

Recent studies have suggested that primary cilia of renal tubular epithelial cells may function to maintain normal tubular architecture, and that ciliary abnormalities may be a cause of cyst formation in polycystic kidney disease (PKD). PKD results from loss-of-function mutations in either the PKD1 or PKD2 gene (1). Cysts develop in PKD patients at a very young age and grow inexorably over time to very large size, eventually overwhelming the kidney. Each cyst is thought to be initiated by the abnormal proliferation of a single tubular epithelial cell, which eventually expands that region of the tubule until it pinches off as an isolated cyst. At that point, continued cyst growth is thought to depend on net fluid secretion into the cyst lumen. One of the central problems in the PKD field has been to understand the cellular mechanisms that trigger cyst formation. The paper by Lin et al. (2) in a recent issue of PNAS provides new insight into this problem by showing that kidney-specific Cre-loxP inactivation of the gene for the KIF3A subunit of kinesin-II, an anterograde (outward moving) ciliary motor protein, causes PKD, thus directly implicating cilia in the cyst-forming mechanism.
These studies by Lin et al. (2) follow previous work in which Cre-loxP conditional mutagenesis was used to inactivate the KIF3A subunit in photoreceptor cells (3). The vertebrate photoreceptor cell has a modified cilium, composed of a connecting axonemal segment that bridges the cell body to the outer segment of the photoreceptor cilium. Photoreceptor components must pass along the connecting cilium to supply the photoreceptor disks in the outer segment. Targeted somatic deletion of the KIF3A subunit was found to prevent anterograde protein movement from the inner to the outer segment, leading to an accumulation of material in the cell body and eventually to apoptotic cell death, similar to what is seen in human retinal degeneration diseases. For these experiments, and for those of Lin et al., tissue-specific Cre-loxP mutagenesis was required, because systemic deletion of KIF3A is embryo lethal.
The paper by Lin et al. is the latest in a series of discoveries that have reported connections between PKD and the assembly and function of cilia of mice and the nematode, Caenorhabditis elegans, and of flagella of the unicellular green alga, Chlamydomonas (for previous reviews, see refs. 42 and 43). The first connection was the discovery of the mouse Tg737 gene in an insertional mutagenesis screen (4). The original Tg737 hypomorphic mutation was found to cause a recessive form of PKD in orpk mice. Later, deletion of the Tg737 gene was found to give rise to an embryo-lethal phenotype that is due to an absence or stunting of cilia on ventral node epithelial cells (5, 6), causing disruption of early morphological left–right axis determination (5). The protein product of this gene, polaris, was localized in the axoneme and basal body of primary cilia (6, 7) and was found to be required for ciliary assembly (6, 8). Another mouse mutant affecting left–right patterning and also causing PKD, inv (9), also has a ciliary defect, which in this case appears to be a functional rather than structural problem (10). The inv gene encodes the novel, calmodulin-binding protein inversin (11, 12). The recessive cpk mouse model of PKD also has a ciliary connection. The cpk gene encodes a protein, cystin, that was found to reside in the axonemal region of primary cilia, partially overlapping with polaris (13). This too may be a functional rather than structural defect because no obvious structural abnormalities were noted by electron microscopy in the cilia of cpk mutant mice (14). At the same time these studies were being carried out, work was also underway to examine mutations affecting male mating behavior in C. elegans, which identified homologs of the PKD genes lov-1 (for location of vulva, a homolog of PKD1) and pkd-2, whose protein products were found to localize in cilia of sensory neurons (15, 16). These proteins also colocalize with another protein, OSM-5, which is a homolog of polaris (17, 18) and IFT88, an intraflagellar transport (IFT) protein of Chlamydomonas (6). The lov-1 and pkd-2 mutations seem to affect ciliary function (16), whereas the osm-5 mutations affect ciliary structure (17, 18). To extend these studies to mammals, Pazour et al. (19) localized the PKD2 product, polycystin-2, to cilia of renal epithelial cells. Then, Yoder et al. (20) reported that the PKD1 product, polycystin-1, as well as polycystin-2, polaris, and cystin all localize to primary cilia of cultured mouse cortical collecting duct cells. What is remarkable about all these studies is the variety of PKD models and the numerous connections between PKD proteins and cilia.
Primary cilia (Fig. 1) have an axonemal structure consisting of a circular array of nine pairs of microtubules (9 + 0 arrangement; ref. 21). They can be found on most eukaryotic cells (22) and are visible on cells in all kidney tubule segments with the exception of collecting duct intercalated cells (23). Primary cilia, which are usually nonmotile, are thought to serve either a chemosensory or mechanosensory function. These cilia often contain high concentrations of receptors and are ideally positioned to interact with their environment, as with the odorant receptors on olfactory cilia (24) or the highly specialized cone and rod photoreceptors that respond to light. Cilia also contain the requisite signaling components, including heterotrimeric G proteins and ion channels. In fact, a recent proteomics study of motile (9 + 2) respiratory tract cilia found evidence for >200 potential axonemal proteins, including the Tg737 protein, polaris (25).
Figure 1.
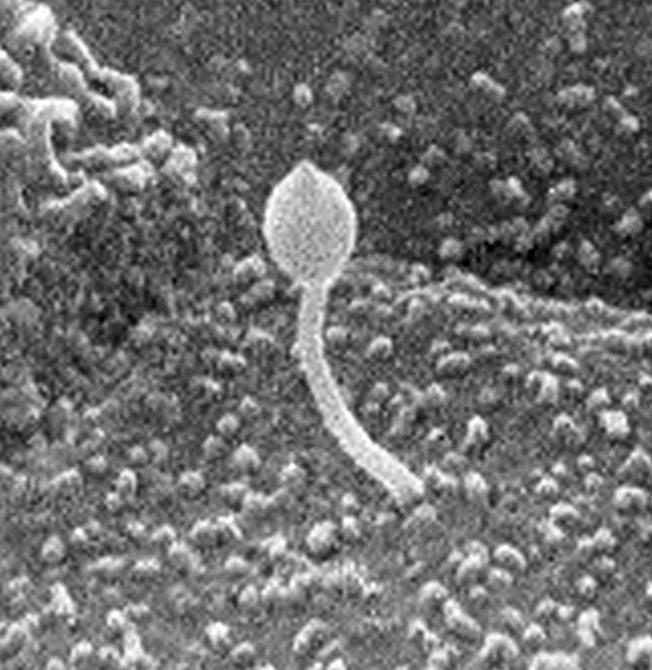
Scanning electron micrograph of a rat kidney collecting duct cilium with a bulbous tip. [Reproduced with permission of John Wiley & Sons, Inc., from ref. 23 (Copyright 1974, Wiley–Liss).]
Primary cilia on cultured renal epithelial cells have been shown to reversibly bend in response to lateral fluid shear forces, as visualized by real-time video recording under conditions that resemble actual tubular flow rates (26, 27). Indeed, fluid shear-force bending of the primary cilia of Madin–Darby canine kidney (MDCK) cells has been shown to increase intracellular calcium, from both extracellular and intracellular sources, and to transmit these signals to adjacent cells through gap junctions (28). Deciliation by chloral hydrate treatment abolished flow-induced calcium increases but did not affect calcium mobilization in response to mechanical cell-surface stimulation, indicating that cilia are mechanosensory flow sensors (29). This flow-induced increase in intracellular calcium also caused membrane hyperpolarization and increased potassium conductance (30).
Polycystin-1 and -2 have now been shown to mediate the mechanosensory function of cilia (31). This was demonstrated by showing that the calcium transients induced by fluid-flow ciliary bending did not occur in polycystin-1 null cells or when cells were treated with polycystin-1 or -2 blocking antibodies. Thus, it appears that the ciliary polycystins may be responsible for mechanosensory calcium signaling. How the elevated calcium affects cell function is still unknown; however, it has been speculated that flow sensing may regulate intratubular luminal diameters and that a failure to do this might lead to compensatory growth of the tubular cells in an attempt to overcome what seems to be a lack of fluid flow. Polycystin-1 and -2 are multipass membrane-spanning proteins that are thought to directly interact via their cytosolic C-terminal tails. Polycystin-1 has been shown to activate a number of signaling pathways and may be a receptor, whereas polycystin-2 has been shown to be a transient receptor potential (TRP)-related calcium channel. Although the cellular functions of the polycystins are still unknown, there is evidence suggesting that polycystin-1 couples with heterotrimeric G proteins (32–34) and that it regulates polycystin-2 calcium channel activity (33, 35, 36). There is also evidence that, together, polycystin-1 and -2 up-regulate the cyclin-dependent kinase inhibitor p21, thus directly inhibiting the cell cycle (37). Lin et al. (2) showed that the KIF3A mutation led to inhibition of p21, as well as to increased expression of β-catenin and c-Myc, which have both been associated with PKD. There is also evidence that polycystin-1 regulates the response of cells to cAMP by ensuring that cell proliferation is not stimulated by increased levels of intracellular cAMP (38–40). Thus, polycystin-1, acting as a ciliary mechanoreceptor, may sense luminal flow rates and signal morphologically relevant information through a number of cellular signaling pathways to establish and maintain appropriate luminal dimensions. Polycystin-1 anchored into the ciliary plasma membrane may be configured to sense ciliary bending. Polycystin-2, acting as a tension-gated ion channel, would then respond to signals from polycystin-1, induced by ciliary flexing, to generate an influx of calcium. Calculations have suggested that as few as one polycystin-2 molecule per cilium (and there is only one cilium per cell) is sufficient to trigger the measured calcium influx in MDCK cells (29).
The experiments of Lin et al. raise the question of whether PKD is caused by loss of cilia per se or by loss of the ciliary polycystins. In other words, is the common thread in the cystic diseases caused by mutations of polycystin-1, polycystin-2, polaris, cystin, and inversin, and now KIF3A, an inability to deliver functionally active polycystins to the ciliary membrane? In the human disease, there is inactivation of the polycystins; however, nonciliary as well as ciliary polycystins are lost, so it is not possible to tell which of these pools of polycystin is critical to the disease (or whether both are). A number of studies have localized polycystin-1 and -2 to lateral membrane junctional complexes, in particular to desmosomes, thus implicating the polycystins in cell–cell communication (41). Furthermore, the bulk of polycystin-2 is found in cytosolic membranes, especially the ER, where it is thought to act as a calcium-activated calcium-release channel (36).
Is PKD caused by loss of cilia or by loss of the ciliary polycystins?
It would seem that a key experiment might be to attempt to block the cilia-targeted polycystins from reaching their destination in what, otherwise, would be normal cilia. It was observed in the photoreceptor experiments that some, but not all, outer segment proteins were cargoed by kinesin-II (3). The KIF3A mutants were unable to transport the aporeceptor opsin; however, there was no abnormality seen in the inner vs. outer segment distribution of its G protein partner, transducin. As such, it is possible that the polycystins might be transported by an active, kinesin-II-dependent mechanism similar to that used for opsin transport. If so, this would suggest that there may be specific cilia-localization signals on the polycystin proteins that would allow their attachment to the IFT machinery for anterograde transport. If these signals could be identified, they could be mutated by using knock-in technology to specifically eliminate ciliary polycystin function only, while sparing nonciliary polycystin function, to determine whether the specific inactivation of only the ciliary polycystin is sufficient to cause PKD.
Footnotes
See companion article on page 5286 in issue 9 of volume 100.
References
- 1.Igarashi P, Somlo S. J Am Soc Nephrol. 2002;13:2384–2398. doi: 10.1097/01.asn.0000028643.17901.42. [DOI] [PubMed] [Google Scholar]
- 2.Lin F, Hiesberger T, Cordes K, Sinclair A M, Goldstein L S B, Somlo S, Igarashi P. Proc Natl Acad Sci USA. 2003;100:5286–5291. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marszalek J R, Liu X, Roberts E A, Chui D, Marth J D, Williams D S, Goldstein L S. Cell. 2000;102:175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- 4.Moyer J H, Lee-Tischler M J, Kwon H Y, Schrick J J, Avner E D, Sweeney W E, Godfrey V L, Cacheiro N L, Wilkinson J E, Woychik R P. Science. 1994;264:1329–1333. doi: 10.1126/science.8191288. [DOI] [PubMed] [Google Scholar]
- 5.Murcia N S, Richards W G, Yoder B K, Mucenski M L, Dunlap J R, Woychik R P. Development (Cambridge, UK) 2000;127:2347–2355. doi: 10.1242/dev.127.11.2347. [DOI] [PubMed] [Google Scholar]
- 6.Pazour G J, Dickert B L, Vucica Y, Seeley E S, Rosenbaum J L, Witman G B, Cole D G. J Cell Biol. 2000;151:709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taulman P D, Haycraft C J, Balkovetz D F, Yoder B K. Mol Biol Cell. 2001;12:589–599. doi: 10.1091/mbc.12.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoder B K, Tousson A, Millican L, Wu J H, Bugg C E, Jr, Schafer J A, Balkovetz D F. Am J Physiol. 2002;282:F541–F552. doi: 10.1152/ajprenal.00273.2001. [DOI] [PubMed] [Google Scholar]
- 9.Mochizuki T, Saijoh Y, Tsuchiya K, Shirayoshi Y, Takai S, Taya C, Yonekawa H, Yamada K, Nihei H, Nakatsuji N, et al. Nature. 1998;395:177–181. doi: 10.1038/26006. [DOI] [PubMed] [Google Scholar]
- 10.Okada Y, Nonaka S, Tanaka Y, Saijoh Y, Hamada H, Hirokawa N. Mol Cell. 1999;4:459–468. doi: 10.1016/s1097-2765(00)80197-5. [DOI] [PubMed] [Google Scholar]
- 11.Yasuhiko Y, Imai F, Ookubo K, Takakuwa Y, Shiokawa K, Yokoyama T. Dev Growth Differ. 2001;43:671–681. doi: 10.1046/j.1440-169x.2001.00604.x. [DOI] [PubMed] [Google Scholar]
- 12.Morgan D, Goodship J, Essner J J, Vogan K J, Turnpenny L, Yost H J, Tabin C J, Strachan T. Hum Genet. 2002;110:377–384. doi: 10.1007/s00439-002-0696-4. [DOI] [PubMed] [Google Scholar]
- 13.Hou X, Mrug M, Yoder B K, Lefkowitz E J, Kremmidiotis G, D'Eustachio P, Beier D R, Guay-Woodford L M. J Clin Invest. 2002;109:533–540. doi: 10.1172/JCI14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ricker J L, Gattone V H, 2nd, Calvet J P, Rankin C A. J Am Soc Nephrol. 2000;11:1837–1847. doi: 10.1681/ASN.V11101837. [DOI] [PubMed] [Google Scholar]
- 15.Barr M M, Sternberg P W. Nature. 1999;401:386–389. doi: 10.1038/43913. [DOI] [PubMed] [Google Scholar]
- 16.Barr M M, DeModena J, Braun D, Nguyen C Q, Hall D H, Sternberg P W. Curr Biol. 2001;11:1341–1346. doi: 10.1016/s0960-9822(01)00423-7. [DOI] [PubMed] [Google Scholar]
- 17.Qin H, Rosenbaum J L, Barr M M. Curr Biol. 2001;11:457–461. doi: 10.1016/s0960-9822(01)00122-1. [DOI] [PubMed] [Google Scholar]
- 18.Haycraft C J, Swoboda P, Taulman P D, Thomas J H, Yoder B K. Development (Cambridge, UK) 2001;128:1493–1505. doi: 10.1242/dev.128.9.1493. [DOI] [PubMed] [Google Scholar]
- 19.Pazour G J, San Agustin J T, Follit J A, Rosenbaum J L, Witman G B. Curr Biol. 2002;12:R378–R380. doi: 10.1016/s0960-9822(02)00877-1. [DOI] [PubMed] [Google Scholar]
- 20.Yoder B K, Hou X, Guay-Woodford L M. J Am Soc Nephrol. 2002;13:2508–2516. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 21.Pazour G J, Rosenbaum J L. Trends Cell Biol. 2002;12:551–555. doi: 10.1016/s0962-8924(02)02410-8. [DOI] [PubMed] [Google Scholar]
- 22.Wheatley D N. Pathobiology. 1995;63:222–238. doi: 10.1159/000163955. [DOI] [PubMed] [Google Scholar]
- 23.Andrews P M, Porter K R. Am J Anat. 1974;140:81–115. doi: 10.1002/aja.1001400107. [DOI] [PubMed] [Google Scholar]
- 24.Ronnett G V, Snyder S H. Trends Neurosci. 1992;15:508–513. doi: 10.1016/0166-2236(92)90104-g. [DOI] [PubMed] [Google Scholar]
- 25.Ostrowski L E, Blackburn K, Radde K M, Moyer M B, Schlatzer D M, Moseley A, Boucher R C. Mol Cell Proteomics. 2002;1:451–465. doi: 10.1074/mcp.m200037-mcp200. [DOI] [PubMed] [Google Scholar]
- 26.Roth K E, Rieder C L, Bowser S S. J Cell Sci. 1988;89:457–466. doi: 10.1242/jcs.89.4.457. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz E A, Leonard M L, Bizios R, Bowser S S. Am J Physiol. 1997;272:F132–F138. doi: 10.1152/ajprenal.1997.272.1.F132. [DOI] [PubMed] [Google Scholar]
- 28.Praetorius H A, Spring K R. J Membr Biol. 2001;184:71–79. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- 29.Praetorius H A, Spring K R. J Membr Biol. 2003;191:69–76. doi: 10.1007/s00232-002-1042-4. [DOI] [PubMed] [Google Scholar]
- 30.Praetorius H A, Frokiaer J, Nielsen S, Spring K R. J Membr Biol. 2003;191:193–200. doi: 10.1007/s00232-002-1055-z. [DOI] [PubMed] [Google Scholar]
- 31.Nauli S M, Alenghat F J, Luo Y, Williams E, Vassilev P, Li X, Elia A E, Lu W, Brown E M, Quinn S J, et al. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 32.Parnell S C, Magenheimer B S, Maser R L, Rankin C A, Smine A, Okamoto T, Calvet J P. Biochem Biophys Res Commun. 1998;251:625–631. doi: 10.1006/bbrc.1998.9514. [DOI] [PubMed] [Google Scholar]
- 33.Delmas P, Nomura H, Li X, Lakkis M, Luo Y, Segal Y, Fernandez-Fernandez J M, Harris P, Frischauf A M, Brown D A, Zhou J. J Biol Chem. 2002;277:11276–11283. doi: 10.1074/jbc.M110483200. [DOI] [PubMed] [Google Scholar]
- 34.Parnell S C, Magenheimer B S, Maser R L, Zien C A, Frischauf A M, Calvet J P. J Biol Chem. 2002;277:19566–19572. doi: 10.1074/jbc.M201875200. [DOI] [PubMed] [Google Scholar]
- 35.Hanaoka K, Qian F, Boletta A, Bhunia A K, Piontek K, Tsiokas L, Sukhatme V P, Guggino W B, Germino G G. Nature. 2000;408:990–994. doi: 10.1038/35050128. [DOI] [PubMed] [Google Scholar]
- 36.Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, Ehrlich B E, Somlo S. Nat Cell Biol. 2002;4:191–197. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 37.Bhunia A K, Piontek K, Boletta A, Liu L, Qian F, Xu P N, Germino F J, Germino G G. Cell. 2002;109:157–168. doi: 10.1016/s0092-8674(02)00716-x. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi T, Pelling J C, Ramaswamy N T, Eppler J W, Wallace D P, Nagao S, Rome L A, Sullivan L P, Grantham J J. Kidney Int. 2000;57:1460–1471. doi: 10.1046/j.1523-1755.2000.00991.x. [DOI] [PubMed] [Google Scholar]
- 39.Hanaoka K, Guggino W B. J Am Soc Nephrol. 2000;11:1179–1187. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- 40.Sutters M, Yamaguchi T, Maser R L, Magenheimer B S, St. John P L, Abrahamson D R, Grantham J J, Calvet J P. Kidney Int. 2001;60:484–494. doi: 10.1046/j.1523-1755.2001.060002484.x. [DOI] [PubMed] [Google Scholar]
- 41.Scheffers M S, Le H, van der Bent P, Leonhard W, Prins F, Spruit L, Breuning M H, de Heer E, Peters D J. Hum Mol Genet. 2002;11:59–67. doi: 10.1093/hmg/11.1.59. [DOI] [PubMed] [Google Scholar]
- 42.Calvet J P. J Am Soc Nephrol. 2002;13:2614–2616. doi: 10.1681/ASN.V13102614. [DOI] [PubMed] [Google Scholar]
- 43.Calvet J P. Nat Genet. 2003;33:113–114. doi: 10.1038/ng1078. [DOI] [PubMed] [Google Scholar]