

Abstract
Electron paramagnetic resonance (EPR) spectroscopy using site-directed spin-labeling is an appropriate technique to analyze the structure and dynamics of flexible protein regions as well as protein-protein interactions under native conditions. The analysis of a set of protein mutants with consecutive spin-label positions leads to the identification of secondary and tertiary structure elements. In the first place, continuous-wave EPR spectra reflect the motional freedom of the spin-label specifically linked to a desired site within the protein. EPR spectra calculations based on molecular dynamics (MD) and stochastic dynamics simulations facilitate verification or refinement of predicted computer-aided models of local protein conformations. The presented spectra simulation algorithm implies a specialized in vacuo MD simulation at 600 K with additional restrictions to sample the entire accessible space of the bound spin-label without large temporal effort. It is shown that the distribution of spin-label orientations obtained from such MD simulations at 600 K agrees well with the extrapolated motion behavior during a long timescale MD at 300 K with explicit water. The following potential-dependent stochastic dynamics simulation combines the MD data about the site-specific orientation probabilities of the spin-label with a realistic rotational diffusion coefficient yielding a set of trajectories, each more than 700 ns long, essential to calculate the EPR spectrum. Analyses of a structural model of the loop between helices E and F of bacteriorhodopsin are illustrated to demonstrate the applicability and potentials of the reported simulation approach. Furthermore, effects on the motional freedom of bound spin-labels induced by solubilization of bacteriorhodopsin with Triton X-100 are examined.
INTRODUCTION
Proteins provide a large diversity of functions due to their complex intrinsic structures. The structural hierarchy of these macromolecules comprises distinct internal dynamics that span a wide range of different timescales: Atomic fluctuations, leading to bond-stretching and angle-bending motions, have periods of several femtoseconds, whereas collective motions of associated atoms show correlation times in the picosecond time range. The correlation times of side-chain motions as well as local backbone fluctuations can reach up to a few nanoseconds, whereas rotational correlation times of entire monomeric proteins are in the order of 10−8–10−6 s. Finally, conformational switching and associated movements of protein domains generally occur in the nano- to millisecond time range (1–3).
Electron paramagnetic resonance (EPR) spectroscopy is a sensitive tool to detect molecular dynamics in almost all of these time ranges. In the special case of continuous-wave X-band EPR spectroscopy, as utilized here, reorientational correlation times of nitroxide radicals between ∼100 ps and 200 ns are detectable. In particular, EPR in combination with site-directed spin-labeling (4) is appropriate to study the dynamics of proteins under physiological conditions providing important insights into their functional mechanisms. Spin-label side chains are introduced at selected sites via cysteine substitution mutagenesis followed by modification of the thiol group with a specific paramagnetic nitroxide reagent. To assure unique labeling, accessible native cysteines have to be replaced by similar substitutes like serines. A widely used nitroxide reporter group is (1-oxy-2,2,5,5-tetramethylpyrrolinyl-3-methyl)-methanethiosulfonate (MTS) (5), which reacts with cysteines under formation of a disulfide bond. The bulkiness of the generated side chain, in the following designated as “R1” (Fig. 2), resembles that of a large native amino acid. Nevertheless, both the correct folding and the functional properties of the spin-labeled protein have to be verified experimentally.
FIGURE 2.

Polar histogram plots of the dihedral angle distributions are assigned to all bonds of the cysteine-bound MTS spin-label (R1) essential for the reorientational motion of R1. The shaded stick-representation shows the structure of R1 (hydrogens neglected). The unpaired electron is located in the π-orbital of the nitroxide bond (NX-OX). To estimate the internal flexibility of a bound spin-label during MD simulations under different conditions, important rotameric states of R1 attached to the center of a pentadeca glycine helix (model system) are visualized. The angular scales of all histogram plots are similarly oriented, and a dihedral angle of zero defines the cis (syn-periplanar) configuration. The shaded lines indicate the results of an MD simulation of the model system with explicit water performed at 300 K (50 ns). These plots are compared with the data of two in-vacuo simulations performed at 600 K (black lines, 6 ns) and 300 K (light-shaded solid graphs, 40 ns), respectively.
The shape of the continuous-wave EPR spectrum reflects the reorientational motion of the nitroxide side chain, which arises from different contributions: internal dynamic modes of the R1 side chain, local backbone fluctuations, interaction with neighboring side chains, and conformational changes and rotational diffusion of the entire protein. Spectral effects due to the latter contribution can be easily suppressed by increasing the viscosity of the protein solution (6). Hence, side-chain motions and local backbone fluctuations are considered to be the predominant influences on the EPR spectral features. The residual mobility of the spin-label side chain leads to partial averaging of the anisotropic contributions of the g- and the hyperfine tensors. Both the anisotropic amplitudes and rates of the reorientational motion are reflected in the line shape of the EPR spectrum, providing direct information on the local structure and dynamics of the protein. Multifrequency EPR approaches (7) and molecular dynamics (MD) simulations provide the means by which to separate these two contributions. In this article, we show that the X-band spectra at different spin-label positions in the protein are mainly determined by the specific reorientational freedom of the nitroxide group.
In principle, a molecular dynamics simulation of a bound spin-label within the specific protein environment provides all required dynamic information to calculate an EPR spectrum. To perform such a simulation, a virtual spin-label has to be introduced at the desired site in a model of the protein structure. However, a direct spectrum calculation, using the method first published by Robinson et al. (8), requires a large set of trajectories, each with a length of several hundred nanoseconds, due to the long transversal relaxation time of the nitroxide. For proteins, the calculation of MD trajectories with the required length is beyond the limits of acceptable technical and temporal expense. A first-generation EPR spectra simulation method was developed by Steinhoff and Hubbell (9), which avoids unrealistic computational effort, nevertheless taking into account the complex motion of a spin-label side chain in a realistic protein environment. This method consists of three steps:
An MD simulation to determine an effective potential of the global reorientational motion of the nitroxide.
Several single particle stochastic dynamics (SD) simulations with consideration of this potential.
The calculation of the EPR spectrum on the basis of the SD trajectories.
The MD simulation is performed at high temperatures (600 K) to focus on the amplitude and anisotropy of the spin-label motion. The required length of the trajectory of atomic positions amounts to a few nanoseconds only, during which the complete accessible conformational space of the nitroxide side chain is covered. Since the essential information is the reorientational motion of the nitroxide, expressed in the Euler angle space, an effective potential energy function is derived from the Euler angle trajectory Ω(α,β,γ)(t). The final set of reorientation trajectories, each >700-ns long, is calculated by an essentially faster SD simulation algorithm. In the present approach, this is achieved by solving the Langevin equation for reorientational motion of a single particle restricted by the potential energy topology. One-hundred-sixty Euler trajectories, obtained from the single particle simulation, are used to calculate the Larmor frequencies and magnetization trajectories. The EPR absorption spectrum is finally given by the real part of the Fourier transform of the averaged magnetization trajectory.
This article presents an improved simulation pathway with special consideration of MD simulations to justify the sampling at 600 K. The simulation approach is further applied to study the dynamics of nitroxide side chains bound to several positions of the membrane protein bacteriorhodopsin (BR) and to shed light on the relation between the nitroxide dynamics and the binding site structure. BR is a light-driven proton pump located in the cytoplasmic membrane of, e.g., Halobacterium salinarium (10). It consists of seven transmembrane helices enclosing the chromophore retinal (Fig. 1 a). In the native purple membrane, BR molecules are present as trimers, which further aggregate to form a two-dimensional lattice (11). Since the invention of crystallization techniques to promote three-dimensional BR/lipid crystals, the structure of BR has been determined by different groups at adequate resolution. Besides analyses of the MD simulation approach, the presented method for EPR spectra simulation is verified here by comparison of calculated spectra, based on the E-F loop conformation consistently presented in different crystal structures of BR, to those spectra experimentally obtained under physiological and solubilized conditions. For this purpose, the EPR spectra of 18 single mutants with a nitroxide side chain at positions 157–171 (Fig. 1 a) were studied. The EPR experiments, performed at 9.5 GHz (X-band), and the interpretation of the experimental data of these mutants, were described elsewhere (12).
FIGURE 1.

Structure of the bacteriorhodopsin monomer (1BRR, Chain B) as published by Essen et al. (13). (a) All investigated positions (157–171) are located in the cytoplasmic part of helices E and F or in the connecting loop. Retinal, the linear chromophoric group, is visualized in gray. (b) Superpositions of six different models of bacteriorhodopsin obtained by x-ray and EM techniques (black: 1BRR (13); blue: 1C3W (32); red: 1CWQ (31); gray: 1QHJ (35); yellow: 2AT9 (54); and green: 1FBB (33)).
METHODS
Molecular dynamics simulations
Apart from results presented in Fig. 9, the structural basis of all spectra simulations is the monomer B (amino acids 2–232, completed side chains) of the homotrimeric BR structure 1BRR published by Essen et al. (13). The virtual R1 side chain was modeled successively at each position investigated by EPR (157–171) preventing atom-atom distances closer than the sum of their van der Waals radii. The structural perturbation caused by the spin-label was in general small and only a few rotameric states of nearby side chains were affected. This is in good agreement with the relatively high tolerance of native backbone conformations regarding amino-acid substitutions (14,15). From the experimental point, Pfeiffer et al. (12) showed that all spin-labeled mutants of BR involved in this study revealed photocycle rates in the same order of magnitude as that of the wild-type BR. Marginal structural perturbations due to the introduced R1 side chain are assumed to induce the slight deviations observed. According to the published data (12), considerable reduction of the photocycle rates was only induced by labeling of the very-buried positions 170 and 171. To save computation time of the MD simulation, atomic subsets were applied containing all amino acids lying within a sphere of at least 16 Å radius around the R1's β-carbon atom. In some cases, residues of the adjacent monomer A had to be considered additionally. Hydrocarbon groups with only steric relevance were combined to extended carbon atoms. This is reasonable because the rotation correlation time of a methyl group amounts to ∼5 ps at 300 K, thus it is unable to affect the continuous-wave X-band EPR spectrum measured at room temperature. After the introduction of R1 in BR, an energy minimization with positionally restricted backbone atoms was applied to relax all side-chain conformations.
FIGURE 9.

Superposition of the wild-type structure (1BRR, shaded ribbon) (13) and the triple-mutant structure (1FBK, black/dark-shaded ribbon) (33) of BR representing the ground state and a structural model of BR with an outwardly tilted helix F, respectively. Results of the motional freedom of R1 bound to the positions 167 (a), 170 (b), and 171 (c) are presented. The Cα atoms of all three positions in both structural models are shown in ball-representation. The outward tilt of helix F in 1FBK facilitates an increased motional freedom of R1 at position 167, 170, and the inwardly oriented R1 side chain at position 171. (a,b) The black mesh contours (smoothed) represent almost the entire accessible space (>90%) of the nitroxyl nitrogen atom (NX) during the in-vacuo MD simulations (600 K, 8 ns) of R1 at positions 167 and 170, whereas the solid contours depict states of higher residence probability. The simulated spectra (shaded line) are compared with the measured spectra of the solubilized spin-labeled BR (solid line) on the right-hand side. (c) The three contours in the left figure indicate almost the entire sampled space of NX during MD simulations (600 K, in vacuo, 8 ns) with different starting orientations of R1. The shaded contour refers to the MD simulation of the inwardly oriented spin-label bound to the wild-type structure of BR (see Fig. 5), whereas the solid contour marks the motional freedom of R1 bound to the triple-mutant structure (inwardly oriented), and the solid dotted contour is the result of the outwardly oriented R1 bound to the triple-mutant structure. Due to a high steric barrier between the in- and outward orientations of position 171, separate simulations for both cases were necessary. Simulated EPR spectra obtained from both MD simulations of the triple-mutant structure (inwardly oriented, shaded dotted; outwardly oriented, shaded line) are compared with the measured spectrum (middle position, solid line) of R1 bound to position 171 of the solubilized BR.
Energy minimizations and MD simulations were performed with the GROMOS96 (BIOMOS, Groningen, the Netherlands) program package. The GROMOS96 simulation engine provides an approved and well-optimized force field. In addition, all chemical and physical properties of the simulated system can be adapted to the operator's demand. An essential prerequisite for realistic sampling of the conformational space of R1 is a correct description of its geometric, electrostatic, and dynamic properties. Hence, a specific parameter set for R1 was introduced manually according to the GROMOS libraries (43A1/43B1) and the crystallographic data (16). The planar conformation of the five-membered nitroxide ring was stabilized by improper dihedral potentials. Different results of several quantum mechanical calculations have revealed that the investigation of well-defined atomic partial charges is a difficult task, especially for radical groups (17–19). The finally assigned values for the R1 atoms had to be furthermore scaled to correspond to the partial charges of the environmental protein atoms. Hence, we utilized the values according to a similar configuration (TEMPO) in the GROMOS library (OX: −0.2; NX: −0.04; C3/C4: 0.12). With respect to published values (17,19) and our own data (investigated by ESP calculation of ROHF/6-31G*; program: GAMESS-US), a stronger polarization (OX: −0.3; NX: 0.08; C3/C4: 0.11) was additionally applied in a 300 K explicit water MD simulation (R1 bound to a glycine helix) without significant differences for the R1 mobility. However, recent EPR spectra simulations of R1 in positively charged protein environments showed better agreement with the measured spectra if the second set of partial charges was used even for in vacuo MD simulations. The dynamic behavior of the nitroxide side chain has been extensively analyzed using MD simulations under various conditions:
Simulations with a single neutral-capped R1 amino acid within 755 molecules of water.
Simulations of a pentadeca glycine α-helix with a R1 substitution at position 10 surrounded by 948 water molecules.
In-vacuo simulations of protein subsets at different temperatures.
The used MD simulation conditions of 600 K, in vacuo, and additional restrictions are an outcome of detailed investigations presented in Results and Discussion. The final system temperature was achieved after two heating procedures. A first MD simulation started with atomic velocities assigned randomly from a Maxwell distribution at 400 K. This high initial temperature required the application of position restraints on all atoms to prevent structural bursts. Despite these restrictions, collective vibrational modes were able to establish during a run time of 6 ps. During the following simulation, the temperature was increased from 400 K to 600 K by weak coupling to a 600 K heat reservoir. This final heating step was performed with position restraints assigned only to the backbone atoms N, C, O (see Fig. 2) of all amino acids. Although the desired temperature was generally achieved within the first 15 ps, the applied heating time of 30 ps ensured a sufficient equilibration determined by converging total energy values.
For the main simulations of at least 6-ns length, time steps of 2 fs were used while all bond lengths were kept constant using the SHAKE algorithm (20). Due to the recent gain in computational power, simulation periods of 10–20 ns are recommended (see also Fig. 3 b). To reach rare conformational states and to assure statistical sufficient sampling within 6 ns, all force constants and Lennard-Jones coefficients were kept unchanged. All heavy atoms of the polypeptide main chain except the α-carbons (backbone N, C, O) of helices or β-sheets were restricted by harmonic position restraints. The restraining force constant was chosen to allow a mean deviation of 0.01 nm from the reference position at 600 K. For soluble proteins, weaker position restraints are suggested. If a crystal structure obtained at room temperature is available, realistic force constants can be estimated from the Debye-Waller factors of the backbone atoms. MD simulations (GROMACS) at 600 K with different force constants were applied to analyze the mean-square fluctuations of the backbone atoms and to relate these values to experimental B-factors considering the equation B = 8π2〈Δx2〉 [Å2]. Applied position restraint force constants of 500 and 100 [kJ mol−1 nm−2] yield mean-square fluctuations that correlate with B-factors of ∼27 and 74 [Å2], respectively. The position restraints are not only necessary to keep the peptide fragments of the protein subsets at their physiological positions but also to inhibit rotations of the whole subset or helical components. The samples utilized for EPR investigation in this article consist of native purple membrane sheets including a high concentration of BR trimers. Hence, rotation of entire BR molecules as well as significant tumbling of complete helices is negligible below 1 μs. The probable spectral contribution of the overall rotation of solubilized BR is discussed later. We further assume that deformation modes of helices, due to a simultaneous loss of some H-bonds, hardly occur within the EPR timescale (<200 ns). The high reestablishing capability of such hydrogen bonds should at least prevent distinct perturbations of the motional modes of R1. Hence, the position restraints applied to N, C, O were additionally used to properly stabilize the peptide planes and hydrogen bonds within the helical backbone. On the other hand, the side chains and all backbone atoms of the core of the EF loop (residues 160–164, Fig. 1 a) were kept unrestricted to allow free conformational sampling. RMSD analyses revealed that the nonrestriction of α-carbons of helical amino acids do not significantly perturb the backbone structure. Further, the flexible α-carbons are expected to distinctly improve the authenticity of side-chain motions.
FIGURE 3.

(a) Front and side view of the model system containing a cysteine-bound MTS spin-label (black sticks) within a helix of 15 glycine amino acids (black ribbon). The contour surfaces cover the space that can be occupied by the nitroxyl nitrogen atom NX in the headgroup of the spin-label during a 6-ns, in-vacuo MD simulation at 600 K. The enclosing transparent area (light shaded, smoothed) limits the entire accessible space of the nitrogen, whereas the inner contour (dark-shaded, smoothed) qualitatively indicates the area with distinctly higher residence probabilities, which are located close to the peptide surface. (b) Each line or solid graph represents the accumulated covered volume of NX during different MD simulations. The covered volume was calculated as the sum of voxels (size: 1 Å3) sampled at least once during the prior simulation time. Results of MD simulations (600 K, in vacuo) of the spin-label bound to the most exposed positions within the E-F loop of BR (161–165) are given in the left panel. In increasing order of the final value: 163 (black solid), 164 (dark-shaded solid), 161 (shaded solid), 165 (light-shaded solid), and 162 (black line). The right panel shows the covered volumes by NX within the model system obtained under three different simulation conditions: 600 K, in vacuo, 20 ns (shaded line); 300 K, in vacuo, 40 ns (black line); and 300 K, explicit water, 50 ns (light-shaded solid graph). Both 300 K plots are sums of two independent simulations with -S-S- dihedral angles ∼90° (first half) and 270° (second half) each. The vertical lines at 20 ns and 25 ns mark the transitions between these simulations. The ordinates of both figures are identical. Specific conditions of the 600 K MD simulations are given in the text.
The MD simulations result in position coordinate trajectories of all atoms. The trajectories of in vacuo simulations and simulations with explicit water were collected in steps of 200 fs. To enable a proper statistical analysis, the coordinates of the R1 nitroxide ring were interpolated up to a final step size of 10 fs or 5 fs in the case of very exposed spin-label positions. Trajectories of the reorientational dynamics of the radical group are necessary to calculate the spectrum. Hence, the respective coordinates had to be transformed into the Euler angle space to obtain this specific information. The nitroxide frame has been defined using the atomic coordinates of the nitrogen and the two adjacent carbons C3 and C4 of the ring. Due to the increased bending vibration of the nitroxide oxygen at 600 K, its coordinates were not involved in these calculations. Nevertheless, strong improper dihedral potentials prevented larger distortions of the plane ring structure and of the NX, OX, C3, C4 plane. According to a right-handed nitroxide frame definition, the y axis is given by the vector connecting C3 and C4, the z axis is calculated from the vector product of the distance vectors C3-NX and NX-C4, and the x axis is then oriented perpendicular to both (approximately parallel to NX-OX). The conversion into the Euler angle space was performed by the transformation matrix of the y-convention (21), and the resulting trajectory consisted of Euler angle triplets Ω = (α,β,γ), where α,γ ∈ [−π,π) and β ∈ [0,π).
Apart from the final spectra calculation the mean-square fluctuation of the Euler angle β, mainly reflecting the reorientation of the nitroxide π-orbital, has been used to estimate the mobility of R1 (22). A more detailed comparison of simulated and experimental mobility parameters are given in Results and Discussion.
The potential energy topology
An effective potential energy function that determines the nitroxide reorientational motion was calculated from the simulated orientation probabilities. For this purpose the Euler angle space was divided into 500,000 unit cells (Θ) enabling a resolution of 3.6° in α, β, and γ. This resolution has been chosen to assure that the orientation distribution of the most rigid spin-label position led to the occupation of at least five cells in each angle dimension. The hits within every unit cell (Θ) were counted, normalized with respect to the total number of hits, and assigned to the center of the cell. The three-dimensional histogram, thus obtained, reflects the complex population distribution of the nitroxide orientations. Due to the limited sampling time of 6 ns, the topology is distinctly modulated by noise. This noise can cause large fluctuations of the potential-dependent motion during the single particle simulation and should be eliminated without influencing the global topology. Smoothing algorithms that utilize averaging techniques tend to an undesirable broadening of sharp peaks caused by that noise. To avoid this problem we applied the MEDIAN smoothing algorithm (23) with the smallest filter matrix (3x3x3) to fully eliminate the sharp noise peaks. The perturbation of the global topology due to the MEDIAN filtering has been proven to be less than that caused by an averaging square filter. Since the MEDIAN technique can only substitute original values with already existing adjacent values, it tends to form small steplike plateaus. However, this effect is less significant when smoothing is performed in high dimensions. If necessary, an additional square filtering could be applied to straighten out these remaining plateaus. The edges of the grid can be easily smoothed taking advantage of the periodic boundary conditions in α, β, and γ.
In the next step, the probability topology W(Θ) was converted into a free energy topology Eeff(Θ),
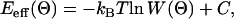 |
(1) |
where Θ(Ω) denotes the α, β, γ triple of the centers of the unit cells; kB is the Boltzmann constant, T the temperature (300 K), and C an arbitrary constant.
The single particle simulation
The calculation of a proper EPR spectrum requires reorientation trajectories with lengths of a few hundred nanoseconds (8). Such trajectories were obtained by the numerical solution of the potential-dependent Langevin equation for a single particle undergoing rotational diffusion (24):
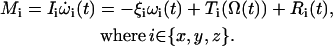 |
(2) |
The equation provides three contributions to the general torque Mi of a particle having a moment of inertia Ii. The first term on the right side represents the friction torque on a particle with the drag coefficient ξi and an angular velocity ωi(t) in the x, y, or z direction. The drag coefficient is related to the rotational diffusion coefficient Di by the Einstein equation:
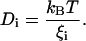 |
(3) |
The expression Ti(Ω(t)) is the torque due to an effecting potential and is given by the gradient of the free energy topology for an orientation Ω:
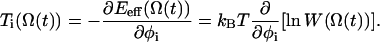 |
(4) |
The value of Eeff at a distinct orientation Ω is obtained from the discrete free energy topology Eeff(Θ) by trilinear interpolation in the Euler angle space. By means of the last term Ri of Eq. 2, a stochastic torque is implemented to mimic the coupling with the solvent. The value Ri satisfies the fluctuation-dissipation theorem
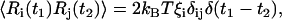 |
(5) |
where δij is the Kronecker and δ(t) the Dirac delta function. According to the proposed algorithm (20,25) for solving Eq. 2 and for Ii/ξi ≪ Δt, the reorientation φi(tn+Δt) is given by
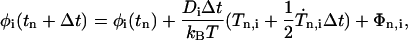 |
(6) |
with the torques Tn,i = Ti(Ω (tn)). The value Tn,i is further assumed to be approximately constant during each time step. Thus, the derivative of Tn,i can be neglected. Under consideration of Eqs. 3 and 4 we get a conventional Brownian dynamics equation:
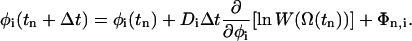 |
(7) |
The last term Φn,i of Eqs. 6 and 7 is a random variable that reflects the contribution of Ri. The value Φn,i meets the rules of a random walk with
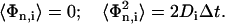 |
(8) |
The probability distribution Pi of Φi is given by the Fick's second law leading to a Gaussian distribution with zero mean and a mean-square value of 2Dit:
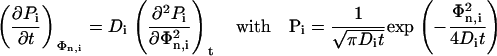 |
(9) |
The arithmetic implementation of this Gaussian probability distribution was made using a normally distributed random number generator.
We generated a set of 160 single particle trajectories for each label position with lengths of 720 ns (18,000 data points per trajectory). The time step per iteration of the simulations was 2.5 ps. Starting orientations were obtained from randomly chosen angle triplets of the Euler angle trajectory of the MD simulation. The average W(Θ) histogram of these 720-ns trajectories showed an excellent agreement with that of the original MD trajectory for all analyzed R1 positions.
Spectra calculation
The calculation of the EPR spectrum is based on the approach described in detail by Steinhoff and Hubbell (9). Due to the anisotropy of the g- and A-tensors, the Larmor frequency ω depends on the orientations Ω(t) of the nitroxide group with respect to the external magnetic field B0, which is defined to be parallel to the z-direction of the laboratory frame. The Larmor frequency trajectory is determined from the energy eigenvalues of the spin Hamiltonian in the laboratory frame. An appropriate expression of the spin Hamiltonian is
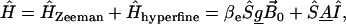 |
(10) |
with g-tensor g and hyperfine tensor A, both diagonal in the nitroxide frame. The value βe is the Bohr magneton, S and I are the electron and nuclear spin operators. This Hamiltonian is converted from the nitroxide frame into the molecular frame of the protein using a transformation matrix L, which is a function of the Euler angles Ω, where index t denotes transposition:
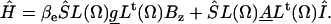 |
(11) |
Considering the protein reorientation with respect to the B-field, a second transformation matrix L′ is necessary to finally transform the Hamiltonian into the laboratory frame. In the case of BR embedded in membrane sheets, the rotational correlation time τBR of the protein is distinctly larger than the upper limit of the sensitive timescale of X-band EPR of ∼200 ns. Hence, the molecular frame can be treated as static within the laboratory frame in the presented EPR spectra simulations.
To determine the eigenvalues of the Hamiltonian, two convenient approximations of Eq. 11 are utilized taking into account that the local nuclear magnetic field is small compared to the external field. The electron spin is, therefore, mainly quantitized along the z axis, whereas the x- and y-contributions of S can be neglected (intermediate field treatment). In the absence of motion, the Hamiltonian is then given by
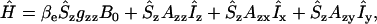 |
(12) |
with the expressions for gzz and Aij given in Steinhoff and Hubbell (9).
An approximation for fast reorientational motion assumes that the influence of the electron magnetic dipole on the nuclear spin becomes less significant in relation to the influence by the external magnetic field (26). Both the electron and nuclear spin are then quantized along the axis of the external magnetic field. The pseudosecular terms SzIx and SzIy of Eq. 12 vanish (high field treatment). Considering the autocorrelation function of the Euler angle β, a weight function is applied to define a correct approximation between the two motional limits for a given spin-label mobility. This function facilitates a smooth transition between the high-field and intermediate-field treatments. A detailed description of this approach is presented elsewhere (9,26,27). From the energy eigenvalues, the energies of the allowed transitions are calculated for the spin-label orientations Ω(t), which finally yield the Larmor frequency trajectory ω(t). For the presented spectra calculations, two different datasets for the principal values of g and A were applied, depending on the polarity of the predominant environment of R1 during the MD simulation. According to fits of several low-temperature EPR spectra of well-characterized positions in BR (e.g., 46, 100, 101, 129, 171, 193), we assigned principle values for water-exposed spin-label positions (g: 2.0083; 2.0065; 2.0026 and A: 0.56; 0.48; 3.65 [mT]) and for protein intrinsic R1 orientations (g: 2.0085; 2.0065; 2.0027 and A: 0.57; 0.41; 3.58 [mT]). A data set containing 320 trajectories of the complex magnetization M+(t) was subsequently calculated from ω(t) by a numerical recursive, approximate solution of the Bloch equation:
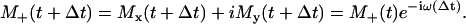 |
(13) |
This equation includes the assumption that the Larmor frequencies can be discretized over a small time step Δt, equal to the time step of the Euler angle trajectory (40 ps). The assumption was initially discussed by Itzkowitz (26) and indirectly justified via comparison of resulting EPR spectra with the results of Freed's MOMD model (9). To consider additional line broadening, the magnetization trajectories M+(t) were multiplied by exp(−t/T2). The relaxation time T2 depends on the local environment of the spin-label. However, the variation is small, and T2 values in the range of (20 ± 2) ns revealed agreement between simulated and measured spectra in almost every case. In addition, a convolution with a Gaussian function may be applied to account for inhomogeneous line broadening, e.g., due to unresolved hyperfine interactions with the methyl protons of the nitroxide ring.
To simulate the spectrum of a sample of randomly oriented spin-labeled protein molecules, the final magnetization trajectory was calculated as an average of 320 trajectories of a representative set of 256 different protein orientations (∼82,000 trajectories). The corresponding magnetization amplitudes were scaled by the sine of the angle between the protein z-axis and the magnetic field. The EPR absorption spectrum is then given by the real part of the Fourier transform of the averaged magnetization trajectory. To allow a convenient comparison of simulated and experimental spectra, all spectra are presented as the first derivative of the related EPR absorbance spectrum.
An EPR-related mobility parameter determined from MD simulations
A basic treatment is given here to obtain an EPR-related mobility parameter directly from the MD trajectories. Due to the definition of the nitroxide coordinate system (z-axis parallel to the p-orbital of NX) and the rotation sequence of the Euler transformation (around z, y′, z″), the Euler angle β specifies the angle between the orientation of the p-orbital of NX and the magnetic field vector, fixed to the z-axis of the laboratory frame. To allow comparisons between MD simulations showing different mean orientations of R1, the z-axis of the laboratory frame is further arbitrarily defined to be parallel to the average orientation of the p-orbital of NX (β′ = β−〈β〉). The population distributions in the Euler angle space are then centered around β′ = 0 for each spin-labeled mutant. This is reasonable under the assumption that the distributions of molecule orientations are isotropic in all measured BR samples. The hyperfine tensor of the used spin-label is nearly axially symmetric and the principle values can be approximated by Axx = Ayy = A⊥ and Azz = A‖, where A‖ is 7–8 times larger than A⊥ for R1. Since we focus our attention to the spectral contributions of the largest spectral splitting, the spatially averaged value of A‖ in the laboratory frame has to be considered. According to Steinhoff and Karim (28), this value can be approximated for rapid reorientational motion by
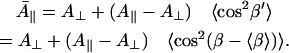 |
(14) |
As discussed by Steinhoff and Karim (28), Eq. 14 includes the simplification that the main orientations of the p-orbital of NX are roughly isotropically distributed around the magnetic field vector or show rotationally symmetric characteristics. This is treated to be reasonable because most of the investigated R1 sites revealed roughly spherical or ellipsoidal topologies of the mainly populated orientation states in the Euler angle space. The variance of the cosine of β′ (Eq. 14) is used here as an EPR-related mobility parameter of the MD simulations.
Solubilization of BR
The investigated samples of membrane-embedded BR mutants were prepared by M. Pfeiffer according to Pfeiffer et al. (12). Solubilization of selected mutants in Triton X-100 was performed as follows.
After 5 min of ultra-sonification to initially reduce the size of the membrane sheets, extraction of BR from the membrane-embedded state was performed by adding at least 300 Triton X-100 detergent molecules per BR monomer corresponding to a concentration of ∼6 mM Triton X-100. This procedure causes the loss of all trimer-trimer and monomer-monomer contacts of BR (29) and inhibits the ability of lipids to form large membrane bilayers. Additional EPR spectra of spin-labeled BR samples with higher excess of Triton X-100 (6–20 mM) were obtained to investigate the structural stability of solubilized BR. Triton-containing samples were prevented from intense radiation to avoid photobleaching. After the solubilization procedures, a characteristic shift of the absorbance maximum of the protein-bound retinal from 570 nm to ∼553 nm (30) was observed. Precipitation of solubilized BR mutants was not observed after prolonged centrifugation (1 h with 16,000 g).
RESULTS AND DISCUSSION
A representative selection of recently published structures of BR is superimposed in Fig. 1 b. The major part of the protein structure is well defined by x-ray and cryo-electron microscopy studies. Although the published BR structures agree well in the transmembrane region of the protein, including positions 165–171, they show distinct differences particularly in the conformation of the cytoplasmic loop between helices E and F. Both presented cryo-EM models (Fig. 1 b; 2AT9, 1CWQ) represent the highest conformational diversity of the loop. Especially the loop structure presented in 1CWQ (31) reveals at least four positions with distinct discrepancies between the predicted degrees of reorientational freedom of R1 and the measured EPR spectra. The expected mobility of R1 bound to the two most exposed E-F loop positions (160, 161) of this structure disagree with the respective EPR spectra, which indicate relatively immobilized R1 side chains, whereas the partially buried positions 164 and 165 show EPR spectra of high R1 dynamics. Furthermore, the low collision frequency between the nitroxide attached to positions 157–161, 167 and the polar quencher chromium-oxalate do not correspond to the water-exposed residue orientations in this loop conformation (12). Most of the x-ray structures are characterized by uncertain E-F loop structures due to diffuse or absent electron densities in this region (13,32,33). However, the loop structure published by Essen et al. (1BRR) reveals no obvious discrepancies to the EPR spectra (13,34). This loop structure is furthermore almost identical to the E-F loop of the structures 1QHJ (35) and 1FBB (33) (Fig. 1 b) and the structure 1IW6 (36) (not shown). The present EPR spectra simulation algorithm is not expected to reveal a clear preference for only one of these very similar conformations. Hence, in this study a detailed comparison between experimental and simulated spectra on the basis of the conformation reported by Essen et al. (13) is performed to elucidate the capabilities of the presented EPR spectra simulation method. Since this structure consists of a complete trimer, the adjacent monomers were involved in the MD simulations as well. Approximately 10 C-terminal amino acids could not been adequately resolved in any model of BR, thus they were not considered.
MD simulations at different temperatures
The MD simulations were performed to sample solely the complex anisotropy of the reorientational motion of the nitroxide ring. It has been shown by LaConte et al. (37) that in vacuo MD simulations at 300 K are sufficient to calculate high order parameters, reflecting a high degree of immobilization of the spin-label. Although the applied simulations seem to yield realistic librational motion of the bound spin-label FDNASL, the question of whether the complete accessible conformational space is sampled within the 14-ns simulation time has not been addressed.
Here, we provide evidence that R1 side chains in exposed loop positions reveal much larger motional amplitudes in the time-range of EPR sensitivity. Special simulation parameters were chosen to focus on an entire conformational scanning, in which all significant restrictions are included. R1 bound to the loop position 165 of BR was selected to analyze the sampled Cartesian and Euler angle space at different temperatures, where the covered Euler angle space represents the reorientational freedom of the nitroxide. This initial investigation is based on the three-dimensional topologies of population densities calculated for each temperature (data not shown). Position 165 provides a solvent-exposed location and considerable opportunities for nitroxide contacts to the protein surface. Fifteen in-vacuo MD simulations (20 ns each) in the temperature range from 300 K to 900 K were performed, and the results were confirmed by simulations (300–750 K) for further positions with minor spatial restrictions (E161R1 and S162R1). We observed a drastic increase in the covered Euler angle space up to 550 K followed by only slight changes of the angle distributions between ∼550 K and 700 K. More frequent transitions over activation barriers of subsets of attractive van der Waals interactions between the nitroxide and the protein surface at 500–550 K, in-vacuo, are suggested as an explanation for the observed behavior. This enables a less hindered diffusion of the nitroxide headgroup along the protein surface. In fact, the revealed orientation distributions below ∼550 K are narrow and shift drastically their locations within the Euler angle space induced by temperature change. As already known, simulations at 300 K without explicit water molecules show a distinct tendency to enhance the internal density of the protein and exhibit an unrealistically strong surface tension. This effect is due to the presence of only protein-intrinsic, therefore anisotropic, nonbonded interactions and the absence of compensatory, mainly attractive protein-water interactions (e.g., H-bond and attractive van der Waals forces). Hence, particularly peripheral side chains, like those of the E-F loop amino acids, show an obviously restricted motion in 300 K in vacuo MD simulations. The observed restrictions seem to be too large even in relation to the hydrophobic effect resulting from real water environments. Increasing the temperature to 600 K weak short-range forces, like attractive van der Waals interactions, are assumed here to lose significance due to the related higher kinetic energy applied to each atom. In fact, on the basis of the used Lennard-Jones potentials, an increased mean separation of only 0.1 nm between the R1 headgroup and the protein surface could roughly halve the energy necessary to break a set of attractive van der Waals interactions. Above 550 K a global distribution is established with almost constant positions of the highest population densities for all analyzed R1 binding sites. In particular for in vacuo simulations, the degree of surface association of exposed side chains at 600 K is suggested to accomplish a more realistic behavior than at 300 K. The presented suggestions are substantiated by a detailed inspection of the side-chain dynamics at 300 and 600 K, given in the following sections. Above ∼700 K a tendency in the reorientational motion of R1 becomes dominant toward more uniform population densities in the entire accessible conformational space. At these temperatures, attractive van der Waals interactions are assumed to lose the ability to bias the motion of the spin-label.
Transition rates and EPR timescale
Although we neglect all kinetic information of the MD simulations for EPR spectra simulation purposes, an analysis of the transition rates is nevertheless important to estimate whether the populations of the found conformational states of R1 are equilibrated within the MD simulation time. Moreover, additional restrictions have to be implemented, if essential potential barriers are too small with respect to the kinetic energy at 600 K. The EPR spectral shape of a nitroxide at X-band frequencies is sensitive to reorientational motions with correlation times of ∼100 ps to 200 ns. Analyzing the reorientational dynamics of the R1 side chain during simulations at several temperatures, we identified two categories of transitions that are not within the EPR time window but nevertheless affect either the general diffusion rate or the maximum reorientation amplitude of R1.
The first category includes transitions over small energy barriers (in the order of 10 kJ/mol) established by individual attractive van der Waals, H-bond, and most of the Coulomb interactions involving the R1 side chain. An explicit-water MD simulation of R1 utilizing the OPLSAA force field and the TIP4P water implementation of GROMACS (38) was performed to study the lifetime of H-bonds between the nitroxide oxygen and surrounding water molecules. Using a polarization of the nitroxide group of OX: −0.3; NX: 0.08; C3/C4: 0.11 (partial charges) we could estimate an average lifetime of <1 ps. This value correlates with a transition barrier of ∼6 kJ/mol estimated by the Eyring equation and considering a symmetric bidirectional decay of the transition state. Due to the weak polarity of R1, the lifetimes of such individual interactions are usually shorter than 100 ps (barrier height of a transition between two states below 18 kJ/mol). Hence, we assume that a further increase in such high transition rates due to an MD simulation at 600 K do not alter the reorientation potential of R1 obtained by the MD simulation. However, the frequent appearance of these interactions leads to a significant contribution to the friction on R1 (e.g., diffusion along the protein surface). This effect is considered macroscopically using a realistic diffusion rate during the single particle simulation. Nevertheless, the cumulative strength of their collective occurrence, especially in the case of van der Waals interactions, causes the main restrictions even to the motional amplitude of the spin-label within the time range of EPR sensitivity. This will later be analyzed on a more macroscopic level and compared with the features of the hydrophobic effect. Strong Coulomb interactions could significantly bias the EPR spectrum. Hence, two different sets of partial charges of the nitroxide atoms (see Methods) were investigated empirically. Although surface-associated partial charges already appear too strong in in-vacuo MD simulations due to the neglecting of a surrounding dielectricum, the larger charge separation (OX: −0.3; NX: 0.08; C3/C4: 0.11) revealed slightly more consistent results for R1 in polar protein environments. However, further investigations might be necessary to optimize the settings. It is noteworthy that the tested charge separations have the same order of magnitude as the previously mentioned uncertainty in the quantum-mechanical determination of the partial charges for the nitroxide atoms.
The second category includes all strong restrictions concerning bond lengths, angle bending, repulsive van der Waals forces, etc. which cannot be overcome within 200 ns at 300 K (beyond the rigid limit). These barriers determine the principal restrictions of the R1 motion. Due to the very steep slope of such potential barriers, the gain in conformational freedom during MD simulations at 600 K with respect to 300 K is relatively small. Only slight corrections of the MD potentials were necessary to avoid unrealistic motion amplitudes at 600 K. The SHAKE algorithm was used to eliminate variations of all bond lengths. Improper dihedral potentials stabilized, e.g., the nitroxide headgroup and all amid planes, whereas weak position restraints, applied to the backbone atoms N, C, O of all helices, sustained the integrity of the helical H-bond network. Finally, a relatively high charge separation, especially in the nitroxide group, was used to facilitate sufficient Coulomb interactions involving R1.
To address the question of whether the helical backbone fluctuations significantly influence the spectral features, we determined the linewidth of the most buried positions on helix E (154,155) and helix F (170, 171). Experimental values of between eight and nine Gauss, which are similar to the linewidths of powder spectra, indicate a very rigid protein environment. Especially for position 171 the effective Azz value of 3.47 mT measured at room temperature was found to be close to the Azz value of the solid state (3.51 mT) obtained at 160 K. With the assumption that the backbone fluctuations through an extended part of a helix are of the same order of magnitude, the motion of the helical backbone in the vicinity of the E-F loop of BR can be considered negligible.
Intrinsic flexibility of the R1 side chain
An absolute prerequisite for a proper motion of the bound spin-label in 600 K MD simulations is a still-realistic intrinsic flexibility. To analyze the internal degrees of freedom of R1 under different conditions, we focus on the dihedral potentials—the weakest type of bonded interactions between covalently bound atoms. Fig. 2 presents histogram plots of the torsional states of all flexible bonds within the R1 side chain. The revealed torsional restrictions, based on the implemented dihedral potentials, are in addition partially amplified by, e.g., the angle-bending potentials and the 1–4 interactions. Using improper dihedrals, all atoms of the five-membered ring including the nitroxyl oxygen were restricted in the widely accepted coplanar conformation (39). MD simulations of our model system were used to compare the distributions of the essential dihedral angles under different conditions. The flexibility of R1 bound to a short glycine helix was determined from simulations at 300 K, both in vacuo (40 ns) and with consideration of 948 water molecules (50 ns), and the proposed in vacuo simulation at 600 K (6 ns). The results represent the dynamical behavior of an exposed protein-bound spin-label within a hydrophilic peptide environment.
In the case of the Cα-Cβ (χ1) bond, the familiar three-peaked plot was obtained showing the favored gauche/trans states of sp3-hybridized carbons. The peak positions are in perfect agreement with results of other χ1 dihedral analyses of native side chains, especially methionines and cysteines (40,41). A first indication for overestimated restrictions at 300 K in vacuo is that the 60° state hardly occurs during the entire 40-ns simulation time. Both -C-S- plots reveal almost ideally staggered conformations, which is also consistent with previously published results (42,43). The torsion angle distributions of the -S-S- bond show two characteristic states at ∼90° or 270°. In these cases the lone-pair electrons in the 3pπ atomic orbitals of both sulfur atoms are orthogonal to each other and their repulsion is minimized. An exhaustive survey about the electronic properties of the disulfide conformations is given by Boyd (44). As discussed later, transitions between these two low-energy orientations are unlikely to occur at room temperature within a few nanoseconds, whereas they were observed in all 600 K MD simulations (average lifetime is ∼1 ns). Hence, all presented histograms obtained at 300 K are 1:1 superpositions of two simulations with different initial values of the disulfide dihedral angle (90° or 270°). Apart from the high transition barrier, Jiao et al. (45) and Maxfield et al. (43) found that the energy penalty for distorting both ground states within 30° in each direction is small. The angle distributions determined here from 600 K MD simulations are in adequate agreement with these results. Rotation around the last bond -Cɛ-C1- shows the highest flexibility and most frequent transitions. With exception of the syn- and anti-periplanar conformations, almost all orientations are populated. If the entire R1 side chain is located in a more restrictive environment, the rotation around -Cɛ-C1- is dominant. Since this bond is nearly parallel to the nitroxide bond, it mainly affects the reorientational motion of R1 around the x-axis of the spin-label system.
Comparing the torsional distributions obtained under the three different conditions (Fig. 2), it is evident that the general positions of highest and lowest populated states are in good agreement. Even at 600 K the relatively weak dihedral potentials lead to acceptable anisotropic distributions. Nevertheless, as visible particularly for both -C-S- bonds, the sparsely populated energy-rich orientations are slightly more frequently occupied at 600 K. This indication of an increased internal flexibility of R1 does not necessarily result in a significantly higher orientational freedom of the nitroxide headgroup since particular orientations of the nitroxide can generally be achieved with different sets of dihedral angles. The dihedral angle distributions of R1 attached to exposed positions of BR display shapes similar to that of the presented 600 K MD simulation of the model system. However, the population ratios of the frequently occupied states differ due to the specific environmental restrictions. Buried or motionally highly restricted R1 side chains provide very narrow distributions of the dihedrals. A predominant steric hindrance can induce several torsion angles to occupy energy-rich values. Such limited gains in the potential energy of R1 are assumed not to be in contradiction to the behavior under experimental conditions.
Due to the implemented additional restrictions in the 600 K MD simulations mentioned previously, we assume that only a few conformational changes might occur during a 600 K simulation which are probably not observable by X-band EPR spectroscopy (lifetimes above 100 ns at 300 K). Referring to the time range of EPR sensitivity, the dihedral reorientation of the disulfide bond is the most critical remaining transition. Both energetic barriers between the two -S-S- ground states at 90° and 270° seem to be very high compared to other single bond dihedral potentials in proteins. Most of the published values are in the order of 25–40 kJ/mol and 37–50 kJ/mol for transitions via the trans and the cis conformations, respectively (44–46). For the preferred trans, transition lifetimes of several hundred nanoseconds can be estimated for both ground states at 300 K. On the other hand, the average lifetime determined by in vacuo MD simulations at 600 K amounts to ∼1 ns only. This is the result of seven trajectories of the most exposed label positions within the loop of BR. The -S-S- dihedral potential is approximated by the GROMOS force-field using a sinusoidal function with a potential barrier of 33.4 kJ/mol. Additional contributions are repulsion forces due to angle bending and the 1–4 Lennard-Jones interaction between Cβ and Cɛ, which lead to an asymmetric potential with a less favored cis transition. Thus, the cis/trans energy barriers used by GROMOS seem to be close to the upper limits of the published values. The expected discrepancy in the dihedral flexibility of disulfide bonds at 300 K and 600 K cannot be fixed without distinct expense. A higher potential barrier may avoid transitions during the 600 K simulations. In this case we have to perform two independent simulations—one for each dihedral ground state. In addition, a third 600 K MD simulation with unaltered or lower barrier height would be necessary to obtain the fractions of R1 side-chain orientations at 90° and 270° within a macroscopic EPR sample. However, torsional transitions around the disulfide bond stimulate compensating collective reorientations of almost all other dihedral angles within the R1 tail. This is necessary to prevent drastic and sudden displacement of the R1 headgroup. Furthermore, a comparison of the covered space of the nitroxide nitrogen during simulations with pure 90° or 270° dihedral orientations shows large overlap. Hence, the total influence on the location of the nitroxide group is relatively weak; see also Fig. 4 (300 K in water simulations) and Fig. 5. According to these results we applied 600 K MD simulations without amplification of the -S-S- dihedral potential. The lifetimes of all other dihedral orientations, shown in Fig. 2, are distinctly shorter due to much smaller potential barriers.
FIGURE 4.

The three parameters of the DTL system are illustrated on the structure of R1: The first parameter is the length of the vector CB-NX. The spreading angle θ is defined by the vectors CA-CB and CB-NX. The dihedral angle λ is the angle between the plane through the vectors N-C and CA-CB and the plane through the vectors CA-CB and CB-NX. θ, λ-histogram plots (TL plots) are efficient for revealing the reorientational behavior of a side chain. Here, TL plots (interval resolution: 1.8°) obtained from R1 of the model system are compared. The outer open areas in the TL plots were not sampled, whereas the gray-scale gradients indicate the histogram counts from black (at least one hit) to white (maximum amount of hits). The upper graph presents R1 orientations obtained from the 600 K, in-vacuo MD simulation (6 ns). Data from 300 K MD simulations with explicit water are visualized in the second line, whereas the results of 300 K, in-vacuo MD simulations are shown in the third line. For both 300 K conditions independent MD simulations for each dihedral ground state of the disulfide bond (∼90° and 270°) were performed (left and middle panel) followed by a superposition of both (right panel). Due to observed transitions between the ground states of the disulfide dihedral within the 600 K MD simulation (see Fig. 2 for the population ratio) the upper graph should be compared with the superpositioned (rightmost) panels of the 300 K MD simulations.
FIGURE 5.

Simulated and experimental EPR spectra of the exposed R1 side chain bound to position 165 of BR. The simulated spectra in panels a–c were calculated based on the reorientational restrictions of R1 during long-time in-vacuo MD simulations at 300 K. Separate spectra simulations (20 ns each) were performed considering R1 conformations in one of both ground states of the disulfide dihedral, i.e., -S-S- of ∼90° (a) or ∼270° (b) (see text). Spectrum calculations based on the superposition of the orientation state populations of both 300 K MD simulations are presented in panel c. For each 300 K MD simulation (a–c), spectra are calculated considering three different rotational diffusion constants during the single particle simulations: D = 0.05 ns−1 (black dashed), 0.2 ns−1 (shaded line), and 0.5 ns−1 (black line). In panel d the measured spectrum (black line) and the simulated spectrum based on a 600 K MD simulation (shaded line) (D = 0.048 ns−1) are given for comparison.
The mobility of the bound R1 side chain
We suggest that in vacuo MD simulations at 600 K might be more appropriate to simulate room temperature EPR spectra than in vacuo simulations at 300 K or MD simulations with explicit water of limited simulation time. This assumption is justified here by a detailed inspection of the conformational space of R1 under different conditions. In this section a small model system containing an R1 side chain within a pentadeca glycine helix has been used to analyze the dynamics of an exposed peptide-bound spin-label under various conditions. Larger structures would require much higher computational effort to achieve simulation times of >50 ns under explicit consideration of surrounding water molecules. The long-time explicit water MD simulations were performed using the efficient SPC/E water model. An estimation of the sampled area and favored conformations of R1 within the model system is visualized in Fig. 3 a.
Accessible sites for spin-labeling are usually in close vicinity to the protein surface. Apart from a small fraction of completely buried label orientations, the motion of the bound spin-label is therefore directly influenced by the hydrophobic effect. Less polar and mainly aprotic groups, like several side chains, perturb the hydrogen-bond network within the surrounding water layer. Thus, a driving force to reduce the surface/volume ratio leads to a compact aggregation of such groups. In particular, the distinctly apolar R1 side chain should generally be in close contact to the protein surface (47). The analysis of the sampled space for the nitroxyl nitrogen atom NX in the model system yields a qualitative representation of the mobility of R1 at 600 K (Fig. 3 a), which is in agreement with the expected motion behavior.
Due to a distinctly decreased diffusion rate at the protein surface in simulations with explicit water, an equilibrated orientation distribution had not been reached within 25 ns. In general, global modes of R1 motion are strongly perturbed by explicit water molecules. This is affirmed by the plot of the covered Cartesian volume of NX, which is a measure of the general sampling rate of R1 conformations, during both 25-ns MD simulations (Fig. 3 b, right panel). As already mentioned, transitions between both ground states of the disulfide dihedral were not observed in all 300 K MD simulations (maximum length: 25 ns). For positions without pronounced steric hindrance, R1 side chains with a disulfide dihedral angle of 90° or 270° should be almost equally represented in a macroscopic sample. Hence, simulations for both R1 species were done for every 300 K MD simulation. Since rare transitions of the disulfide dihedral are expected in long-time dynamics at 300 K, the covered volume of the consecutively summarized trajectories of both species is presented in Fig. 3 b (right panel). In contrast to the in vacuo MD simulations, a nearly constant level of the covered volume, indicating the shortest time to reach a conformational equilibration, was not achieved within the evaluated time window of 50 ns. Furthermore, a comparison of the sampled volumes during all simulations under different conditions clearly reveals that the final value of the covered volume at 300 K in water exceeds the highest level of the screened volume during the 300 K in vacuo simulations. A rough extrapolation of the maximum level for 300 K in water results in a value similar to those of the 600 K in vacuo simulation between 10 and 20 ns.
To perform a detailed inspection of the preferred conformational states within the covered space of R1, we introduce the DTL (distance, theta, lambda) coordinate system shown in Fig. 4. It is represented by
The length of the distance vector V between the atoms Cβ and NX.
The angle θ between the Cα-Cβ-direction and V.
A virtual dihedral angle λ, which is the angle between the plane defined by the vector N-C (backbone orientation) and Cα-Cβ and the plane defined by Cα-Cβ and V, respectively.
Using these linearly independent DTL parameters, the side-chain motion can be expressed in relation to the Cα-Cβ bond and to the local backbone orientation. The length of V is generally in the range of 0.4–0.8 nm. Its value depends strongly on the torsional states of the -C-S- bonds. Histograms of the angle θ and the dihedral angle λ (TL plots) are sufficient to observe all possible orientations of R1 (Fig. 4). The analyzed TL plots of R1 of the model system reveal a high probability of NX positions located within a sinusoidal belt that is defined by the largest possible values of angle θ. This upper limit of θ is given by the steric hindrance due to the helical backbone. Therefore, the frequently occupied belt indicates R1 orientations with close surface contact of the R1 headgroup. It corresponds to the dark-shaded ring of mainly occupied NX positions in the Cartesian space (Fig. 3 a). The sinusoidal shape in the TL plots results from the fact that the plane of this circular distribution of mainly occupied NX positions is not completely perpendicular to the Cα-Cβ bond. R1 orientations without contact to the helix are represented by θ angle values below the belt.
The two TL plots of the 300 K in-vacuo simulations (Fig. 4 below) illustrate the previously suggested strong motional restriction due to an exaggerated surface tension. Here, the simulation with a dihedral angle of ∼270° revealed the highest observed flexibility of R1 at 300 K in vacuo. Although the spin-label is mainly oriented in one direction, diffusion along the surface and weak sampling of most of the feasible orientations are nevertheless visible for both simulations. All performed 300 K MD simulations of R1 on more complex protein surfaces showed distributions similar to the 90° dihedral state behavior (Fig. 4, below-left panel) due to a much larger amount of buried conformational states with higher numbers of van der Waals interactions involving R1 than in the glycine helix model (data not shown).
The motion behavior during the MD simulations with explicit water showed a more uniform screening along the surface, indicative for significantly weaker interactions of R1 to local spots on the peptide surface. Further, the sampling rate of orientations apart from the surface was higher than that of the 300 K in vacuo simulations. In addition to the results of the covered volume analyses, another clear indication for a still not equilibrated conformational screening in the presented explicit water MD simulations is given by distinctly differing TL plots obtained after 16, 19, 22, and 25 ns simulation time. On the other hand, all in vacuo simulations show negligible changes in the TL plots during the last 25% of simulation time (data not shown).
As already discussed, transitions of the disulfide dihedral states have been observed in all 600 K MD simulations but not in 300 K simulations within 25 ns. Hence, superpositions of the obtained probability topologies of both dihedral states were considered for all 300 K simulations (Fig. 4, rightmost panels). These superpositions imply frequent transitions between 90° and 270° within the timescale of EPR sensitivity (≤200 ns). Since this behavior is not realistic, the real conformational restriction of R1 is in between the discrete and superpositioned topologies. Nevertheless, the TL plot obtained from the 600 K MD simulation should be compared with the superpositions (Fig. 4, rightmost panels). Even at 600 K the lack of compensational interactions, due to the absence of solvent molecules, generates a remaining force toward the surface of the peptide. Therefore, as in the presence of a hydrophobic effect, the major orientations of R1 are those with distinct surface association. On the other hand, the high kinetic energy counteracts unrealistic conditions of surface tension and solute density. The ratio of NX positions aggregated and separated from the peptide surface is smaller at 600 K compared to the 300 K MD simulations. However, we assume a similar ratio in the case of the 300 K MD simulations with explicit water when a complete conformational equilibration is achieved (see Fig. 3 b).
The main peak in the TL plot of the 300 K in-vacuo MD simulation (-S-S- = 90°) is identical to the position with the highest occupation probability of the 600 K simulation, but this position was weakly sampled in the explicit water MD simulations (Fig. 4). The area of the peak is characterized by the maximum possible values of θ and a large contact surface to the helix atoms (represented by the surrounding open area). An R1 side chain with such a conformation is wrapped round the glycine helix along the helical groove, yielding the highest number of van der Waals interactions between R1 and the atoms of the helix. The corresponding Cβ-NX distances (first DTL parameter) show the highest variation (0.44–0.76 nm) in relation to regions with other λ-values. Hence, the mentioned area in the TL plots represents a large variety of surface-attached R1 conformations. Interestingly, a 600 K in-vacuo MD simulation with neglected Coulomb interactions (all partial charges of R1 set to zero) reveals a shift of the most probable R1 orientation away from the mentioned area to smaller λ-values. Thus, it seems that a combination of all nonbonded interactions possibly leads to overestimated attractive forces for R1 orientations with intense surface contact in in-vacuo MD simulations even at 600 K.
Due to the relatively high polarizability of sulfur atoms, van der Waals interactions under participation of the disulfide sulfurs are relatively strong. This fact is implemented in the MD force field via the Lennard-Jones parameters for sulfur. In addition, a relevant hydrogen-bond interaction between the Sδ of R1 and the hydrogen atom bound to Cα has been suggested (48). A common method to estimate the strength of hydrogen bonds is a comparison of the measured atom-atom distances in low-temperature crystal structures with the corresponding sum of their van der Waals radii. If sulfur atoms are involved, this method might fail (49). Atoms, like sulfur, with occupied high level electronic orbitals (due to hybridization) and/or lone pairs are not well defined by an isotropic electron density distribution provided by the spherical van der Waals model. Thus, the Lennard-Jones potential should be considered as rather nonspherical. Because hydrogen bonds are predominantly electrostatic interactions, the small permanent partial charges of both disulfide sulfur atoms as well as Cα-hydrogen atoms (absolute values <0.2 e) indicate a weak ability to form significant hydrogen bonds at 300 K (50). This is even valid in the case of the stronger polarized cysteines (51). Neither the 600 K in-vacuo MD simulations nor the explicit water simulations revealed any distinct attractive interactions between the sulfur atoms of the R1 side chain and surrounding hydrocarbon groups or other weak hydrogen donors. In the similar case of methionine, with an even more polarized sulfur at the δ-position, Zhao et al. (41) quantified a high side-chain flexibility at both exposed and even buried positions.
Our results justify the assumption that the nitroxide head of exposed R1 side chains mainly performs a rolling motion along the protein surface at 600 K in vacuo and during a long-time explicit water MD simulation at 300 K, whereas 300 K in-vacuo simulations reveal unrealistically restricted conformational sampling of R1. The strong motional restrictions of R1 in 300 K in-vacuo MD simulations lead to simulated EPR spectra showing drastic immobilization. As an example, spectra of R1 bound to the exposed position 165 of BR are given in Fig. 5. Here, in-vacuo MD simulations (300 K: two trajectories of 20 ns each, 600 K: 10 ns) were used to calculate the EPR spectra. Spectra directly calculated from the 300 K simulations with different disulfide dihedral angles are presented in Fig. 5, a and b, whereas the spectra of Fig. 5 c are based on the superposition of the reorientational distributions of both simulations. As already mentioned, this superposition implies frequent transitions between the two dihedral states and presents, therefore, a probably unrealistic upper limit of the R1 mobility at 300 K. Spectra calculations were done using three different rotational diffusion constants D during the single particle simulation (always performed at 300 K). The highest value of D (0.5 ns−1) corresponds to a correlation time of τ = 330 ps in case of isotropic reorientational motion. Lower correlation times are not expected for protein-bound spin-labels. A comparison between all spectra based on the 300 K MD simulations and the measured spectrum displays distinct differences for all values of D applied, whereas the spectrum based on the 600 K simulation agrees with the experiment (Fig. 5 d).
Another promising result of the presented MD simulation technique concerns the determination of spin-spin distances between two bound spin-labels. According to previously published data, we were able to simulate distance distributions, which are in good agreement with the distributions determined from the measured spectra (52,53).
MD simulation time length
The final question is whether the applied length of the 600 K in vacuo MD simulations is sufficient to screen the entire accessible space of the nitroxide group. It is shown that the nitroxyl nitrogen NX of R1 in the model system samples almost the entire accessible space during a simulation time of 6 ns (Fig. 3 a). Furthermore, the almost complete manifestation of the areas with high residence probability (Fig. 3 a, dark-shaded ring) is already present after the first 3–4 ns. However, a quantitative analysis of the very large covered space of NX in the model system revealed that, despite a rapid convergence, an absolutely constant level could not be reached within 6 ns (Fig. 3 b, right panel). Plots of the covered volume of NX of R1 bound to the most mobile region of the E-F loop (positions 161–165) are presented in Fig. 3 b (left panel). Based on two-exponential fits of the last 200 ps, the still remaining slopes (derivations at 6 ns) were calculated. The relative slopes at 6 ns with respect to the final absolute values are reduced to levels between only 2.0% and 2.8% volume gain per nanosecond for all analyzed positions (Fig. 3 b, left panel). The results presented below are based on MD simulations with 6-ns time length. Nevertheless, simulation times of 10–20 ns are suggested as a better tradeoff between accuracy and computational effort for future EPR spectra simulations.
Comparison of MD and EPR-related mobility parameters
The use of simple mobility parameters, as presented in Fig. 6, facilitate direct comparisons between the motion restrictions of R1 during the applied MD simulations and the shape of the respective experimental EPR spectra. The variation of the cosine of the Euler angle β′ (see Eq. 14), as a mobility parameter determined from the MD simulations, is compared here with the square-root of the second central moment (Ispec)1/2 of the EPR absorbance spectra. The second-moment value is very sensitive for shifts of spectral contributions in the outer parts of the spectrum. The largest spectral splitting depends on the degree of spatial averaging of the hyperfine interaction, which is proportional to 〈cos2(β′)〉. An approximately linear correlation between 〈cos2(β′)〉 and (Ispec)1/2 is given for R1 sites with small spatial averaging of the A-tensor. The value Ispec was in general obtained from spectra of BR embedded in purple membrane. As discussed in the next section, Ispec values determined for solubilized BR are presented for positions 157, 161, 168, and 169 to provide better analogy to the MD simulation conditions. The presented comparison confirms an approximately linear behavior for R1 positions with restricted mobility (Fig. 6). The high deviations for exposed R1 positions indicate that especially for large reorientational amplitudes the reorientational rates are not in the fast motional limit. Further, shifts of spectral contributions caused by A⊥ are not negligible concerning Ispec in this motion regime.
FIGURE 6.

Comparison of mobility parameters obtained from MD simulations and EPR measurements of the BR loop region (157–171). The variance of the cosine of (β–〈β〉) (see Eq. 14) calculated from MD trajectories (600 K, in vacuo) is used to visualize the reorientational restrictions of the nitroxide headgroup during the simulations. These values are compared with the square-root of the second central moments (Ispec)1/2 of the respective measured EPR spectra. The experimental mobility parameters are in general taken from spectra of trimeric BR embedded in the purple membrane (▪), whereas spectra measured under solubilized conditions are applied for positions 157, 161, 168, and 169 (□) (see text). A linear dependency is indicated by the straight regression line. The MD simulations of 160 and 171i revealed two distinctly separated agglomerations in the histograms of the Euler angle triplets offering different average β-angles. In both cases, the variances were calculated separately, and the population-weighted averages are shown here. The difference between both, almost equally populated variances of position 160 amounts to 0.07, whereas the difference of 171i is negligible (0.002).
Simulated EPR spectra
Fig. 7 represents calculated EPR spectra based on the molecular and stochastic dynamics simulations described. The simulation algorithm is verified here, using the conformation of the E-F loop of BR as published in 1BRR (13). Simulated EPR spectra of spin-labels bound to positions 157–171 are compared with the respective measured spectra. Small remaining fractions (<5%) of unbound spin-labels cause an additional spectral contribution, visible only in the measured spectra. This contribution is characterized by sharp peaks close to the zero-crossings of the low-, middle-, and high-field EPR lines. The degree of the observed motional freedom of R1 ranges from very restricted, as, e.g., for the buried positions 170 and 171, to mobile, as revealed for exposed loop positions (e.g., 162, 164, 165). Spin-labels characterized by large reorientational freedom show spectra with smaller effective hyperfine splittings and narrow linewidths. The shape of the positive peak of the low-field line is appropriate to distinguish between mobile and immobile components. For every odd residue Fig. 7 depicts a structural representation with two cutoff contours for the population density of the nitroxide nitrogen position determined from the related MD simulation at 600 K. These images are further assumed to illustrate the expected accessible space of the nitroxide during a long-timescale MD simulation under physiological conditions. They clearly indicate a high probability (black area) of nitroxide orientations with considerable contact to the protein surface (e.g., figures of positions 161, 163, and 165). Hence, a regular surface association occurred in consistence with the behavior expected due to the hydrophobic effect. However, it should be mentioned that this visualization is not able to give information about some rotational degrees of freedom, especially concerning the torsion around Cɛ-C1, which additionally bias the spectrum simulation.
FIGURE 7.

Simulated EPR spectra based on in-vacuo MD simulations at 600 K compared to EPR spectra measured at room temperature of R1 at positions 157–171 of bacteriorhodopsin. The B-field range, which covers all components of the low-field peak from the most immobile (left limit) to the most mobile (right limit) case, is indicated by a shaded bar. The spectra labeled by an asterisk show distinct differences between measurement and simulation, which can be explained by intermolecular contacts (see Fig. 8 and text). A representation of the R1 side chain and its binding site is shown on the left for each odd position. These images additionally visualize the motion behavior of the nitroxide group as determined from the MD simulation (6 ns, 600 K). The shaded meshes mark >90% of the complete accessible space (smoothed) of the nitroxide nitrogen (NX), whereas the solid contours (smoothed) delimit all areas with significantly higher residence probability of NX.
During the modeling of the initial spin-label orientations at positions 160, 170, and 171, two distinct conformations of R1 could be identified. Due to high steric barriers between these predicted conformations, transitions are assumed to be unlikely. Hence, separate simulations with both starting orientations were performed and compared to the measurements. The outwardly oriented spin-label at position 160 and the internal orientation at 170 reveal best agreement with the experimental counterparts, whereas both orientation states at position 171 (shown in Fig. 7) lead to nearly identical EPR line shapes. The conformation of the native residue at position 171, phenylalanine, is similar to the inward orientation of R1.
In general, all simulated spectra with the exception of those of positions 157, 161, 168, and 169 agree with their measured counterparts as particularly revealed by comparison of the shapes of the low-field absorption lines. The main reason for the discrepancies in the high-field region might be a still too simple model of the single particle reorientational motion.
A comparison of the spectra for positions 157, 161, 168, and 169 shows distinct differences between the measured and calculated mobilities. As visible in Fig. 1, all four residues are outwardly oriented with respect to the trimeric complex. The large fraction of the immobile component visible in the four measured spectra (Fig. 7) implies further aggregation apart from the trimer-intrinsic interactions, in line with the fact that BR forms a two-dimensional lattice of trimers within the native purple membrane (11). This additional aggregation results in close contacts particularly between helices F and E of adjacent trimers. The distance between both helical centers is estimated to be <2 nm. Additional steric interactions might further arise from stacking of purple membrane sheets.
To study these trimer-trimer interactions we performed solubilization experiments on several spin-labeled BR mutants. Fig. 8 shows a selection of EPR spectra measured under native and solubilized conditions. Several solubilization procedures of mutants 157, 170, and 171 with higher concentrations of Triton X-100, as mentioned in Methods, were performed without significant changes of the presented spectral shapes. We conclude that the solubilization is almost complete under the conditions described in Methods, and the achieved structure of all solubilized BR mutants is stable. A comparison of the spectra (Fig. 8) reveals a slight general increase in the mobility of all analyzed side chains as shown, e.g., for R1 bound to positions 158, 159, and 162. According to Meyer et al. (30), at least 64 Triton X-100 molecules are directly involved covering one BR monomer (molar mass of this complex: 66 ± 1.5 kDa). A rough estimation of the radius of the complex, treated as a sphere, yields 2.7 nm (average density of a protein: 1.37 g/cm3). Considering Debye's equation, the rotational correlation time of the overall tumbling of one solubilized BR amounts to ∼20 ns, which most likely explains the observed slight general mobilization. However, a limited loss of the structural integrity, which would correlate with the small blue-shift of the optical absorbance maximum of the BR-bound retinal, cannot be excluded.
FIGURE 8.

Overlaid EPR spectra of several labeled positions of bacteriorhodopsin embedded in the native membrane bilayer (shaded dashed) and after solubilization with Triton X-100 (black line).
After solubilization a significant increase in mobility can be observed for buried R1 side chains at helix F, positions 167, 170, and 171. This is evidence for a permanent outward movement of the N-terminal moiety of helix F. To substantiate this assumption, the triple-mutant structure 1FBK (33) of BR (mutations: D96G/F171C/F219L) is used as a model of BR with an outward-tilted, probably relaxed, helix F. EPR spectra simulations of R1 bound to the positions 167, 170, and 171 of this structure are presented in Fig. 9. The revealed increases of the mobility of R1 at positions 170 and 171 (inwardly oriented conformation) lead to an excellent agreement between the simulated spectra and the measurements of the solubilized samples. Even for R1 bound to position 167, the simulated results based on the structures 1BRR and 1FBK display a similar gain in mobility as observed by the experimental spectra under physiological and solubilized conditions (Fig. 8). However, the analyses of these three spin-labeled positions are not sufficient to postulate a detailed model of the solubilized structure. An outward tilt of helix F seems nevertheless to be consistent with our results.
The third effect of the solubilization is a considerable increase in mobility of R1 side chains oriented toward the adjacent BR trimer as well as toward a possibly attached second layer of the purple membrane. The four most prominent examples within the E-F loop region are those positions that show distinct differences between simulation and measurement (157, 161, 168, and 169). Hence, the drastic change in their spectral shapes upon solubilization (Fig. 8) is additional evidence of close trimer-trimer contacts in this area. The simulated spectra of these positions should be compared to the measured spectra of the solubilized samples, because trimer-trimer interactions were not taken into account during the MD simulations. This leads to a distinctly better agreement between simulation and measurement for all four mentioned positions.
The applied value of the rotational diffusion coefficient Di of 0.05 ± 0.005 ns−1 during the single particle simulations was found to provide agreement between calculated and measured spectra for all positions from 157 to 171. The value D mainly represents the friction due to the interaction with the surrounding solvent molecules and/or the lifetimes of particular attractive van der Waals interactions restricting R1 to the protein surface. Hence, the variation of the friction for R1 at different binding sites is treated to be small. However, due to the complex structure of the free energy potential we assume that different effective correlation times can appear during the single particle simulation. We conclude that environmental influences on the nitroxide motion can be satisfyingly described by the reorientational potential and an isotropic diffusion constant. The presented spectra simulation approach implies the assumption that the reorientational dynamics of the nitroxide group is hardly biased by sudden rotational transitions in the R1 tail. This could be reasonable due to the four very unrestricted torsional potentials (see Fig. 2). Interestingly, the strong restriction of the disulfide dihedral angle, leading to a permanently almost perpendicular orientation of both carbon-sulfur bonds, increases the effects of torsions around these carbon-sulfur bonds on the nitroxide reorientation. To provide a more complex dynamic model that includes influences by internal transitions, the implementation of a jump diffusion term to the Brownian dynamics of the single particle simulation is in progress. Another further improvement will consider the different direction-dependent moments of inertia of the nitroxide headgroup. This will lead to an anisotropic diffusion coefficient in the spin-label frame.
CONCLUSION
In this article we have presented an improved method for structure-based simulations of EPR spectra that combines sufficient accuracy with an acceptable computational effort. This approach requires the knowledge of a structural model of the protein, which further include the bound nitroxide spin-label. The EPR spectrum can be directly determined from a large set of reorientation trajectories calculated from a potential-dependent simulation of the reorientational motion of a single particle. This stochastic dynamics simulation combines a realistic motional rate with a potential representing the specific reorientational restrictions of the bound spin-label (R1). To obtain this site-specific potential, it is essential not only to screen the complex accessible space of the nitroxide group within the protein environment but also to ensure realistic preference states of the spin-label conformation. Analyses of MD simulations at different temperatures show that in-vacuo simulations at room temperature do not provide realistic mobility of R1 at any timescale. Nevertheless, MD simulations performed at 600 K and coupled with additional restrictions reveal an acceptable estimation of the orientation distribution of long-time R1 dynamics under physiological conditions. Particularly, the strength of the protein surface aggregation of R1 seems to be sufficiently rendered. In this context the presented DTL parameterization was found to be useful to analyze side-chain motions. The orientation distribution of R1 bound to a model system obtained from a 600 K in-vacuo MD simulation shows similar features compared to long-timescale 300 K MD simulations with explicit water, whereas unrealistically restricted R1 mobility was obtained from 300 K in-vacuo simulations. Due to a sufficient simulation time at 600 K of only 6–10 ns, a spectrum simulation can usually be completed within 2–3 days.
Based on the structure of one trimer of BR molecules the developed simulation approach was used to calculate EPR spectra of BR spin-labeled at positions 157–171. A comparison between simulated and measured spectra of membrane-embedded BR shows good agreement in the low field region, which is very sensitive to the reorientational dynamics of R1. Beside the transmembrane state, solubilized BR samples of mutants with exposed R1 positions represent conditions that are partially closer to the simulation conditions, e.g., there are no trimer-trimer contacts. Thus, revealed discrepancies concerning positions 157, 161, 168, and 169 of membrane-embedded BR could be traced to trimer-trimer contacts. In these cases, measurements on solubilized BR samples yield satisfying agreement between experimental and simulated spectra. On the other hand, clear indications for a local structural change due to solubilization are found. The solubilized spectra of the buried positions 167, 170, and 171 present a distinct increase in mobility, which is explained by an outward tilt of helix F.
Our results provide evidence that the observed spectral diversity of R1 in different protein environments can mainly be dedicated to the motional freedom of the R1 side chain. However, permitted fluctuations of exposed loop regions and of all Cα atoms during the MD simulations cause definite improvement toward a realistic motion of all side chains.
Important applications of the presented spectra simulation technique could be the validation of theoretical models of unresolved protein structures or conformational changes and analyses of intermolecular interactions in the vicinity of the bound spin-label. In addition, this simulation procedure might be an interesting tool to confirm theoretical models obtained from refinements based on homologous protein structures.
Acknowledgments
We gratefully acknowledge the financial support of the Deutsche Forschungsgemeinschaft grant No. STE 640/5-2 and the Volkswagenstiftung grant No. I/78668.
References
- 1.Fayer, M. D. 2001. Fast protein dynamics probed with infrared vibrational echo experiments. Annu. Rev. Phys. Chem. 52:315–356. [DOI] [PubMed] [Google Scholar]
- 2.Frauenfelder, H., S. G. Sligar, and P. G. Wolynes. 1991. The energy landscapes and motions of proteins. Science. 254:1598–1603. [DOI] [PubMed] [Google Scholar]
- 3.McCammon, J. A., and M. Karplus. 1980. Simulation of protein dynamics. Annu. Rev. Phys. Chem. 31:29–45. [Google Scholar]
- 4.Cornish, V. W., D. R. Benson, C. A. Altenbach, K. Hideg, W. L. Hubbell, and P. G. Schultz. 1994. Site-specific incorporation of biophysical probes into proteins. Proc. Natl. Acad. Sci. USA. 91:2910–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berliner, L. J., J. Grunwald, H. O. Hankovszky, and K. Hideg. 1982. A novel reversible thiol-specific spin-label—papain active-site labeling and inhibition. Anal. Biochem. 119:450–455. [DOI] [PubMed] [Google Scholar]
- 6.Timofeev, V. P., and V. I. Tsetlin. 1983. Analysis of mobility of protein side-chains by spin-label technique. Biophys. Struct. Mech. 10:93–108. [Google Scholar]
- 7.Liang, Z. C., Y. Lou, J. H. Freed, L. Columbus, and W. L. Hubbell. 2004. A multifrequency electron spin resonance study of T4 lysozyme dynamics using the slowly relaxing local structure model. J. Phys. Chem. B. 108:17649–17659. [Google Scholar]
- 8.Robinson, B. H., L. J. Slutsky, and F. P. Auteri. 1992. Direct simulation of continuous wave electron paramagnetic resonance spectra from Brownian dynamics trajectories. J. Chem. Phys. 96:2609–2616. [Google Scholar]
- 9.Steinhoff, H. J., and W. L. Hubbell. 1996. Calculation of electron paramagnetic resonance spectra from Brownian dynamics trajectories: application to nitroxide side chains in proteins. Biophys. J. 71:2201–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haupts, U., J. Tittor, and D. Oesterhelt. 1999. Closing in on bacteriorhodopsin: progress in understanding the molecule. Annu. Rev. Biophys. Biomol. Struct. 28:367–399. [DOI] [PubMed] [Google Scholar]
- 11.Grigorieff, N., T. A. Ceska, K. H. Downing, J. M. Baldwin, and R. Henderson. 1996. Electron-crystallographic refinement of the structure of bacteriorhodopsin. J. Mol. Biol. 259:393–421. [DOI] [PubMed] [Google Scholar]
- 12.Pfeiffer, M., T. Rink, K. Gerwert, D. Oesterhelt, and H. J. Steinhoff. 1999. Site-directed spin-labeling reveals the orientation of the amino acid side-chains in the E-F loop of bacteriorhodopsin. J. Mol. Biol. 287:163–171. [DOI] [PubMed] [Google Scholar]
- 13.Essen, L. O., R. Siegert, W. D. Lehmann, and D. Oesterhelt. 1998. Lipid patches in membrane protein oligomers: crystal structure of the bacteriorhodopsin-lipid complex. Proc. Natl. Acad. Sci. USA. 95:11673–11678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim, W. A., D. C. Farruggio, and R. T. Sauer. 1992. Structural and energetic consequences of disruptive mutations in a protein core. Biochemistry. 31:4324–4333. [DOI] [PubMed] [Google Scholar]
- 15.Matthews, B. W. 1995. Studies on protein stability with T4 lysozyme. Adv. Protein Chem. 46:249–278. [DOI] [PubMed] [Google Scholar]
- 16.Lajzerowicz-Bonneteau, J. 1976. Molecular structures of nitroxides. In Spin Labeling I—Theory and Applications. L. J. Berliner, editor. Academic Press, New York. 239–249.
- 17.Barone, V., A. Bencini, M. Cossi, A. Di Matteo, M. Mattesini, and F. Totti. 1998. Assessment of a combined QM/MM approach for the study of large nitroxide systems in vacuo and in condensed phases. J. Am. Chem. Soc. 120:7069–7078. [Google Scholar]
- 18.Schwartz, R. N., M. Peric, S. A. Smith, and B. L. Bales. 1997. Simple test of the effect of an electric field on the N-14-hyperfine coupling constant in nitroxide spin probes. J. Phys. Chem. B. 101:8735–8739. [Google Scholar]
- 19.Improta, R., A. di Matteo, and V. Barone. 2000. Effective modeling of intrinsic and environmental effects on the structure and electron paramagnetic resonance parameters of nitroxides by an integrated quantum mechanical/molecular mechanics/polarizable continuum model approach. Theor. Chem. Acc. 104:273–279. [Google Scholar]
- 20.van Gunsteren, W. F., and H. J. C. Berendsen. 1982. Algorithms for Brownian dynamics. Mol. Phys. 45:637–647. [Google Scholar]
- 21.McClung, R. E. D. 1968. Transformation of spin coordinates and solution of spin Hamiltonian for an orthorhombic paramagnetic center in a rigid lattice. Can. J. Phys. 46:2271–2275. [Google Scholar]
- 22.Steinhoff, H. J., M. Mueller, C. Beier, and M. Pfeiffer. 2000. Molecular dynamics simulation and EPR spectroscopy of nitroxide side chains in bacteriorhodopsin. J. Mol. Liquids. 84:17–27. [Google Scholar]
- 23.Press, W. H., B. P. Flannery, S. A. Teukolsky, and W. T. Vetterling. 1986. Numerical Recipes. Cambridge University Press, Cambridge, UK.
- 24.Wolynes, P. G., and J. M. Deutch. 1977. Dynamical orientation correlations in solution. J. Chem. Phys. 67:733–741. [Google Scholar]
- 25.Branka, A. C., and D. M. Heyes. 1998. Algorithms for Brownian dynamics simulation. Phys. Rev. E. 58:2611–2615. [DOI] [PubMed] [Google Scholar]
- 26.Itzkowitz, M. 1967. Monte Carlo simulation of the effects of molecular motion on the EPR spectrum of nitroxide free radicals. J. Chem. Phys. 46:3048–3056. [Google Scholar]
- 27.Freed, J. H. 1976. Theory of slow tumbling ESR spectra for nitroxides. In Spin Labeling—Theory and Applications. L. J. Berliner, editor. Academic Press, New York. 53–132.
- 28.Steinhoff, H. J., and C. Karim. 1993. Protein dynamics and EPR-spectroscopy—comparison of molecular dynamic simulations with experiments. Ber. Bunsen. Phys. Chem. 97:163–171. [Google Scholar]
- 29.Sasaki, T., M. Sonoyama, M. Demura, and S. Mitaku. 2005. Photobleaching of bacteriorhodopsin solubilized with Triton X-100. Photochem. Photobiol. 81:1131–1137. [DOI] [PubMed] [Google Scholar]
- 30.Meyer, O., M. Ollivon, and M. T. Paternostre. 1992. Solubilization steps of dark-adapted purple membrane by Triton-X-100—a spectroscopic study. FEBS Lett. 305:249–253. [DOI] [PubMed] [Google Scholar]
- 31.Sass, H. J., G. Bueldt, R. Gessenich, D. Hehn, D. Neff, R. Schlesinger, J. Berendzen, and P. Ormos. 2000. Structural alterations for proton translocation in the M state of wild-type bacteriorhodopsin. Nature. 406:649–653. [DOI] [PubMed] [Google Scholar]
- 32.Luecke, H., B. Schobert, H. T. Richter, J. P. Cartailler, and J. K. Lanyi. 1999. Structure of bacteriorhodopsin at 1.55 Ångstrom resolution. J. Mol. Biol. 291:899–911. [DOI] [PubMed] [Google Scholar]
- 33.Subramaniam, S., and R. Henderson. 2000. Molecular mechanism of vectorial proton translocation by bacteriorhodopsin. Nature. 406:653–657. [DOI] [PubMed] [Google Scholar]
- 34.Rink, T., M. Pfeiffer, D. Oesterhelt, K. Gerwert, and H. J. Steinhoff. 2000. Unraveling photoexcited conformational changes of bacteriorhodopsin by time-resolved electron paramagnetic resonance spectroscopy. Biophys. J. 78:1519–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belrhali, H., P. Nollert, A. Royant, C. Menzel, J. P. Rosenbusch, E. M. Landau, and E. Pebay-Peyroula. 1999. Protein, lipid and water organization in bacteriorhodopsin crystals: a molecular view of the purple membrane at 1.9 Ångstrom resolution. Structure. 7:909–917. [DOI] [PubMed] [Google Scholar]
- 36.Takeda, K., Y. Matsui, N. Kamiya, S. Adachi, H. Okumura, and T. Kouyama. 2004. Crystal structure of the M intermediate of bacteriorhodopsin: allosteric structural changes mediated by sliding movement of a transmembrane helix. J. Mol. Biol. 341:1023–1037. [DOI] [PubMed] [Google Scholar]
- 37.LaConte, L. E. W., V. Voelz, W. Nelson, M. Enz, and D. D. Thomas. 2002. Molecular dynamics simulation of site-directed spin-labeling: experimental validation in muscle fibers. Biophys. J. 83:1854–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Mod. 7:306–317. [Google Scholar]
- 39.Makinen, M. W., D. Mustafi, and S. Kasa. 1998. ENDOR of spin-labels for structure determination: from small molecules to enzyme reaction intermediates. In Biological Magnetic Resonance. L. J. Berliner, editor. Plenum Press, New York. 181–245.
- 40.Pal, D., and P. Chakrabarti. 1998. Different types of interactions involving cysteine sulfhydryl group in proteins. J. Biomol. Struct. Dyn. 15:1059–1072. [DOI] [PubMed] [Google Scholar]
- 41.Zhao, S. R., D. S. Goodsell, and A. J. Olson. 2001. Analysis of a data set of paired uncomplexed protein structures: new metrics for side-chain flexibility and model evaluation. Proteins Struct. Funct. Gen. 43:271–279. [DOI] [PubMed] [Google Scholar]
- 42.Jorgensen, F. S., and J. P. Snyder. 1979. Disulfide conformational analysis—the nature of the S-S rotation barrier. Tetrahedron. 35:1399–1407. [Google Scholar]
- 43.Maxfield, F. R., and H. A. Scheraga. 1977. Raman spectroscopic investigation of disulfide conformation in oxytocin and lysine vasopressin. Biochemistry. 16:4443–4449. [DOI] [PubMed] [Google Scholar]
- 44.Boyd, D. 1972. Conformational dependence of the electronic energy levels in disulfides. J. Am. Chem. Soc. 94:8799–8804. [Google Scholar]
- 45.Jiao, D., M. Barfield, J. E. Combariza, and V. J. Hruby. 1992. Ab initio molecular-orbital studies of the rotational barriers and the S-33 and C-13 chemical shieldings for dimethyl disulfide. J. Am. Chem. Soc. 114:3639–3643. [Google Scholar]
- 46.Fraser, R. R., G. Boussard, J. K. Saunders, J. B. Lambert, and C. E. Mixan. 1971. Barriers to rotation about sulfur-sulfur bond in acyclic disulfides. J. Am. Chem. Soc. 93:3822–3823. [Google Scholar]
- 47.Steinhoff, H. J., O. Dombrowsky, C. Karim, and C. Schneiderhahn. 1991. Two-dimensional diffusion of small molecules on protein surfaces—an EPR study of the restricted translational diffusion of protein-bound spin-labels. Eur. Biophys. J. Biophys. Lett. 20:293–303. [DOI] [PubMed] [Google Scholar]
- 48.Langen, R., K. J. Oh, D. Cascio, and W. L. Hubbell. 2000. Crystal structures of spin-labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 39:8396–8405. [DOI] [PubMed] [Google Scholar]
- 49.Jeffrey, G. A., and W. Saenger. 1991. Hydrogen Bonding in Biological Structures. Springer Verlag, New York.
- 50.Burley, S. K., and G. A. Petsko. 1988. Weakly polar interactions in proteins. Adv. Protein Chem. 39:125–189. [DOI] [PubMed] [Google Scholar]
- 51.Scheiner, S., T. Kar, and Y. L. Gu. 2001. Strength of the C(α)-H…O hydrogen bond of amino acid residues. J. Biol. Chem. 276:9832–9837. [DOI] [PubMed] [Google Scholar]
- 52.Kensch, O., T. Restle, B. M. Woehrl, R. S. Goody, and H. J. Steinhoff. 2000. Temperature-dependent equilibrium between the open and closed conformation of the p66 subunit of HIV-1 reverse transcriptase revealed by site-directed spin-labeling. J. Mol. Biol. 301:1029–1039. [DOI] [PubMed] [Google Scholar]
- 53.Steinhoff, H. J. 2004. Inter- and intra-molecular distances determined by EPR spectroscopy and site-directed spin-labelling reveal protein-protein and protein-oligonucleotide interaction. Biol. Chem. 385:913–920. [DOI] [PubMed] [Google Scholar]
- 54.Mitsuoka, K., T. Hirai, K. Murata, A. Miyazawa, A. Kidera, Y. Kimura, and Y. Fujiyoshi. 1999. The structure of bacteriorhodopsin at 3.0 Ångstrom resolution based on electron crystallography: implication of the charge distribution. J. Mol. Biol. 286:861–882. [DOI] [PubMed] [Google Scholar]