

Abstract
To evaluate the proficiency of phosphatases as catalysts, the rate of the uncatalyzed hydrolysis of simple phosphate monoester dianions was estimated by extrapolating rates measured over a range of high temperatures. The rate of spontaneous hydrolysis of phenyl phosphate dianion indicates that a linear free energy relationship reported earlier is reliable for leaving groups whose conjugate acids have pKa values up to at least 10. Using Teflon reaction vessels, it proved possible to follow the hydrolysis of methyl phosphate and 3-(4-carboxy)-2,2-dimethylpropyl phosphate in strong alkali. Even in 1 M KOH, the reaction was found to be specific acid catalyzed. These results establish an upper limit for dianion reactivity, which had been overestimated earlier as a result of the leaching by alkali of silicic acid from quartz reaction vessels. The present findings indicate that the half-time for attack by water on alkyl phosphate dianions is 1.1 × 1012 years (k = 2 × 10−20 s) at 25°C and that phosphatases involved in cell signaling and regulation produce the largest rate enhancements that have been identified thus far. Protein phosphatase-1 and inositol 1-phosphatase exceed all other known enzymes in their affinities for the altered substrates in the transition state.
Most enzyme reactions proceed with kcat/Km values in the neighborhood of 107 M−1⋅s−1 and appear to be similarly efficient according to that criterion. However, to assess the proficiency of an enzyme as a catalyst, and its corresponding affinity for the altered substrate in the transition state, it is necessary to compare kcat/Km with the rate constant of the corresponding reaction under the same conditions in the absence of a catalyst. In contrast to the relatively narrow range of values of kcat/Km observed for enzyme catalyzed reaction rates, the rates of these uncatalyzed reactions span a range of at least 19 orders of magnitude (1). Thus, differences in enzyme proficiency tend to reflect differences in the rates of the uncatalyzed reactions rather than the catalyzed reactions, and enzymes differ greatly from one another in their prowess as catalysts. Enzymes that catalyze the slowest reactions are of practical interest, in that they offer sensitive targets for inhibition by transition-state analogues. Their mechanisms of action are also particularly challenging to rationalize. Conspicuous among these slow reactions is the hydrolysis of monoesters of phosphoric acid.
In biological systems, phosphate monoesters are cleaved by two groups of monoesterases that act by different mechanisms.
One group of phosphomonoesterases, typified by bacterial alkaline phosphatase (2) and protein tyrosine phosphatases (3), uses an active-site nucleophile to displace the alcohol-leaving group from the substrate to form a phosphoryl-enzyme intermediate, which is subsequently hydrolyzed. Rate enhancement by enzymes that act through a double-displacement mechanism is not simple to analyze in terms of transition-state affinity or transition-state stabilization (4), although rates of reaction with model nucleophiles can be used to estimate equilibrium constants for transition-state interchange between an enzyme and the model nucleophile (5).
A second group of phosphatases catalyzes direct attack by water on phosphorus to displace the alcohol-leaving group and therefore lends itself directly to the estimation of transition-state affinities. Enzymes that bring about direct water attack include protein phosphatase-1 of which numerous variants with differing specificities have been found within the human proteome; fructose 1,6-bisphosphatase, which catalyzes the rate-determining step in carbohydrate catabolism; and inositol 1-phosphatases, which are involved in the transmission of hormonal signals. Here, we report that these enzymes exceed all other known enzymes in their powers as catalysts and in their affinities for the altered substrate in the transition state.
In the investigation of very slow reactions, simple model substrates are helpful in avoiding side reactions that might obscure the reaction of interest. The spontaneous hydrolysis of methyl phosphate (MeP) is relatively rapid at low pH where it is present mainly as the monoanion, but proceeds more slowly with increasing pH as the abundance of the monoanion (MeP−) declines (6). In experiments in sealed quartz tubes, reported earlier (7), the rate of hydrolysis of MeP appeared to reach a constant value more than pH 10, corresponding to the reaction of the dianion (MeP−2) with water. Slow as it was, that reaction proceeded considerably more rapidly than had been expected by extrapolation of rate constants for hydrolysis of monoester dianions with much better leaving groups (Fig. 1) (8, 9). Reinvestigating that discrepancy, we have established that the spontaneous hydrolysis of MeP−2 proceeds much more slowly than was previously reported and that phosphatases are even more proficient as catalysts than had been suspected.
Figure 1.

Linear free energy relationship between leaving-group pKa values and rate constants for hydrolysis of phosphate monoester dianions at 39°C. ○, benzoyl (9) and aryl (12) phosphates. ●, phenyl phosphate (present work). □, MeP (7). ■, present data. The line shown is corresponds to that in ref. 9 and is described by the equation 0.862 − 1.23 pKa.
Materials and Methods
Phenyl phosphate was purchased from Aldrich and used as received. MeP was prepared by the method of Bailly (10). Compound 1 (Scheme S1) was synthesized from 3-(4-carboxyphenyl)-2,2-dimethylpropan-1-ol (11) by using an excess of phosphoryl chloride in tetrahydrofuran and pyridine as base. Hydrolysis with KOH gave the monoester 1, which was extracted into ethyl acetate after acidification of the aqueous phase: δH (250 MHz; D2O) 0.87 (6H, s), 2.67 (2H, s), 3.52 (2H, d, J 4.6 Hz), 7.36 (2H, d, J 8.8 Hz), 7.93 (2H, d, J 8.8 Hz); δP (100 MHz; D2O) 0.83; MS(ES−) 287 (M−). HPLC retention time was 6.2 min [flow rate 0.75 ml⋅min−1; eluant: 25% MeOH, 75% 20 mM ammonium phosphate buffer (pH 5.5) on a Synergi 4-μ polar-RP 80A,150 × 4.6-mm column] monitored at 236 nm.
Scheme 1.

Compound 1.
In a typical experiment, MeP, 1, or phenyl phosphate was incubated, along with toluic acid as an internal standard, in 1 M KOH in Teflon-lined stainless-steel reaction vessels immersed in a circulating oil bath. Aliquots were analyzed by HPLC or proton NMR in 2 H2O, using added pyrazine as an integration standard. First-order rate constants were obtained by analysis of the initial rate or by following the reaction for three half-times and fitting the results to the first-order rate equation.
Results and Discussion
Reexamining the experimental conditions used earlier to observe the hydrolysis of MeP−2 in alkali (7), we came to suspect that the rate of hydrolysis might have been overestimated because of undetected leaching of silicic acid from the quartz reaction vessels by the alkaline solutions. With a pKa value of ≈9.8, the silicic acid released might have passed undetected because it did not change the total amount of titratable alkali that was present, but might have buffered the reaction mixture in such a way that the actual pH range extended only as far as pH ≈10. The apparent leveling of the reaction rate might thus have been caused by a leveling of the pH.
To circumvent that difficulty, we conducted the hydrolysis of MeP and 3-(4-carboxy)-2,2-dimethylpropyl phosphate (1) in Teflon reaction vessels at high pH over the temperature range from 160°C to 240°C. We found that in 1 M KOH the rate of hydrolysis was significantly slower than at pH 10, showing that the previous data had indeed led to an overestimate of the rate of the dianion reaction. From the Arrhenius plot (Fig. 2), the enthalpy of activation is 50.1 ± 0.6 kcal⋅mol−1 and the entropy of activation is 23 ± 1 cal⋅mol−1⋅°K−1 for the hydrolysis of 1. The results obtained for MeP, also plotted in Fig. 2, were closely similar. Extrapolating those data to 39°C yields a rate constant of 2 × 10−17 s−1, still somewhat higher than predicted by the linear free energy relationship in Fig. 1 but no longer in serious disagreement. Moreover, varying the KOH concentration in the range from 0.05 to 1 M, we found that the rate of reaction varied inversely with the concentration of KOH. Thus, even in 1 M KOH, the reaction that is observed is specific acid catalyzed, and MeP−, not the dianion, is the kinetically active species.
Figure 2.

Observed first-order rate constants for hydrolysis in 1 M KOH of phenyl phosphate (○), compound 1 (□), and MeP2− (▵), plotted as a function of reciprocal temperature (Kelvin).
As the original linear free energy relationship (9) only included leaving groups with pKa values of 7 or less, we decided to extend that range to include the less acidic alcohols that are typical of biological phosphate esters. Using the same experimental procedures to monitor phenyl phosphate hydrolysis in 1 M KOH between 107°C and 191°C, we again obtained linear Arrhenius plots. The hydrolysis of phenyl phosphate in 0.1, 0.3, and 1.0 M KOH (ionic strength adjusted to 1.0 with KCl) showed no differences in rate within experimental error, indicating that these data represent the pH-independent reaction of the phenyl phosphate dianion. The enthalpy of activation (38 ± 2 kcal⋅mol−1) and the small, positive, entropy of activation (7 ± 2 cal⋅mol−1⋅°K−1) are consistent with unimolecular decomposition via a loose transition state, a mechanism that has been widely (12, 13), although not universally (22), accepted for phosphate monoester dianions in aqueous solution. Extrapolation of these data yielded a value of 1 × 10−12 s−1 at 39°C, in satisfactory agreement with the linear free energy relationship shown in Fig. 1. A least-squares fit of the data in Fig. 1, including the value reported here for phenyl phosphate, yields Eq. 1:
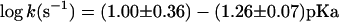 |
1 |
in which pKa refers to the conjugate acid of the leaving group.
The results obtained for MeP and compound 1 establish only an upper limit for dianion reactivity, and it is of interest to inquire how much lower the actual rate constant for MeP−2 hydrolysis is likely to be. Eq. 1 leads to the prediction that kobs should be 5 × 10−19 s−1 at 39°C, for a leaving alcohol with a pKa value of 15.5. To estimate the rate at 25°C, it seems reasonable to assume that the entropy of activation for spontaneous hydrolysis of MeP−2 is equivalent to the value determined above for the phenyl phosphate dianion. That leads to an estimated enthalpy of activation of 47 kcal⋅mol−1 and indicates that the rate would be ≈30-fold slower at 25°C than at 39°C. Accordingly, our best estimate of the rate constant for dianion hydrolysis at 25°C is 2 × 10−20 s−1. That value is in reasonable agreement with the upper limit obtained by extrapolating our Arrhenius plot to 25°C and with an earlier estimate based on thermodynamic data (14). Evidently the reaction of the dianion is only just below the limit of detection.
These results imply that simple phosphate monoesters are much less reactive and that phosphatases are even more proficient catalysts than had been recognized (7). Table 1 shows some kinetic constants that have been reported for type 1 protein phosphatase (EC 3.1.3.17; 15), fructose bisphosphatase (EC 3.1.3.11; 16), and inositol phosphatase (EC 3.1.3.25; 17). Each of these enzymes, which catalyze direct water attack on the substrate (18, 19), is seen to enhance the rate of water attack on the dianionic substrate by a factor of ≈1021 and to bind the altered substrate in the transition state with a formal dissociation constant of ≈10−26 M. These rate enhancements and catalytic proficiencies exceed even those of decarboxylases acting on orotidine 5′-phosphate (OMP) and amino acids, the most proficient enzymes identified thus far (Fig. 3). It should be added that, within this group, OMP decarboxylase retains the distinction of acting as a pure protein catalyst without the assistance of cofactors. The phosphatases mentioned above all require metal ion cofactors for activity.
Table 1.
Enzymatic hydrolysis of phosphate monoesters by direct water attack at 25°C
| Enzyme | Phosphorylated substrate | kcat (s−1) | kcat/Km (M−1⋅s−1) |
|---|---|---|---|
| PP115 | Phosphoryl-phosphorylase a | 39 | 4 × 106 |
| FBPase16 | Fructose 1,6-bisphosphate | 21 | 1.5 × 107 |
| IP17 | Inositol-1-phosphate | 22 | 3 × 105 |
| None | MeP and compound 1 | 2 × 10−20 (knon) |
Figure 3.

Half-times of biological reactions proceeding spontaneously in water at 25°C in the absence of a catalyst (for earlier references, see ref. 1).
As noted in the Introduction, transition-state affinity cannot be evaluated directly for those phosphatases that react through a covalent phosphoryl-enzyme intermediate. For that group of phosphatases, a recent investigation of Escherichia coli alkaline phosphatase (APase, EC 3.1.3.1), acting on MeP under conditions where product inhibition was carefully avoided (20), has shown that kcat/Km is similar in magnitude to those of type 1 protein phosphatase, fructose bisphosphatase, and inositol phosphatase (Table 1). Thus, APase matches those mentioned above in its formal proficiency [(kcat/Km)/knon] as a catalyst. Among enzymes of this latter type, a high value of kcat (1.5 × 104 s−1) has been recorded for a human protein tyrosine phosphatase (PTP; EC 3.1.3.48), acting on a model peptide through a phosphorylated cysteine residue as an intermediate (21). Comparison with the present rate constant for spontaneous hydrolysis of the phenyl phosphate dianion indicates that PTP enhances the rate of hydrolysis of phosphorylated tyrosine residues by ≈17 orders of magnitude.
Acknowledgments
We thank the Biotechnology and Biological Sciences Research Council and Avecia for a studentship (to C.L.) and the National Institutes of Health for Grant GM-18325 (to R.W.).
Abbreviation
- MeP
methyl phosphate
References
- 1.Wolfenden R, Snider M J. Acc Chem Res. 2001;12:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz J H, Lipmann F. Proc Natl Acad Sci USA. 1963;149:871–877. [Google Scholar]
- 3.Jackson M D, Denu J M. Chem Rev. 2001;101:2313–2340. doi: 10.1021/cr000247e. [DOI] [PubMed] [Google Scholar]
- 4.Wolfenden R. Acc Chem Res. 1972;5:10–18. [Google Scholar]
- 5.Lienhard G E. Science. 1973;180:149–155. doi: 10.1126/science.180.4082.149. [DOI] [PubMed] [Google Scholar]
- 6.Bunton C A, Llewellyn D R, Oldham K G, Vernon C A. J Chem Soc. 1958;1958:3574–3587. [Google Scholar]
- 7.Wolfenden R, Ridgway C, Young G. J Am Chem Soc. 1998;120:833–834. [Google Scholar]
- 8.Di Sabato G, Jencks W P. J Am Chem Soc. 1961;83:4400–4405. [Google Scholar]
- 9.Kirby A J, Varvoglis A G. J Am Chem Soc. 1967;89:415–423. [Google Scholar]
- 10.Bailly O. Bull Soc Chim. 1919;25:240–253. [Google Scholar]
- 11. Williams, N. H. & Wyman, P. (2001) Chem. Commun. 1268–1269.
- 12.Hengge A C. In: Comprehensive Biological Catalysis: A Mechanistic Reference. Sinnott M, editor. Vol. 1. New York: Academic; 1998. pp. 517–542. [Google Scholar]
- 13.Thatcher G R J, Kluger R. Adv Phys Org Chem. 1989;25:99–265. [Google Scholar]
- 14.Guthrie J P. J Am Chem Soc. 1977;99:3991–4001. [Google Scholar]
- 15.Huang H-B, Horiuchi A, Goldberg J, Greengard P, Nairn A C. Proc Natl Acad Sci USA. 1997;94:3530–3535. doi: 10.1073/pnas.94.8.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelley N, Giroux E L, Lu G, Kantrowitz E R. Biochem Biophys Res Commun. 1996;219:848–852. doi: 10.1006/bbrc.1996.0321. [DOI] [PubMed] [Google Scholar]
- 17.Ganzhorn A J, Lepage P, Pelton P D, Strasser F, Vincendon P, Rondeau J-M. Biochemistry. 1996;35:10957–10966. doi: 10.1021/bi9603837. [DOI] [PubMed] [Google Scholar]
- 18.Sanvoisin J, Gani D. Bioorg Med Chem Lett. 2001;11:471–474. doi: 10.1016/s0960-894x(00)00694-6. [DOI] [PubMed] [Google Scholar]
- 19.Gani D, Wilkie J. J Chem Soc Rev. 1995;24:55–63. [Google Scholar]
- 20.O'Brien P J, Herschlag D. Biochemistry. 2002;41:3207–3225. doi: 10.1021/bi012166y. [DOI] [PubMed] [Google Scholar]
- 21.Harder K W, Owen P, Wong L K H, Aebersold R, Clark-Lewis I, Jirik F R. Biochem J. 1994;298:395–401. doi: 10.1042/bj2980395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Åqvist J, Kolmodin K, Florian J, Warshel A. Chem Biol. 1999;6:R71–R80. doi: 10.1016/S1074-5521(99)89003-6. [DOI] [PubMed] [Google Scholar]