interaction of cyt f+Rieske subcomplex-close with cyt c6
The interactions of cyt f+Rieske subcomplex-close with cyt c6 are shown in Fig. 3 B and summarized in Table 2. As can be seen, a similar number of complexes are observed for cyt f+Rieske subcomplex-close as for cyt f alone. When all of the Rieske charges in the model structure were set to zero, there was a decrease in the number of close-distance complexes formed with respect to both cyt f+Rieske subcomplex-close and cyt f alone (Table 2).
The interaction of cyt f+Rieske subcomplex-close with cyt c6 resulted in a smaller interaction rate than for cyt f alone (1.6 ± 0.2 and 3.0 ± 0.3 × 108 M−1s−1, respectively). However, this rate was larger than that observed for cyt f+Rieske subcomplex-far under the same conditions (Table 1). Removal of the charges on the Rieske protein in the close structure reduced the interaction rate to 0.68 ± 0.15 × 108 M−1s−1, which is similar to that observed on removing the charges on the Rieske protein on cyt f+Rieske subcomplex-far.
Overall, when the Rieske protein was moved closer to cyt f, the interaction rates of the complex with both PC and cyt c6 increased compared to those for cyt f+Rieske subcomplex-far.
The interaction of cyt f-modified+Rieske subcomplexes with PC
There are seven available C. reinhardtii cyt f crystal structures. In our previous work (38), we showed that each of these structures differed in its ability to form complexes with PC and cyt c6. In the complex formations with PC, structure B from Chi et al. (PDB code 1CFM (15)) was the best, and the one from the cyt b6f complex (PDB code 1Q90 (2)) was the worst of the seven structures. The major difference in the seven structures lies in a small, flexible loop on the small domain of cyt f consisting of residues 184–191 (Fig. 4). When this loop on cyt f from the cyt b6f complex was replaced with the corresponding loop from the cyt f structure B in the PDB code 1CFM, the number of complexes formed between the modified cyt f and either PC or cyt c6 was greatly increased. These results reinforce the importance of positively charged residues 188 and 189 on the interactions of cyt f with its reaction partners (Fig. 4).
To test the effect of the conformation of this loop on the ability of the cyt f+Rieske subcomplexes to form complexes with PC and cyt c6, we replaced residues 184–191 on the cyt f portion of cyt f+Rieske subcomplexes with the corresponding residues from the cyt f structure B of the PDB code 1CFM and studied their interactions with both PC and cyt c6. For the sake of clarity, the cyt f structure from the cyt b6f complex is called cyt f-unmodified alone, and the one with the replaced loop is called cyt f-modified alone. Also, the subcomplexes containing the modified cyt f are termed cyt f-modified+Rieske subcomplex-far and cyt f-modified+Rieske subcomplex-close (Tables 1 and 2).
When cyt f-modified+Rieske subcomplex-far interacted with PC, a greater number of complexes were observed than for cyt f-modified alone (528 ± 10 vs. 449 ± 8; Fig. 2 A and Table 1). The calculated interaction rates were similar for these two structures and were ∼3-fold higher than those for cyt f-unmodified alone. Removing the charges selectively from the Rieske protein resulted in a smaller number of complexes formed, and the corresponding interaction rate decreased (Table 1). However, the calculated rate (10.9 ± 0.5 × 108 M−1s−1) was still higher than the interaction rate of cyt f-unmodified alone with PC.
The cyt f-modified+Rieske subcomplex-close showed the highest rate of interaction (14.9 ± 0.5 × 108 M−1s−1) with PC of all of the cyt f or cyt f+Rieske complexes (Table 1 and Fig. 3 A). However, when the charges on the Rieske protein were set to zero, the interaction rate was the same as for the cyt f-modified+Rieske subcomplex-far under the same conditions. Once more, the position of the Rieske protein did not affect complex formation when the electrostatic field of the Rieske protein was absent.
The interactions of the cyt f-modified+Rieske subcomplexes with cyt c6
As was the case for PC, when the cyt f-modified+Rieske subcomplex-far interacted with cyt c6, the number of complexes formed increased compared to those for cyt f-modified alone (551 ± 8 vs. 493 ± 6, Fig. 2 B and Table 2). This was not reflected in the interaction rates, which were the same to within the limit of error. These rates were five times larger than the cyt f-unmodified alone interaction rate. Removal of the charges on the Rieske protein decreased both the number of complexes formed and the interaction rates.
In the case of the close complex, the number of complexes formed was less than that for cyt f-modified alone, as were the interaction rates. Removal of the charges on the Rieske protein decreased the number of complexes formed and the interaction rates still further (Table 2). However, both of these rates were still higher than the rate of cyt f-unmodified alone interacting with cyt c6.
Overall these structural studies showed that the cyt f structure is a more important factor in the rate of complex formation than is the presence or the absence of the Rieske protein or its position with respect to cyt f.
DISCUSSION
The use of MacroDox to study electrostatic interactions
See Gross and Pearson (21) and Gross (33) for a detailed discussion of the advantages and disadvantages of the program MacroDox. One of the disadvantages of MacroDox is that the proteins are treated as rigid bodies. This means that we cannot study changes in conformation as the two proteins approach each other and dock. On the other hand, it is an advantage in that we can examine individual conformations of both proteins, as was done comparing the effect of changing the conformation of the 184–191 loop on cyt f on the binding of PC and cyt c6 (38). MacroDox has previously proved very useful in studying the effects of mutations and in elucidating the structures of the complexes formed. For example, MacroDox predictions of the relative order of the effects of the mutations of charged residues in C. reinhardtii PC (29) agree well with experimental results from higher plant PCs (51–53). Furthermore, the effects of mutations of charged residues on the cyanobacterium PC from Phormidium laminosum (33) agree well with the experimental results from that species (54). Also, the structures obtained from BD simulations were very similar to those obtained from NMR experiments (55–58). Our typical complex structure between PC and cyt f had an RMS difference value of 0.7 Å with the complex structure No. 1 of Ubbink et al. (55). For a detail discussion of this, see Gross and Pearson (21).
The effect of the presence of the Rieske FeS protein on the interactions of cyt f with PC and cyt c6
The observation that the number of complexes formed by cyt f+Rieske subcomplex-far with both PC and cyt c6 was less than that for cyt f alone can be explained either by a decrease in the rotational diffusion coefficient for the cyt f+Rieske subcomplexes compared to cyt f alone, by electrostatic differences, or by the bulk structure of the Rieske protein preventing access to the cyt f heme.
The effect of the rotational diffusion coefficient on the number of complexes formed
Based on the MacroDox program, the cyt f+Rieske subcomplex has a rotational diffusion coefficient of 0.80 × 10−5 ps−1, which is four times smaller than the rotational diffusion coefficient of cyt f alone (0.33 × 10−4 ps−1). This difference, which is caused by the larger size of the cyt f+Rieske subcomplex, causes a smaller rate of rotation for the subcomplex compared to cyt f alone. The following experiment was conducted to determine the effect of the rotational diffusion coefficient on the number of complexes formed: The number of complexes formed between either PC or cyt c6 and cyt f was determined using two values for the rotational diffusion coefficient. The results are presented in Table 3. For the runs labeled cyt f (a), the rotational diffusion coefficient was 0.33 × 10−4 ps−1, the value calculated by MacroDox for cyt f alone, and for the run labeled cyt f (b), the rotational diffusion coefficient was 0.8 × 10−5 ps−1, the value calculated for the cyt f+Rieske subcomplex. The results show that decreasing the rotational diffusion coefficient had only a small effect on the number of complexes formed and the corresponding association rates.
TABLE 3.
Effect of cyt f rotation rate on the number of complexes formed and the interaction rates with PC and cyt c6
| Interactions with PC
|
Interactions with cyt c6
|
|||
|---|---|---|---|---|
| Number of complexes/10,000 trajectories† | ka† (×108) M−1 s−1 | Number of complexes/10,000 trajectories† | ka† (×108) M−1 s−1 | |
| cyt f (a)* | 137 ± 4 | 4.5 ± 0.3 | 91 ± 3 | 3.0 ± 0.3 |
| cyt f (b)* | 127 ± 2 | 4.2 ± 0.5 | 72 ± 1 | 2.4 ± 0.3 |
Cyt f (a) is the molecule with the rotational diffusion coefficient of 0.33 × 10−4 ps−1, which is the value computed by the MacroDox program for the cyt f molecule alone and has been used for cyt f in the simulations listed in Tables 1 and 2. Cyt f (b) is also cyt f alone but with its rotational diffusional coefficient replaced by that calculated by MacroDox for the cyt f+Rieske subcomplex-far (i.e., a value of 0.80 × 10−5 ps−1).
The complexes formed were those with Cu-Fe distances ≤17 Å for PC interactions and those with heme-heme distances ≤14.5 Å for cyt c6 interactions. These complexes are considered electron transfer-active. The second-order association rate constants, ka, were calculated for the formation of these complexes, as described in the Methods section. Five sets of 10,000 trajectories each were carried out to obtain the error values.
The effect of the presence of the Rieske protein on the electrostatic field seen by PC and cyt c6
A second explanation for the lower number of complexes formed is that the electrostatic field of the Rieske protein in the original crystal structure (far) position affects the electrostatic field of the cyt b6f complex so as to decrease the number of complexes formed for the cyt f+Rieske subcomplex-far compared to cyt f alone. Fig. 5 compares the electrostatic fields of isolated cyt f (A) with those of cyt f+Rieske subcomplex-far (B) and cyt f+Rieske subcomplex-close (C). In all three cases, note the positive electrostatic field (blue) in the neighborhood of five lysine residues (Lys-58, 65, 66, 188, and 189) shown at the top of the cyt f molecules. These residues are involved in the binding of PC and cyt c6 (see Figs. 7 and 8) (21). However, when the Rieske FeS protein is in the far position (B), it presents a negatively charged face toward the PC binding site, decreasing the positive electrostatic field and thereby decreasing both the number of complexes formed and the interaction rates. In contrast, when the Rieske protein is in the close position (C), a positive face is presented toward the PC binding site, increasing the interactions.
FIGURE 5.

Electrostatic fields of reduced cyt f (A), reduced cyt f plus oxidized Rieske (B) in the cyt f+Rieske subcomplex-far, and reduced cyt f plus oxidized Rieske in the cyt f+Rieske subcomplex-close (C) from the C. reinhardtii cyt b6f complex. The electrostatic field contours at +1 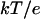 (blue) and –1
(blue) and –1 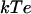 (red) were calculated at 10 mM ionic strength and pH 7.0 using the program GRASP (41). The heme and the FeS cluster are shown as space-filling models. The backbone of cyt f is colored in black and that of the Rieske protein in green.
(red) were calculated at 10 mM ionic strength and pH 7.0 using the program GRASP (41). The heme and the FeS cluster are shown as space-filling models. The backbone of cyt f is colored in black and that of the Rieske protein in green.
FIGURE 7.

Overlays of the peptide backbones of five complexes each, for the cyt f+Rieske subcomplex-far (A) and the cyt f+Rieske subcomplex-close (B) with PC. These complexes were chosen at random from those complexes with the Fe-Cu distances less than the peak distances in the plots of the complexes formed. The overlays were constructed using the program GRASP (41). The heme and the Cu atoms are shown as space-filling models and are in black.
To test the effect of the electrostatic field of the Rieske protein, we set all of the charges on the Rieske protein to zero in the charge file used for MacroDox simulations while keeping them on cyt f and either PC or cyt c6. This increased the complex formations slightly for PC and had no effect for cyt c6 (Tables 1 and 2). Based on these results, some of the decrease in complex formation observed in the presence of the Rieske protein must be related to its bulk structure rather than its electrostatic field. MacroDox checks the overlap between all of the atoms of the mobile molecule and the target molecule and prevents overlapping at each step of the trajectory by rejecting that step. The cyt f+Rieske subcomplex is a larger target molecule (therefore a larger rejection volume) than cyt f alone, causing some of the incoming trajectories to be blocked. In other words, the incoming molecules have a somewhat lower chance to get close to the cyt f metal center. We have termed this effect “shadowing”.
This Rieske molecule shadowing cyt f in the simulations is evident from Fig. 6, which shows the final positions of the center of mass of PC molecules with respect to cyt f (A) and the cyt f+Rieske subcomplex-far (B) for 400 different trajectories. As can be seen, fewer PC molecules are observed below the cyt f+Rieske complex than for cyt f alone. However, the shadow moves because at each step of the trajectory the cyt f+Rieske subcomplex rotates to a different, random position. Therefore, a particular trajectory is sometimes in the shadow and sometimes not.
FIGURE 6.

Final positions of the center of mass of PC molecules with respect to cyt f (A) and the cyt f+Rieske subcomplex-far (B) for 400 different trajectories. The complexes formed at the closest approach of each trajectory with the Fe-Cu distances ≤90 Å were shown. The program MacroDox outputs the structure of the complexes formed in the form of a PDB file and replaces the PC molecules with their centers of mass. The cyt f and Rieske molecules are shown as space-filling models and are colored as gray and green, respectively. The centers of mass of PC molecules are shown as red spheres. The heme is in black. The five important lysine residues (Lys-58, 65, 66, 188, and 189) on cyt f are shown as blue.
The shadowing effect would not be observed for the cyt b6f complex embedded in the thylakoid membrane because PC and cyt c6 could approach cyt f only from the lumen space (top of Fig. 6). Therefore, the Rieske protein would not physically block the PC and cyt c6 binding on cyt f. However, in our simulations, the difference between the number of complexes formed with cyt f alone and that observed for the cyt f+Rieske subcomplexes when there are no charges on the Rieske protein represents the shadowing effect. Therefore, the following corrections were made. First, the number of complexes formed for cyt f+Rieske subcomplexes with all of the Rieske charges set to zero were subtracted from that obtained for the cyt f-unmodified alone. Second, the difference obtained was added to the number of complexes formed for the cyt f+Rieske subcomplexes with all of the Rieske charges in place. In the case of modified cyt f, the subtractions were made from the number of complexes formed by the cyt f-modified alone structure. The values of the interaction rates were corrected in the same way (Tables 1 and 2).
Based on these corrected values, compared to the cyt f alone interactions, there is a small decrease in complex formations with PC and cyt c6 when the Rieske protein is in the far position because of its negative electrostatic field facing the binding sites of PC and cyt c6 on cyt f.
Does the presence of the Rieske protein affect the structure of the complexes formed?
The exact location of the proximal (close) binding site of Rieske on cyt f is not known. Based on the cyt b6f complex crystal structures, the Rieske protein occupies a cleft between the cyt f large and small domains and faces the hydrophobic surface of cyt f and its heme ligand, His-25 (Fig. 7). This surface is on the opposite side of the cyt f molecule from the positively charged region of cyt f that binds PC and cyt c6. Therefore, it is possible that both the Rieske protein and the redox partners (PC or cyt c6) could bind to cyt f at the same time (3).
To verify this, we compared the structure of the complexes formed in the presence of the Rieske protein with those formed in its absence (29). We also examined the position, orientation, and homogeneity of the complexes formed. The results are shown in Fig. 7. In each case, five complexes were chosen at random from those with Cu-Fe distances less than the peak distances in the plots of the complexes formed (Fig. 2 A), after which their backbones were overlaid. For complexes with the Rieske protein in the far position (Fig. 7 A) and in the close position (Fig. 7 B), the position and orientation of PC is the same as with cyt f alone (see Gross and colleagues (21,29,38)). Both in the presence and in the absence of the Rieske protein, PC docks on cyt f with an incline toward its small domain, which is very similar to the available NMR structures of the complexes between these proteins (55–58). Our typical complex structure between PC and cyt f had an RMS difference value of 0.7 Å with the complex structure No. 1 of Ubbink et al. (55; see Gross and Pearson (21)). Also, the complexes formed are as uniform as for cyt f alone. Therefore, the Rieske protein, in both its far and close positions, did not physically interfere with the binding position or orientation of PC or cyt c6 on cyt f.
The effect of moving the Rieske FeS protein close to cyt f on the interaction rates
As already discussed, in both available crystal structures of the cyt b6f complex (1,2) the FeS cluster of the Rieske protein lies too far away from the heme of cyt f for efficient electron transfer, indicating that the extramembrane domain of the Rieske protein must move closer to the extramembrane domain of cyt f. The important question is: How does moving the Rieske protein to the close position affect the binding rates of PC or cyt c6 on cyt f?
In the case of PC (Table 1), after correcting for the shadowing effect of the Rieske protein, a greater number of complexes formed were observed for cyt f+Rieske subcomplex-close than for cyt f+Rieske subcomplex-far for both control (243 ± 12 for Rieske-close compared to 108 ± 5 for Rieske-far) and modified cyt f (645 ± 14 for Riske-close compared to 566 ± 14 for Rieske-far). The same trend was observed for the corresponding interaction rates. Similar results were obtained for cyt c6 (Table 2).
As described, in the cyt f+Rieske subcomplex-far, the negative face of the Rieske protein faces cyt f, decreasing the positive electrostatic field, thereby decreasing the attraction of negatively charged PC or cyt c6 (Fig. 6 B). In contrast, for cyt f+Rieske subcomplex-close, the positively charged face of the Rieske FeS protein faces cyt f, enhancing the positive electrostatic field, thereby increasing the interaction with PC or cyt c6 (resulting in a increase in the number of complexes formed and the corresponding interaction rates; Fig. 6 C). Thus, in contrast to Rieske in its far position, the electrostatic effects are more pronounced when Rieske was moved close to cyt f.
As was stated before, moving the Rieske protein to the close position had no effect on the position or orientation of the complexes formed. Therefore, the Rieske protein in the close position not only did not physically interfere with the binding of PC or cyt c6 on cyt f, it even enhanced the interaction rates between these proteins.
The same number of complexes were formed in the absence of the charges on the Rieske protein for both the close and far cyt f+Rieske subcomplexes, which showed that both subcomplexes experienced the same shadowing effects. Thus, the difference between the two types of subcomplexes is related to the differences in their electrostatic fields.
The effect of the 184–191 loop on cyt f on complex formation by the cyt f+Rieske subcomplexes
We previously reported that the structure of a flexible loop containing residues 184–191 (including K-188 and K-189) on the small domain of cyt f is very important in determining the interaction of cyt f with its reaction partners, PC and cyt c6 (38; Fig. 5). For example, replacing this loop on the cyt f structure taken from the x-ray structure of the cyt b6f complex (PDB code 1Q90 (2)) with the same loop from the structure B of the truncated cyt f (PDB code 1CFM (15)) increased the number of complexes formed by severalfold. Replacing the same loop on both cyt f+Rieske subcomplex-far and subcomplex-close increased significantly the number of complexes formed as well as the associated interaction rates (Tables 1 and 2). In fact, the effect was greater than that for moving the Rieske protein into the close position.
These data, once more, show the very important role of the orientation of this loop in the interactions of cyt f with PC and cyt c6. Note that the electrostatic field of the cyt f-modified structure is more favorable for the binding of PC and cyt c6 than is the electrostatic field of unmodified cyt f (38). In fact, the electrostatic field of the modified cyt f dominates the electrostatic field of the Rieske protein in both its far and close positions, and thus, the position of the Rieske protein has little effect on complex formations. In other words, the cyt f structure is a more important factor in complex formation than is the presence or the absence of the Rieske protein, or its position with respect to cyt f.
CONCLUSIONS
The goal of these simulations was to study the extent to which Rieske FeS protein affects the binding of PC and cyt c6 on cyt f in both its close and far positions. This is a complicated task because the exact proximal binding site of Rieske on cyt f is not known. Therefore, modeling and simulations could provide some answers that would lead to further experimentations.
Our data showed that the Rieske protein in both the close and the far cyt f+Rieske subcomplexes did not physically interfere with binding position or orientation of PC or cyt c6 on cyt f. PC docked on cyt f with the same orientation in the presence or the absence of the Rieske protein, which matched well with the previously reported NMR structures of complexes between cyt f and PC. When the FeS center of the Rieske protein was moved close to the heme of cyt f, it even enhanced the interaction rates. The structural studies on cyt f showed that the conformation of the 184–189 loop on cyt f is a more important factor in determining the rate of complex formations than is the presence or the absence of the Rieske protein or its position with respect to cyt f.
The fact that the Rieske protein in the close position stimulates the interaction rates of PC and cyt c6 with cyt f and in its far position has a small effect on the rate of complex formations might possibly suggest a biological significance of this protein as a regulatory mechanism in the electron transfer process. Besides transferring the electron to cyt f from cytochrome b6 protein, the Rieske protein may possibly help to coordinate electron transfer between cyt f and PC or cyt c6. This requires more testing and experiments.
All of the studies mentioned could serve as a good guide for future experimental work on these proteins to understand better the electron transfer process between them. Also, these results signified the sensitivity and the power of the BD simulations in the study of the molecular interactions.
Acknowledgments
The authors thank Dr. Justin Wu of the Dept. of Biochemistry, The Ohio State University, for his careful reading of the manuscript.
References
- 1.Kurisu, G., H. Zhang, J. L. Smith, and W. A. Cramer. 2003. Structure of the cytochrome b6f complex of oxygenic photosynthesis: tuning the cavity. Science. 302:1009–1014. [DOI] [PubMed] [Google Scholar]
- 2.Stroebel, D., Y. Choquet, J. L. Popot, and D. Picot. 2003. An atypical heme in the cytochrome b6f complex. Nature. 426:413–418. [DOI] [PubMed] [Google Scholar]
- 3.Smith, J. L., H. Zhang, J. Yan, G. Kurisu, and W. A. Cramer. 2004. Cytochrome bc complexes: a common core of structure and function surrounded by diversity in the outlying provinces. Curr. Opin. Struct. Biol. 14:432–439. [DOI] [PubMed] [Google Scholar]
- 4.Hope, A. B. 2000. Electron transfers amongst cytochrome f, plastocyanin and photosystem I: kinetics and mechanisms. Biochim. Biophys. Acta. 1456:5–26. [DOI] [PubMed] [Google Scholar]
- 5.Xia, D., C.-A. Yu, H. Kim, J.-Z. Xia, A. M. Kachurin, L. Zhang, L. Yu, and J. Deisenhofer. 1997. Crystal structures of the cytochrome bc1 complex from bovine heart mitochondria. Science. 227:60–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang, Z., L. Huang, Y.-I. Chi, K. K. Kim, A. R. Crofts, E. A. Berry, and S.–H. Kim. 1998. Electron transfer by domain movement in cytochrome bc1. Nature. 392:677–684. [DOI] [PubMed] [Google Scholar]
- 7.Iwata, S., J. W. Lee, K. Okada, J. K. Lee, M. Iwata, B. Rasmussen, T. A. Link, S. Ramaswamy, and B. K. Jap. 1998. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 281:64–71. [DOI] [PubMed] [Google Scholar]
- 8.Izrailev, S., A. R. Crofts, E. A. Berry, and K. Schulten. 1999. Steered molecular dynamics simulation of the Rieske subunit motion in the cytochrome bc1 complex. Biophys. J. 77:1753–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heimann, S., M. V. Ponamarev, and W. A. Cramer. 2000. Movement of the Rieske iron-sulfur protein in the p-side bulk aqueous phase: effect of luminal viscosity on redox reaction of the cytochrome b6f complex. Biochemistry. 39:2692–2699. [DOI] [PubMed] [Google Scholar]
- 10.Yan, J., and W. A. Cramer. 2003. Functional insensitivity of the cytochrome b6f complex to structure changes in the hinge region of the Rieske iron-sulfur protein. J. Biol. Chem. 278:20925–20933. [DOI] [PubMed] [Google Scholar]
- 11.De Vitry, C., Y. Ouyang, G. Finazzi, F. A. Wollman, and T. Kallas. 2004. The chloroplast Rieske iron-sulfur protein. At the crossroad of electron transport and signal transduction. J. Biol. Chem. 279:44621–44627. [DOI] [PubMed] [Google Scholar]
- 12.Soriano, G. M., L. W. Guo, C. De Vitry, T. Kallas, and W. A. Cramer. 2002. Electron transfer from the Rieske iron-sulfur protein (ISP) to cytochrome f in vitro. Is a guided trajectory of the ISP necessary for competent docking? J. Biol. Chem. 44:41865–871 [DOI] [PubMed] [Google Scholar]
- 13.Martinez, S. E., D. Huang, A. Szczepaniak, W. A. Cramer, and J. L. Smith. 1994. Crystal structure of the chloroplast cytochrome f reveals a novel cytochrome fold and unexpected heme ligation. Structure. 2:95–105. [DOI] [PubMed] [Google Scholar]
- 14.Martinez, S. E., D. Huang, M. Ponomarev, W. A. Cramer, and J. L. Smith. 1996. The heme redox center of chloroplast cytochrome f is linked to a buried five-water chain. Protein Sci. 5:1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chi, Y. I., L. S. Huang, Z. Zhang, J. G. Fernandez-Velasco, and E. A. Berry. 2000. X-ray structure of a truncated form of cytochrome f from Chlamydomonas reinhardtii. Biochemistry. 39:7689–7701. [DOI] [PubMed] [Google Scholar]
- 16.Pearson, D. C., Jr., E. L. Gross, and E. S. David. 1996. The electrostatic properties of cytochrome f: Implications for docking with plastocyanin. Biophys. J. 71:64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soriano, G. M., M. V. Pomamarev, R. A. Piskorowski, and W. A. Cramer. 1998. Identification of the basic residues of cytochrome f responsible for electrostatic docking interactions with plastocyanin in vitro: relevance to the electron transfer reaction in vivo. Biochemistry. 37:15120–15128. [DOI] [PubMed] [Google Scholar]
- 18.Gong, X. S., J. Q. Wen, N. E. Fisher, S. Young, C. J. Howe, D. S. Bendall, and J. C. Gray. 2000. The role of individual lysine residues in the basic patch on turnip cytochrome f for the electrostatic interactions with plastocyanin in vitro. Eur. J. Biochem. 267:3461–3468. [DOI] [PubMed] [Google Scholar]
- 19.Redinbo, M. R., D. Cascio, M. K. Choukair, D. Rice, S. Merchant, and T. O. Yeates. 1993. The 1.5 Å crystal structure of plastocyanin from the green alga Chlamydomonas reinhardtii. Biochemistry. 32:10560–10567. [DOI] [PubMed] [Google Scholar]
- 20.Gross, E. L. 1996. Plastocyanin: structure, location, diffusion, and electron transfer mechanisms. In Oxygenic Photosynthesis: the Light Reactions, D. Ort and C. Yocum, editors. Kluwer Academic Publishers, Dordrecht, The Netherlands. 413–429.
- 21.Gross, E. L., and D. C. Pearson, Jr. 2003. Brownian dynamics simulations of the interaction of Chlamydomonas cytochrome f with plastocyanin and cytochrome c6. Biophys J. 85:2055–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sigfridsson, K. 1998. Plastocyanin, an electron-transfer protein. Photosynth. Res. 57:1–28. [Google Scholar]
- 23.Durell, S. R., J. K. Labanowski, and E. L. Gross. 1990. Modeling the electrostatic potential field of plastocyanin. Arch. Biochem. Biophys. 277:241–254. [DOI] [PubMed] [Google Scholar]
- 24.Kerfeld, C. A., and D. W. Krogmann. 1998. Photosynthetic cytochromes c in cyanobacteria, algae, and plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 49:397–425. [DOI] [PubMed] [Google Scholar]
- 25.Gupta, R., Z. He, and S. Luan. 2002. Functional relationship of cytochrome c6 and plastocyanin in Arabidopsis. Nature. 417:567–571. [DOI] [PubMed] [Google Scholar]
- 26.Merchant, S., and L. Bogorad. 1986. Regulation by copper of the expression of plastocyanin and cytochrome c552 in Chlamydomonas reinhardtii. Mol. Cell Biol. 6:462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kerfeld, C. A., H. P. Anwar, R. Interrante, S. Merchant, and T. O. Yeates. 1995. The structure of chloroplast cytochrome c6 at 1.9 Å resolution: evidence for functional oligomerization. J. Mol. Biol. 250:627–647. [DOI] [PubMed] [Google Scholar]
- 28.Ullmann, G. M., M. Hauswald, A. Jensen, N. M. Kostic, and E. W. Knapp. 1997. Comparison of the physiologically equivalent proteins cytochrome c6 and plastocyanin on the basis of their electrostatic potentials. Tryptophan 63 in cytochrome c6 may be isofunctional with tyrosine 83 in plastocyanin. Biochemistry. 36:16187–16196. [DOI] [PubMed] [Google Scholar]
- 29.Haddadian, E. J., and E. L. Gross. 2005. Brownian dynamics study of cytochrome f interactions with cytochrome c6 and plastocyanin in Chlamydomonas reinhardtii, plastocyanin and cytochrome c6 mutants. Biophys. J. 88:2323–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ullmann, G. M., E. W. Knapp, and N. M. Kostic. 1997. Computational simulation and analysis of dynamic association between plastocyanin and cytochrome f. Consequences for the electron-transfer reaction. J. Am. Chem. Soc. 119:42–52. [Google Scholar]
- 31.Pearson, D. C., Jr., and E. L. Gross. 1998. Brownian dynamics study of the interaction between plastocyanin and cytochrome f. Biophys. J. 75:2698–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Rienzo, F., R. R. Gabdoulline, M. C. Menziani, P. G. De Benedetti, and R. C. Wade. 2001. Electrostatic analysis and Brownian dynamics simulation of the association of plastocyanin and cytochrome f. Biophys. J. 81:3090–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gross, E. L. 2004. A Brownian dynamics study of the interaction of Phormidium laminosum plastocyanin with Phormidium laminosum cytochrome f. Biophys. J. 87:2043–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reference deleted in proof.
- 35.Musiani, F., A. Dikiy, A. Y. Semenov, and S. Ciurli. 2005. Structure of the intermolecular complex between plastocyanin and cytochrome f from spinach. J Biol Chem. 280:18833–18841. [DOI] [PubMed] [Google Scholar]
- 36.Soriano, G. M., M. V. Ponomarev, G. S. Tae, and W. A. Cramer. 1996. Effect of the interdomain basic region of cytochrome f on its redox reactions in vivo. Biochemistry. 35:14590–14598. [DOI] [PubMed] [Google Scholar]
- 37.Zhou, J., J. G. Fernandez-Velasco, and R. Malkin. 1996. N-Terminal mutants of chloroplast cytochrome f: Effect on redox reactions and growth in Chlamydomonas reinhardtii. J. Biol. Chem. 271:6225–6232. [DOI] [PubMed] [Google Scholar]
- 38.Haddadian, E. J., and E. L. Gross. 2006. A Brownian Dynamics study of the effects of cytochrome f structure and deletion of its small domain in interactions with cytochrome c6 and plastocyanin in Chlamydomonas reinhardtii. Biophy. J. 90:566–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berman, H. M, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, L. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 18: 2714–2723. [DOI] [PubMed] [Google Scholar]
- 41.Nicholls, A., and B. Honig. 1991. A rapid finite-difference algorithm, utilizing successive over-relaxation to solve the Poisson-Boltzmann equation. J. Comp. Chem. 12:435–445. [Google Scholar]
- 42.Matthew, J. B. 1985. Electrostatic effects in proteins. Annu. Rev. Biophys. Chem. 14:387–417. [DOI] [PubMed] [Google Scholar]
- 43.Warwicker, J., and H. C. Watson. 1982. Calculation of the electric potential in the active site cleft due to alpha-helix dipoles. J. Mol. Biol. 157:671–679. [DOI] [PubMed] [Google Scholar]
- 44.Moser, C. C., C. C. Page, R. Farid, and P. L. Dutton. 1995. Biological electron transfer. J. Bioenerg. Biomembr. 27:263–274. [DOI] [PubMed] [Google Scholar]
- 45.Moser, C. C., J. M. Keske, K. Warncke, R. S. Farid, and P. L. Dutton. 1992. Nature of biological electron transfer. Nature. 355:796–802. [DOI] [PubMed] [Google Scholar]
- 46.Northrup, S. H. 1996. Theoretical simulation of protein-protein interactions. In Cytochrome c: A Multidisciplinary Approach. R. A. Scott and A. G. Mauk, editors. University Science Publishers, Sausalito, CA. 543–570.
- 47.Northrup, S. H., J. A. Luton, J. O. Boles, and J. C. Reynolds. 1987. Brownian dynamics simulation of protein association. J. Comput. Aided Mol. Des. 1:291–311. [DOI] [PubMed] [Google Scholar]
- 48.Northrup, S. H., J. O. Boles, and J. C. Reynolds. 1987. Electrostatic effects in the Brownian dynamics of association and orientation of heme proteins. J. Phys. Chem. 91:5991–5998. [Google Scholar]
- 49.Northrup, S. H., J. O. Boles, and J. C. Reynolds. 1988. Brownian dynamics of cytochrome c and cytochrome c peroxidase association. Science. 241:67–70. [DOI] [PubMed] [Google Scholar]
- 50.Northrup, S. H., K. A. Thomasson, C. M. Miller, P. D. Barker, L. D. Eltis, J. G. Guillemette, S. C. Inglis, and A. G. Mauk. 1993. Effect of charged amino acid mutations on the bimolecular kinetics of reduction of yeast iso-1-ferricytochrome c by bovine ferrocytochrome b5. Biochemistry. 32:6613–6623. [DOI] [PubMed] [Google Scholar]
- 51.Kannt, A., S. Young, and D. S. Bendall. 1996. The role of acidic residues of plastocyanin in its interaction with cytochrome f. Biochim. Biophys. Acta. 1277:115–126. [PubMed] [Google Scholar]
- 52.Lee, B. H., T. Hibino, T. Takabe, P. J. Weisbeek, and T. Takabe. 1995. Site-directed mutagenetic study on the role of negative patches on silene plastocyanin in the interactions with cytochrome f and photosystem Int. J. Biochem. (Tokyo). 117:1209–1217. [DOI] [PubMed] [Google Scholar]
- 53.Gong, X. S., J. Q. Wen, N. E. Fisher, S. Young, C. J. Howe, D. S. Bendall, and J. C. Gray. 2000. The role of individual lysine residues in the basic patch on turnip cytochrome f for the electrostatic interactions with plastocyanin in vitro. Eur. J. Biochem. 267:3461–3468. [DOI] [PubMed] [Google Scholar]
- 54.Schlarb-Ridley, B. G., D. S. Bendall, and C. J. Howe. 2002. Role of electrostatics in the interaction between cytochrome f and plasatocyanin of the cyanobacterium Phormidium laminosum. Biochemistry. 41:3279–3285. [DOI] [PubMed] [Google Scholar]
- 55.Ubbink, M., M. Ejdeback, B. G. Karlsson, and D. S. Bendall. 1998. The structure of the complex of plastocyanin and cytochrome f, determined by paramagnetic NMR and restrained rigid-body molecular dynamics. Structure. 6:323–335. [DOI] [PubMed] [Google Scholar]
- 56.Ejdeback, M., A. Bergkvist, B. G. Karlsson, and M. Ubbink. 2000. Side-chain interactions in the plastocyanin-cytochrome f complex. Biochemistry. 39:5022–5027. [DOI] [PubMed] [Google Scholar]
- 57.Crowley, P. B, D. M. Hunter, K. Sato, W. McFarlane, and K. Dennison. 2004. The parsley plastocyanin-turnip cytochrome f complex: a structurally distorted but kinetically functional acidic patch. Biochem. J. 378:4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lange, C., T. Cornvik, I. Diaz-Moreno, and M. Ubbink. 2005. The transient complex of poplar plastocyanin with cytochrome f: effects of ionic strength and pH. Biochim. Biophys. Acta. 1707:179–188. [DOI] [PubMed] [Google Scholar]