

Abstract
In epithelial cells, alternative splicing of fibroblast growth factor receptor 2 (FGFR2) transcripts leads to the expression of the FGFR2(IIIb) isoform, whereas in mesenchymal cells, the same process results in the synthesis of FGFR2(IIIc). Expression of the FGFR2(IIIc) isoform during prostate tumor progression suggests a disruption of the epithelial character of these tumors. To visualize the use of FGFR2 exon IIIc in prostate AT3 tumors in syngeneic rats, we constructed minigene constructs that report on alternative splicing. Imaging these alternative splicing decisions revealed unexpected mesenchymal–epithelial transitions in these primary tumors. These transitions were observed more frequently where tumor cells were in contact with stroma. Indeed, these transitions were frequently observed among lung micrometastases in the organ parenchyma and immediately adjacent to blood vessels. Our data suggest an unforeseen relationship between epithelial mesenchymal plasticity and malignant fitness.
Keywords: alternative splicing, mesenchymal–epithelial transitions, tumor plasticity
Regulation of alternative splicing is essential for normal gene expression (1), and alterations of this regulation are linked to disease (2), as illustrated by the association between cancer and splicing defects (3–7). An elegant example of this is provided by the splicing of transcripts encoding fibroblast growth factor receptor 2 (FGFR2). The status of FGFR2 alternative splicing depends on the interplay between several cis-acting elements in the FGFR2 premRNA and transacting factors, some of which are cell-type-specific (8). In mesenchymal cells, exon IIIb is silenced by the action of an exonic splicing silencer (9) and two flanking intronic splicing silencers (10–12). This silencing is mediated by Polypyrimidine tract-binding protein (PTB), hnRNP A1, and heretofore unknown factors. In epithelial cells, exon IIIb silencing is countered by several intronic elements. The best characterized are the intronic activating sequence 2, the intronic splicing activator and repressor (ISAR, also known as IAS3), and several GCAUG repeats (13–18). These elements have a dual function in epithelial cells, because they are also involved in silencing exon IIIc (14–18).
FGFR2 splicing has been studied in tumors derived from the R-3327 Dunning rat prostate tumor, which arose spontaneously from the dorsal lobe of the prostate in a Copenhagen rat (19). Some R-3327-derived tumors (DT or DT3) express FGFR2(IIIb), which is consistent with their epithelial phenotype (20), whereas AT tumors (e.g., AT3), which have lost epithelial markers and display many mesenchymal indicators (21), express FGFR2(IIIc) (20). The significance of these alternative decisions for tumor behavior is underscored by the fact that forced expression of FGFR2(IIIb) suppresses tumor progression of AT3 tumors (22). Most importantly, however, the differential splicing of FGFR2 transcripts in these two cell types highlights broad differences in gene expression programs. Arguably, monitoring alternative splicing, which has an unrivaled capacity to multiply coding diversity, can provide information that exquisitely discerns between unique cellular states.
These considerations led us to conclude that the state of tumor cells could be effectively reflected by the splicing status of FGFR2 transcripts. To observe this status in tumors in animals, we have developed reporters that can monitor alternative splicing decisions in time and space. In this article, we visualized the alternative inclusion of FGFR2 exon IlIc in cells in culture and in tumors in animals. Studies of AT3 tumors revealed unexpected plasticity of these tumors, which where previously thought to be anaplastic (23). Based on these alternative splicing choices, we concluded that groups of cells within the primary tumors had transitioned toward an epithelial phenotype. This was noted to be associated with contact with stroma. Remarkably, many tumor cells that had invaded the lungs also displayed this mesenchymal to epithelial transition (MET). These data force a reconsideration of the plasticity of this aggressive tumor and suggest a new relationship between epithelial mesenchymal plasticity and malignant fitness.
Results
Imaging Alternative Inclusion of FGFR2 Exon IlIc in Cell Culture.
We set out to visualize the alternative inclusion of FGFR2 exon IlIc in tumor cells in culture. We used two well established Dunning cell lines, DT3 and AT3, and the imaging reporters described in Fig. 1A. The control pRint reporter encodes transcripts that contain strong splice sites expected to promote splicing efficiently in all cell types, and indeed Rint transcripts are efficiently spliced and express high levels of red fluorescent protein (RFP) in both DT3 and AT3 cells (Fig. 1 B and C; Fig. 6, which is published as supporting information on the PNAS web site). RIIIcI2 transcripts, which include the elements necessary for silencing of exon IlIc in epithelial cells, were expected to include exon IIIc in a cell-type-specific fashion. Indeed, expression of pRIIIcI2 in DT3 cells led to high levels of RFP, as detected by epifluorescence microscopy (Fig. 1C) or FACS (Fig. 6), which is accompanied by efficient skipping of exon IIIc in pRIIIcI2 transcripts (Fig. 1B). Efficient inclusion of exon IIIc among pRIIIcI2 transcripts in AT3 cells disrupts the RFP ORF, leading to low levels of RFP (Figs. 1 B and C and 6A). We also tested splicing of RΔ,Δ transcripts, which are identical to RIIIcI2 RNAs, except that the intronic activating sequence 2 and intronic splicing activator and repressor elements have been deleted (Fig. 1A). These two elements have no known function in mesenchymal cells but are required for efficient repression of exon IIIc in epithelial cells (15, 17). As expected, RΔ,Δ transcripts show a disruption of exon IIIc silencing in DT3 cells and express lower levels of RFP than observed with RIIIcI2 RNAs (Fig. 1 B and C). It should be noted that, whereas the RT-PCR shown here can be used to compare between constructs and cells lines, the assay overestimates the levels of exon IIIc skipping, and therefore we calculate that the majority of RΔ,Δ transcripts included this exon in DT3 cells (17). As expected, in AT3 cells, the RΔ,Δ transcripts include exon IIIc as efficiently as do RIIIcI2 transcripts. These results indicated that the RFP constructs described above accurately report on the cell-type-specific use of FGFR2 exon IIIc in tissue culture.
Fig. 1.

Minigene constructs, molecular analysis, and imaging of exon IIIc inclusion in DT3 and AT3 cells. (A) Schematic representation (not to scale) of the minigene reporters used to image alternative splicing. (B) RT-PCR analysis of the reporter in total RNA extracts from the different DT3 and AT3 cell lines was carried out as described in Methods. RT-PCRs for pRint, pRIIIcI2, or pRΔ,Δ transcripts resulted in the expected size products (398 bp for products that include IIIc and 250 bp for the products that skip this exon), and splice junctions were confirmed by sequencing. (C) Imaging FGFR2 exon IlIc inclusion in DT3 and AT3 cells. (Upper) Fluorescence imaging of DT3 and AT3 cells stably transfected with the pRint, pRIIIcI2, or pRΔ,Δ minigenes. (Lower) Phase-contrast pictures of the same fields (images were acquired at ×200 magnification).
Visualizing the Inclusion of Exon IIIc in AT3 Tumors.
Because the pRIIIcI2 reporter provided an accurate readout of splicing decisions in cells in culture, we asked whether it could be used to image these decisions in tumors in animals. AT3 cells harboring either pRint or pRIIIcI2 were injected s.c. into the flanks of syngeneic Copenhagen white 2331 male and female rats. AT3-Rint or -RIIIcI2 tumors were grown for 20–30 days, harvested, and imaged whole in a fluorescence light box before freezing (see Methods). Rint tumors displayed high levels of fluorescence compared with naïve AT3 tumors (not shown). AT3-RIIIcI2 tumors were expected to include exon IIIc and thus prevent synthesis of RFP and, indeed, the fluorescence intensity of these tumors was much lower than that of Rint tumors and just slightly above background (not shown). These data suggested that pRIIIcI2 could accurately report on alternative splicing in tumors in living animals.
To confirm the conclusions reached by gross examination, we inspected histological sections of AT3-Rint and -RIIIcI2 tumors using epifluorescence microscopy. Three sections (15 μm) from different parts of at least three tumors were fixed in paraformaldehyde and imaged as described below. Sections of AT3 tumors appeared as homogenous sheets of malignant cells, as has been described by others (20). Most, if not all, of the cells in Rint tumors expressed high levels of RFP, whereas the great majority of cells in RIIIcI2 tumors had fluorescence that was indistinguishable from background auto fluorescence in naïve AT3 tumors (Fig. 2A). The low level of RFP expression was due to efficient inclusion of exon IIIc in pRIIIcI2 reporter transcripts (Fig. 2B), which paralleled the inclusion of exon IlIc in endogenous FGFR2 transcripts (data not shown). In AT3-RIIIcI2 tumors, a few cells expressing RFP at low levels were observed (Fig. 2A Right), and this was consistent with what had been observed in tissue culture, i.e., the AT3-RIIIcI2 population had few cells expressing RFP above background, but at low levels (see AT3-RIIIcI2 FACS in Fig. 6). All these data indicated that RIIIcI2 transcripts were reporting accurately the status of exon IlIc inclusion in tumors in vivo.
Fig. 2.

Imaging alternative use of exon IlIc in AT3 tumors using fluorescent reporters. (A) Fluorescence imaging of AT3 tumor sections either untransfected (background) or stably transfected with the pRint and pRIIIcI2 constructs (all images were acquired at ×200 magnification). (B) RT-PCR analysis of the reporter was carried out as described in Methods. RT-PCRs for RFP transcripts resulted in the expected size products, and splice junctions were confirmed by sequencing. Analysis of four different regions (denoted 1, 2, 3, or 4) for each type of tumor is shown.
Imaging Alternative Splicing Revealed Surprising Plasticity of AT3 Tumors.
As described above, we noted very low expression of RFP in AT3-RIIIcI2 tumors. Extensive surveys of histological sections from RIIIcI2 tumors, however, revealed the existence of “clusters” of red fluorescence (Fig. 3A). These RFP+ cells must be skipping exon IlIc very efficiently. We examined 14 RIIIcI2 tumors (nine in males and five in females), and 12 contained at least one RFP+ cluster (≈85% frequency; Table 1, which is published as supporting information on the PNAS web site). There was no significant difference between males and females in the frequency of these clusters (Table 1). Within each tumor, the number of RFP+ clusters was low, as indicated by the fact that the clusters were found in ≈34% of the sections surveyed (Table 1). These results indicated that, whereas RFP expression was occurring in a small number of cells in each tumor, it was observed in the majority of AT3-RIIIcI2 tumors.
Fig. 3.

MET in tumors. (A) Fluorescence and phase-contrast images of a section of an AT3-RIIIcI2 tumor close to the tumor capsule. Images were acquired at ×200 magnification. (B) (Upper) Fluorescence images of sections of AT3-Rint, -RIIIcI2, and -RΔ,Δ tumors. (Lower) Phase-contrast pictures of the same fields. Images were acquired at ×400 magnification. (C) E-cadherin counterstaining confirms MET. Example of a section from AT3-RIIIcI2 tumors shows that the red fluorescent cells also stained positive for E-cadherin. Images were acquired at ×400 magnification.
One possibility for this splicing choice between different cells in the tumors could be differences in the reporters (e.g., acquired mutations around exon IlIc). The nonrandom location of these “clusters” (see below) suggested that this explanation was improbable. A more likely possibility was that inclusion of exon IIIc was due to differences in transacting factors secondary to either genetic alterations or phenotypic transitions in response to environmental signals. Indeed, efficient silencing of exon IlIc and concomitant RFP expression would be the expected result of a phenotypic transformation toward a more epithelium-like state, as in MET (24–26). To examine this idea, we tested AT3-RΔ,Δ tumors, which harbor plasmid pRΔ,Δ (Fig. 1B). A systematic examination of four −RΔ,Δ tumors failed to detect any clusters of RFP expression (Fig. 3B Right and Table 1), strongly suggesting that the clusters observed in RIIIcI2 tumors depended on epithelial specific cis elements and thus on transacting factors found in epithelial cells.
To test the possibility that the RFP+ clusters were indeed the result of an epithelial transition, we interrogated these RFP-producing clusters for the expression of E-cadherin, a cell adhesion molecule that is poorly expressed in mesenchymal cells but highly expressed in epithelial cells (24). Of 18 RFP+ clusters from different tumors and different animals (see Table 1), 14 stained positive for E-cadherin, confirming our prediction that the majority of these cells were undergoing MET (Fig. 3C). To further test the idea that RFP+ cells were transitioning to an epithelial state, we tested for the expression of zonula occludens protein-1 (ZO-1), which increases and/or redistributes in the cell during MET, and for vimentin, which decreases during MET (27, 28). RFP+ cells were also ZO-1+ but had poor staining for vimentin, and this contrasted with the bulk of the RIIIcI2 tumors that stained negative for ZO-1 and positive for vimentin (Fig. 7, which is published as supporting information on the PNAS web site). It must be noted that the subcellular localizations of E-cadherin and ZO-1 are not those observed in normal epithelial tissues, suggesting that these RFP+ AT3 cells had transitioned toward, but had not completely arrived to, an epithelial state (29). The staining with vimentin suggested that RFP clusters usually arose within larger areas of the tumors that had lost vimentin staining (Fig. 7). These data indicate that the skipping of exon IlIc, which led to RFP expression, was, in the majority of cases, due to MET. Therefore, we conclude that RFP expression in RIIIcI2 tumors revealed epithelial plasticity of AT3 tumors.
MET Clusters Are Frequently Found Near Stroma.
Examination of the histological sections indicated that the location of the RFP+ clusters was not random. The majority of cases of the RFP+ clusters occurred in the vicinity of structures that appeared to be stroma [e.g., the tumor capsule (Fig. 3A)]. We confirmed this suggestion by counterstaining histological sections with Masson’s Trichrome, which differentially stains stromal components and has been used to identify stroma in human prostate tumors (30). In a survey of tumor sections, we noted that 14 of 16 RFP+ clusters were found in close association with stromal structures containing collagen, which was stained blue by Masson’s Trichrome (Fig. 4 and Table 1). These observations indicated that RFP+ cells (i.e., cells that would silence exon IIIc) predominantly occurred near stroma.
Fig. 4.

MET predominates in stromal regions rich in collagen fibers. Masson’s Trichrome staining confirms that RFP+ cells are situated in stromal regions. Examples are shown from three regions of three different AT3-RIIIcI2 tumors (Left, image obtained with epifluorescence microscopy; Center, phase-contrast picture of the same field; Right, bright field of the same region located after Masson’s Trichrome staining. All images were acquired at ×200 magnification). In the Masson’s Trichrome images, collagen is stained in blue, nuclei in black, and cytoplasm of cells in red.
Alternative Splicing Reporters Visualized MET in Lung Metastases.
The imaging reporters described here permitted a comprehensive survey of AT3 lung metastases in 13 lungs from animals bearing AT3-RIIIcI2 tumors. This survey revealed the presence of RFP+ disseminated tumor cells, which we refer to as micrometastases, in 12 of these lungs (Fig. 5A and Table 1). As in the primary tumors, we observed expression of E-cadherin in the overwhelming majority (Table 1) of RFP+ cells in AT3-RIIIcI2 lung metastases (Fig. 5B). RFP+ micrometastasis in lungs coincided also with expression of ZO-1 (not shown). Although we found these RFP+ cells distributed throughout the lung parenchyma, we noted a propensity of these cells to surround blood vessels (Fig. 5). These data suggested that cells actively involved in lung metastases had undergone a transition to a more epithelium-like state.
Fig. 5.

MET in lung metastasis. (A) Fluorescence and phase-contrast images of a lung section from animals harboring AT3-RIIIcI2 tumors (×200 magnification). Fluorescent cells are situated around a blood vessel (confirmed by hematoxylin/eosin staining; data not shown). (B) Example of a section from lungs of an animal bearing an AT3-RIIIcI2 tumor shows that the great majority of red fluorescent metastatic cells also stained positive for E-cadherin. Images were acquired at ×200 magnification.
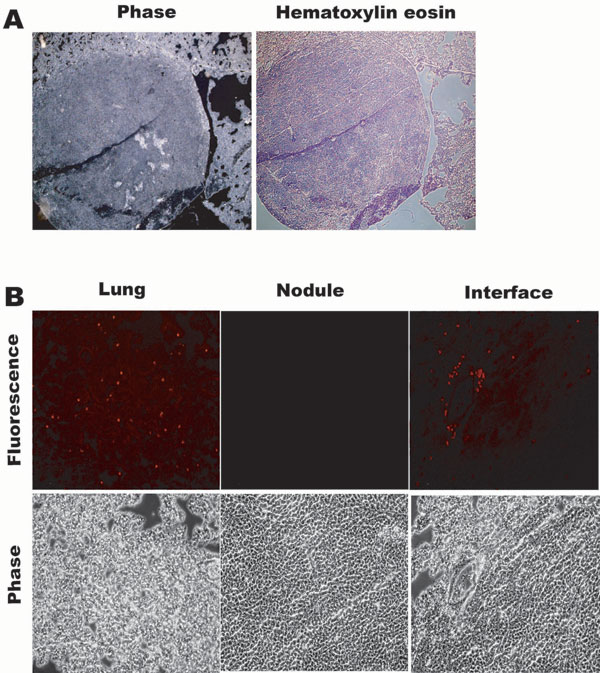
In lung sections, we also observed large metastatic nodules (Fig. 8A, which is published as supporting information on the PNAS web site). Although in the lung tissue in the immediate vicinity of nodules we were able to detect RFP+ micrometastases (Fig. 8B, “lung”), analysis of the central parts of the nodule failed to detect RFP expression (Fig. 8B, “nodule”). Interestingly, RFP+ metastatic cells could be observed at the interface of the lung tissue with the nodule (Fig. 8B, “interface”). These data suggested that, in primary tumors and large nodules, where most tumor cells are in contact with each other, there are few RFP+ tumor cells. On the other hand, RFP+ tumor cells were frequently observed in contact with lung parenchyma and other stromal components, which were confirmed as such by Masson’s Trichrome staining (not shown). All of these observations from primary tumors suggest that a population of metastatic AT3 cells undergo MET upon seeding in the lung stroma.
Discussion
The original goal of the study was to design and construct reporters of an alternative splicing decision in tumors in vivo. We showed here, by macroscopic and microscopic imaging, that this objective was achieved. In this and related work, which describes the effective use of reporters to image splicing decisions in normal tissues in transgenic mice (V.I.B., S.O., R.M.B., and M.A.G.-B., unpublished work), we describe tools and methods that will be generally useful to those wanting to examine alternative splicing in vivo. In addition to this technical goal, we anticipated at the outset that monitoring alternative splicing would provide a sensitive measure of altered tumor cell states or responses. The design of the RIIIcI2 reporters was such that even weak silencing of exon IIIc would result in the reporter protein expressed well above background. Indeed, the existence of AT3 tumor cells that silenced FGFR2 exon IIIc led to the discovery of unexpected plasticity among AT3 cells in primary tumors and metastases. Here we will discuss what we know and what we hypothesize about this plasticity.
What Are These RFP+ Cells?
Alternative silencing of FGFR2 exon IIIc suggested that groups of AT3 cells (RFP+ clusters) were in transition to an epithelium-like state. This suggestion was supported by coexpression of well characterized markers of MET. These transitions were observed in a small minority of the cells in each AT3 tumor, but clusters were found in 12 of 14 tumors (≈85% of tumors). Our data support the conclusion that RFP+ clusters in AT3 RIIIcI2 tumors are the result of epithelial plasticity. The opposite type of plasticity, EMT, has been proposed to occur at the leading edge of colorectal carcinomas (31) and is considered by some a required step in invasion and metastasis (24, 29, 32, 33). Transition to a mesenchymal-like state has also been noted in primary pancreatic carcinomas (34). Less is known about sarcomas; however, phenotypic plasticity has been noted in experimental fibrosarcomas induced by Rous sarcoma virus oncogene (v-src) in athymic mice (35) and in biopsies of Ewing sarcomas in humans (36). We predict that careful examination of sarcomas will reveal plasticity due to MET as observed in the AT3 tumors.
What Is the Origin of MET in AT3 Tumors?
An answer to this question is suggested by the nonrandom spatial distribution of the AT3 cells undergoing MET (RFP+ cells). These clusters of RFP expression predominate near stroma, many near the tumor capsule. We observed a similar phenomenon among lung micrometastases where many RFP+ AT3 cells were seen in direct contact with the lung parenchyma or decorating blood vessels. This was in contrast to large metastatic nodules, which had very few, if any, RFP+ cells. There are two general explanations for the nonrandom distribution of RFP+ cells near stroma: recruitment or induction. Recruitment would suggest that a preexisting population of cells undergoing MET is attracted to or preferentially survives near stroma. There is no evidence for the existence of RFP+ cells among the AT3-RIIIcI2 cell population in culture 1 day before implantation (compare the distribution of RFP expression of AT3-RIIIcI2 and Rint in Fig. 6), and thus we do not favor this idea. Induction, which is the explanation we prefer, implies that the transition to an epithelium-like state is a phenotypic response to signals from the microenvironment. A similar explanation has been offered to explain MET in the central areas of metastases of colorectal cancers in animal models (31). Stromal signals are likely growth factors, and indeed a switch in FGFR2 isoforms was observed in rat bladder carcinoma NBT-II cells in culture during growth-factor-induced EMT (37).
What Are the Implications of MET in Terms of Tumor Progression and Metastasis?
Although distinct roles for EMT in tumor progression and metastasis have been proposed (24, 25, 38–40), the function of MET in malignancy is not clear. MET could be seen as a process of redifferentiation and perhaps a desired outcome. Our data suggest otherwise. The appearance of RFP+ lung metastases in animals harboring AT3-RIIIcI2 tumors indicated that the potential to undergo MET was compatible with the capacity to invade and form metastases. Indeed, the numbers of these RFP+ micrometastases suggest they represent frequent events, especially when compared with the frequency of RFP+ clusters in primary tumors. We propose that what makes AT3 tumors highly aggressive is not their mesenchymal character per se but rather their capacity to undergo phenotypic transitions. We summarize this idea by analogy to a potential energy diagram, where the likelihood to transition from one cell state to another is determined by the equivalent of an activation energy barrier. For instance, a normal polarized epithelial cell is much less likely to reach a mesenchymal state than a transformed cell. We suggest that AT3 cells probably inhabit a metastable state, and the great majority of AT3 cells in a tumor can be induced to undergo MET. We further propose that the increased capacity to transition is related to malignant fitness (41). Finally, we believe this idea can explain the rarity and aggressiveness of tumors of mixed carcinoid and sarcomatoid histology. We propose that in sarcomatoid carcinomas, the epithelioid and fibroblastoid cells, which can arise from the same clone (42), inhabit different phenotypic states very near an unstable transition state. The instability of these phenotypes suggests these states would be relatively uncommon, and indeed a survey of prostate sarcomatoid carcinomas so confirms (43). The ability to readily transition between epithelial and mesenchymal phenotypes makes these tumors fit to conquer the many hurdles to establish a successful metastasis. Thus, for both carcinomas and sarcomas, we postulate that phenotypic versatility leads to increased malignant fitness.
Methods
Plasmids.
Plasmids encoding the RFP were constructed on the backbone of the pcDNA6/V5-HisA vector (Invitrogen, Carlsbad, CA), which has an ampicillin cassette for selection in Escherichia coli and a blasticidin resistance gene for generation of stable transfectants in mammalian cell cultures. The RFP we have used is a DsRed dimer variant (44) kindly provided by Roger Tsien. The plasmid pRint has the intron from plasmid pI12 (11) inserted at position 369 of the RFP ORF (see Fig. 1B). The plasmid pRIIIcI2 is derived from pRint by subcloning the rat FGFR2 exon IIIc and flanking intronic sequences, including the regulatory sequences the intronic activating sequence 2 (IAS2) and intronic splicing activator and repressor (ISAR), into the middle of the pRint intron. pRΔ,Δ is identical to pRIIIcI2, except that the IAS2 and ISAR sequences have been deleted as described for pI12IIIc-ΔΔ in ref. 17. The complete sequence of the plasmids will be provided upon request.
Cell Culture and Transfections.
DT and AT3 rat prostate tumor cells were kindly provided to our laboratory by Wallace McKeehan. We refer to the DT cells as DT3 cells. Cells were transfected by using lipofectamine (Invitrogen), and stable transfectants were selected by using 15 μg/ml blasticidin. Fluorescent cells were analyzed by fluorescence cytometry at the Duke University Cytometry facility, and this analysis revealed that initial blasticidin resistant populations were relatively homogeneous. Nonetheless, we used preparative FACS to exclude low-intensity fluorescence cells in the case of AT3-Rint, DT3-Rint, and DT3-RIIIcI2 and high-intensity fluorescence cells for AT3-RIIIcI2 and -RΔΔ, which were present in very small numbers. Cell populations with high levels of RFP expression (AT3-Rint, DT3-Rint, and DT3-RIIIcI2) contain a small number of cells with background levels of fluorescence (see Fig. 6) that probably represent cells where the reporter is disrupted or lost. Variability of RFP expression among cells in the transfected populations could be deduced from FACS of AT3-Rint cell populations (see Fig. 6). FACS profiles indicated that AT3-RIIIcI2 and AT3-RΔΔ cells have stable low levels of RFP expression over several months (not shown). The FACS analysis of AT3 cells that is shown in Fig. 6 was performed 1 day before implantation of tumor cells in animals.
Animals and Tumor Cell Implantation.
For the studies involving fluorescent reporters, AT3 cells were trypsinized, resuspended in PBS to a final concentration of 107/ml, and kept on ice. Cells (1–2 × 106) were injected s.c. in one flank of Copenhagen 2331 rats (Harlan Labs, Indianapolis, IN; 75–90 g, 2 mo of age). Animals were continuously monitored for tumor growth. All animal procedures were performed according to the Duke University Institutional and Animal Care and Use guidelines.
Histological Sections.
Preparation and imaging.
Excised tumors and lungs were washed in PBS at room temperature. Tumors were dissected in eight smaller sections to provide a thorough survey of the tumor. Depending on the size of the lungs, they were frozen either together or separate. The tumor sections and the lungs were placed in cryomolds, embedded in optimal-cutting-temperature tissue sectioning medium (Sakura Finetek, Torrance, CA), snap-frozen in liquid nitrogen, and stored at −80°C. Slides for fluorescence imaging were prepared as follows: the tissue was incubated for 15 min at −20°C to equilibrate the temperature and then sectioned with a microtome. The sections (15 μm) were placed on glass slides, fixed in 4% (wt/vol) paraformaldehyde for 5 min at room temperature, and rinsed in PBS at room temperature. The slides were mounted with gel/mount media (Biomeda, Foster City, CA). The sections were analyzed by using an Olympus (Melville, NY) IX 71 epifluorescence microscope, and images were acquired by using an Olympus DP70 digital camera. Image processing was done with DP controller software (Olympus). Masson’s Trichrome staining was performed by using a commercially available kit (DBS, Pleasanton, CA). After taking the fluorescent and phase-contrast images of particular regions, the section was stained with the Masson’s Trichrome kit, and the same regions were identified by comparison to the phase contrast and imaged as described above.
Immunofluorescence staining.
Sections were fixed in 4% paraformaldehyde for 1 h before blocking and incubation with antibody. The E-cadherin (BD Biosciences, Franklin Lakes, NJ), ZO-1 (Biogenesis), and vimentin (Invitrogen) antibodies were previously labeled with Zenon Alexa Fluor 488 (Molecular Probes, Eugene, OR), according to the manufacturer’s specifications. The sections were analyzed by epifluorescence microscopy as described above.
Molecular analysis.
After macroscopic imaging, the tumors were snap-frozen and stored at −80°C. The tissue was ground to a powder in liquid nitrogen using a mortar and pestle, and RNA was extracted by using TRIzol as recommended by the manufacturer (Invitrogen). RT-PCR for RFP transcripts was performed by using primers on the R and FP exons (Fig. 1B).
Supplementary Material
Acknowledgments
We thank Dr. Roger Tsien (University of California, San Diego, CA) for the gift of the dimer dsRED (RFP) and Dr. Wallace McKeehan (Texas A&M University System Health Science Center, Houston, TX) for the gift of the DT3 and AT3 cells. We thank members of the M.A.G.-B. laboratory for helpful discussions. M.A.G.-B. acknowledges support from National Cancer Institute Grant R33 CA97502.
Abbreviations
- FGFR2
fibroblast growth factor receptor 2
- MET
mesenchymal to epithelial transition
- RFP
red fluorescent protein.
Footnotes
Conflict of interest statement: M.A.G.-B. is a founder and consultant for Intronn, Inc., which owns and is commercializing the use of transsplicing reactions in gene therapy.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Black DL. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 2.Faustino NA, Cooper TA. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 3.Schwerk C, Schulze-Osthoff K. Mol Cell. 2005;19:1–13. doi: 10.1016/j.molcel.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 4.Kalnina Z, Zayakin P, Silina K, Line A. Genes Chromosomes Cancer. 2005;42:342–357. doi: 10.1002/gcc.20156. [DOI] [PubMed] [Google Scholar]
- 5.Venables JP. Cancer Res. 2004;64:7647–7654. doi: 10.1158/0008-5472.CAN-04-1910. [DOI] [PubMed] [Google Scholar]
- 6.Brinkman BM. Clin Biochem. 2004;37:584–594. doi: 10.1016/j.clinbiochem.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 7.Garcia-Blanco MA, Baraniak AP, Lasda EL. Nat Biotechnol. 2004;22:535–546. doi: 10.1038/nbt964. [DOI] [PubMed] [Google Scholar]
- 8.Baraniak AP, Chen JR, Garcia-Blanco MA. Mol Cell Biol. 2006;26:1209–1222. doi: 10.1128/MCB.26.4.1209-1222.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Del Gatto F, Gesnel MC, Breathnach R. Nucleic Acids Res. 1996;24:2017–2021. doi: 10.1093/nar/24.11.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carstens RP, Wagner EJ, Garcia-Blanco MA. Mol Cell Biol. 2000;20:7388–7400. doi: 10.1128/mcb.20.19.7388-7400.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner EJ, Garcia-Blanco MA. Mol Cell. 2002;10:943–949. doi: 10.1016/s1097-2765(02)00645-7. [DOI] [PubMed] [Google Scholar]
- 12.Wagner EJ, Baraniak AP, Sessions OM, Mauger D, Moskowitz E, Garcia-Blanco MA. J Biol Chem. 2005;280:14017–14027. doi: 10.1074/jbc.M414492200. [DOI] [PubMed] [Google Scholar]
- 13.Del Gatto F, Plet A, Gesnel MC, Fort C, Breathnach R. Mol Cell Biol. 1997;17:5106–5116. doi: 10.1128/mcb.17.9.5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baraniak AP, Lasda EL, Wagner EJ, Garcia-Blanco MA. Mol Cell Biol. 2003;23:9327–9337. doi: 10.1128/MCB.23.24.9327-9337.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carstens RP, McKeehan WL, Garcia-Blanco MA. Mol Cell Biol. 1998;18:2205–2217. doi: 10.1128/mcb.18.4.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Del Gatto-Konczak F, Bourgeois CF, Le Guiner C, Kister L, Gesnel MC, Stevenin J, Breathnach R. Mol Cell Biol. 2000;20:6287–6299. doi: 10.1128/mcb.20.17.6287-6299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagner EJ, Curtis ML, Robson ND, Baraniak AP, Eis PS, Garcia-Blanco MA. RNA. 2003;9:1552–1561. doi: 10.1261/rna.5840803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hovhannisyan RH, Carstens RP. Mol Cell Biol. 2005;25:250–263. doi: 10.1128/MCB.25.1.250-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunning WF. Natl Cancer Inst Monogr. 1963;12:351–369. [PubMed] [Google Scholar]
- 20.Yan G, Fukabori Y, McBride G, Nikolaropolous S, McKeehan WL. Mol Cell Biol. 1993;13:4513–4522. doi: 10.1128/mcb.13.8.4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu X, Jin C, Wang F, Yu C, McKeehan WL. Cancer Res. 2003;63:4936–4944. [PubMed] [Google Scholar]
- 22.Matsubara A, Kan M, Feng S, McKeehan WL. Cancer Res. 1998;58:1509–1514. [PubMed] [Google Scholar]
- 23.Isaacs JT. Current Concepts and Approaches to the Study of the Prostate. New York: Liss; 1987. pp. 513–576. [Google Scholar]
- 24.Thiery JP. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 25.Thiery JP. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Thiery JP, Sleeman JP. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 27.Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, Borok Z. Am J Pathol. 2005;166:1321–1332. doi: 10.1016/s0002-9440(10)62351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khoury H, Naujokas MA, Zuo D, Sangwan V, Frigault MM, Petkiewicz S, Dankort DL, Muller WJ, Park M. Mol Biol Cell. 2005;16:550–561. doi: 10.1091/mbc.E04-07-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson EW, Newgreen DF, Tarin D. Cancer Res. 2005;65:5991–5995. doi: 10.1158/0008-5472.CAN-05-0616. discussion 5995. [DOI] [PubMed]
- 30.Tuxhorn JA, Ayala GE, Smith MJ, Smith VC, Dang TD, Rowley DR. Clin Cancer Res. 2002;8:2912–2923. [PubMed] [Google Scholar]
- 31.Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T. Proc Natl Acad Sci USA. 2001;98:10356–103561. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Radisky DC. J Cell Sci. 2005;118:4325–4326. doi: 10.1242/jcs.02552. [DOI] [PubMed] [Google Scholar]
- 33.Tarin D, Thompson EW, Newgreen DF. Cancer Res. 2005;65:5996–6000. doi: 10.1158/0008-5472.CAN-05-0699. discussion 6000–6001. [DOI] [PubMed]
- 34.Nakajima S, Doi R, Toyoda E, Tsuji S, Wada M, Koizumi M, Tulachan SS, Ito D, Kami K, Mori T, et al. Clin Cancer Res. 2004;10:4125–4133. doi: 10.1158/1078-0432.CCR-0578-03. [DOI] [PubMed] [Google Scholar]
- 35.England JM, Panella MJ, Kopen GC, Wisner TW, Halpern MS. Virchows Arch. 1994;424:83–88. doi: 10.1007/BF00197397. [DOI] [PubMed] [Google Scholar]
- 36.van der Schaft DW, Hillen F, Pauwels P, Kirschmann DA, Castermans K, Egbrink MG, Tran MG, Sciot R, Hauben E, Hogendoorn PC, et al. Cancer Res. 2005;65:11520–11528. doi: 10.1158/0008-5472.CAN-05-2468. [DOI] [PubMed] [Google Scholar]
- 37.Savagner P, Valles AM, Jouanneau J, Yamada KM, Thiery JP. Mol Biol Cell. 1994;5:851–862. doi: 10.1091/mbc.5.8.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang Y, Massague J. Cell. 2004;118:277–279. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 39.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 40.Karreth F, Tuveson DA. Cancer Biol Ther. 2004;3:1058–1059. doi: 10.4161/cbt.3.11.1302. [DOI] [PubMed] [Google Scholar]
- 41.Nowell PC. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 42.Ray ME, Wojno KJ, Goldstein NS, Olson KB, Shah RB, Cooney KA. Urology. 2006;67:423. doi: 10.1016/j.urology.2005.08.013. e5–e8. [DOI] [PubMed] [Google Scholar]
- 43.Fukawa T, Numata K, Yamanaka M, Miyamoto T, Kurokawa Y, Kanayama HO, Kagawa S, Utsunomiya M, Hirokawa M. Int J Urol. 2003;10:108–113. doi: 10.1046/j.1442-2042.2003.00574.x. [DOI] [PubMed] [Google Scholar]
- 44.Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. Proc Natl Acad Sci USA. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}