

Abstract
There are large inter- and intraspecies differences in susceptibility to dioxin-induced toxicities. A critical question in risk assessment of dioxin and related compounds is whether humans are sensitive or resistant to their toxicities. The diverse responses of mammals to dioxin are strongly influenced by functional polymorphisms of the arylhydrocarbon receptor (AHR). To characterize responses mediated by the human AHR (hAHR), we generated a mouse possessing hAHR instead of mouse AHR. Responses of these mice to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and 3-methylcholanthrene were compared with the responses of naturally sensitive (C57BL/6J) and resistant (DBA/2) mice. Mice homozygous for hAHR exhibited weaker induction of AHR target genes such as cyp1a1 and cyp1a2 than did C57BL/6J (Ahrb-1/b-1) mice. DBA/2 (Ahrd/d) mice were less responsive to induction of cyp genes than C57BL/6J mice. hAHR and DBA/2 AHR exhibit similar ligand-binding affinities and homozygous hAHR and Ahrd/d mice displayed comparable induction of AHR target genes by 3-methylcholanthrene. However, when TCDD was administered, a greatly diminished response was observed in homozygous hAHR mice compared with Ahrd/d mice, indicating that hAHR expressed in mice is functionally less responsive to TCDD than DBA/2 AHR. After maternal exposure to TCDD, homozygous hAHR fetuses developed embryonic hydronephrosis, but not cleft palate, whereas fetuses possessing Ahrb-1 or Ahrd developed both anomalies. These results suggest that hAHR may define the specificity of the responses to various AHR ligands. Thus, the hAHR knock-in mouse is a humanized model mouse that may better predict the biological effects of bioaccumulative environmental toxicants like TCDD in humans.
Keywords: human‖C57BL6/J‖DBA/2‖CYP1A1
Polycyclic aromatic hydrocarbons (PAH) and halogenated aromatic hydrocarbons (HAH), including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), benzo[a]pyrene, and polychlorinated biphenyls, are ubiquitous environmental toxicants whose chemical stability and lipophilicity make them highly persistent in the environment and in living organisms. These groups of chemicals cause various toxicological and biological responses, typified by teratogenesis, thymic atrophy, severe epithelial disorders, wasting syndrome, tumor promotion, and induction of xenobiotic-metabolizing enzymes in experimental animals (1, 2). The toxicities of these compounds are mediated by a conserved signaling pathway (1–4) through binding to and activation of the arylhydrocarbon receptor (AHR). AHR activation in turn mediates a transcriptional response for genes regulated by this transcription factor (5–8). Despite strong conservation of this pathway, there are wide inter- and intraspecies differences in the toxicological responses to AHR ligands (9–11). The molecular basis for these species and strain differences appears to relate to polymorphisms in AHR. Factors influencing susceptibility to the toxicity of TCDD have been studied in several animal models. There is a 10-fold difference in susceptibility between the dioxin-sensitive C57BL/6 and the resistant DBA/2 strains of mice that can be explained by polymorphic variations in the ligand-binding domain and in the C-terminal region of the AHR molecule of each strain (9, 12–14). Response to TCDD in the Long–Evans (sensitive) and Han/Wistar rats (resistant) differs by >1,000-fold due to a critical point mutation in the transactivation domain in the AHR of the Han/Wistar rat (15–17).
The effects of TCDD on humans are less well understood, although high incidences of chloracne, teratogenicity, and abortion have been associated with high blood concentrations of dioxin and related compounds in residents of regions where industrial accidents or extensive use of dioxin-containing defoliants have resulted in human exposures (3). Increased levels of dioxin in the body have been reported recently to be associated with abnormal sex ratio of newborns nearly 25 years after the accident in Seveso, Italy (18). Because the AHR primarily mediates the pleiotropic manifestations of dioxin exposure, characterization of the structural and functional properties of the human AHR (hAHR) is critical for understanding the types and magnitudes of human responses to various PAH/HAHs.
To date, in vitro characterization of the hAHR has provided ambiguous insights into human sensitivity to dioxin. The dissociation constant (Kd) of hAHR for TCDD was comparable to that of TCDD-resistant DBA/2 AHR (9, 19), suggesting that humans might be resistant to TCDD. By contrast, high homology of the human receptor to the AHR of the guinea pig, which is the most sensitive animal to TCDD, suggests a high responsiveness of humans to the toxin (20). Ligand specificity of hAHR was also examined and compared with those of zebrafish and rainbow trout AHRs using polychlorinated dibenzo-p-dioxin, dibenzofuran, and biphenyl congeners as test ligands. These studies revealed that mono-ortho polychlorinated biphenyls activated hAHR but were not very effective in activating either zebrafish or rainbow trout AHRs (21).
Assessment of human responses in vivo to unintended exposures to various PAH/HAHs has been hampered by limited exposure assessments and toxicological follow-up. Observational studies after intentional exposures have not been and should not be conducted. To gain stronger insight into the hazards to human health posed by compounds interacting with the hAHR in vivo, we generated a mouse model that harbors the hAHR cDNA instead of the mouse AhR gene. This mouse may reveal a humanized susceptibility to chemical toxicities. In response to challenges with 3-methylcholanthrene (3-MC) and TCDD, two prototypical AHR ligands, the hAHR knock-in mouse displayed a distinct response profile compared with control animals harboring either the C57BL/6 Ahr allele (Ahrb-1) (TCDD-sensitive C57BL/6J AHR) or the DBA/2 Ahr allele (Ahrd) (TCDD-resistant DBA/2 AHR) in the same C57BL/6J genetic background. Although gene expression responses mediated by hAHR from 3-MC were comparable to that by DBA/2 AHR, the homozygous human AHR knock-in allele (hAHR) mouse was the weakest responder to TCDD among the three strains examined. These results suggest that hAHR molecules expressed in mice retain a functional human specificity that can be distinguished from the murine AHR and provide important insights into the toxicological susceptibility of humans to AHR ligands released into the environment.
Materials and Methods
Construction of the hAHR Knock-in Vector.
The hAHR knock-in vector was constructed by using 129SV/J mouse Ahr genomic clones and hAHR cDNA as described (22). A 2-kb BamHI/HphI fragment containing the 129SV/J Ahr promoter was ligated to the hAHR cDNA (9, 22). The neo gene cassette was fused to the 3′ end of the hAHR cDNA in a reverse orientation, followed by a 6.5-kb HindIII/EcoRI fragment of the 129SV/J Ahr gene. This construct was ligated to the thymidine kinase cassette on the 5′ end.
Generation of hAHR Knock-in Mice.
The knock-in vector was electroporated into E14 embryonic stem (ES) cells (23). A pair of primers (sense, GTATGCATTACCATGCTCCCATTCTGCTGG; antisense, ACATCTTGTGGGAAAGGCAGCAGGCTAGCC) was used for PCR screening. After confirmation by Southern blot analysis, positive clones were injected into blastocysts. Heterozygous hAHR knock-in mice were backcrossed into a C57BL/6J background up to the seventh generation and interbred to yield heterozygous and homozygous hAHR and wild-type Ahrb-1/b-1 mice. The genotype of each pup was determined by PCR, with a common sense primer; 5′-ATGAGCAGCGGCGCCAACAT-3′, an antisense primer for endogenous Ahr allele; 5′-GCTAGACGGCACTAGGTAGG-3′, and an antisense primer for targeted allele; 5′-CAGGTAACTGACGCTGAGCC-3′. PCR amplification was carried out for 30 cycles under the following conditions; 94°C for 30 sec, 62°C for 30 sec, and 72°C for 30 sec.
Chemicals and Animals.
TCDD (99.5% pure) and 3-MC were purchased from Cambridge Isotope Laboratories (Andover, MA) and Wako Pure Chemical (Osaka), respectively. D2N-Ahrd mice and inbred C57BL6/J mice were procured from The Jackson Laboratory. Ahr-null mutant mice used in this study were generated by Y.F.-K (22).
RNA Blotting Analyses.
We isolated total RNA by using ISOGEN (Nippon Gene, Tokyo) and purified polyA RNA by using an Oligotex-MAG mRNA purification kit (Takara Biotechnology, Tokyo). For detection of Ahr mRNA, 5 μg of polyA RNA per lane was applied, and a portion of mouse Ahrb-1 cDNA (Bpu1102I-KpnI; 734-bp) encoding the PAS domain was used for a probe. This nucleotide sequence is conserved with 83% homology to the corresponding hAHR cDNA (12, 24). To examine the inducibility of CYP1A1 and CYP1A2, 6-week-old littermates (Ahrb-1/b-1 and homozygous hAHR) and D2N-Ahrd (Ahrd/d) mice were given a single i.p. injection of 80 mg/kg 3-MC or 100 μg/kg TCDD. Mice were killed by cervical dislocation 24 h after injection. Ten micrograms of total RNA per lane was hybridized with the appropriate mouse cDNA probes (25).
RT-PCR Analyses of hAHR and Murine Ahr mRNA Expression in Embryos.
Total RNA was isolated from palate and kidney of gestation day (GD)18.5 fetus by using ISOGEN. One microgram of the total RNA was reverse-transcribed into cDNA with Superscript-II reverse transcriptase (Life Technologies, Gaithersburg, MD) and random hexamers at 42°C for 50 min. The resulting cDNAs were subjected to 30 cycles of PCR by using the specific primers for the gene for the hAHR (5′ primer, 5′-GTAAGTCTCCCTTCATACC-3′; 3′ primer, 5′-AGGCACGAATTGGTTAGAG-3′), mouse Ahr (5′ primer, 5′-CTTTGCTGAACTCGGCTTGC-3′; 3′ primer, 5′-TTGCTGGGGGCACACCATCT-3′) and GAPDH (5′ primer, 5′-CCCCTTCATTGACCTCAACTACATGG-3′; 3′ primer, 5′-GCCTGCTTCACCACCTTCTTGATGTC-3′). The reaction was performed under the following conditions: 94°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec.
Immunohistochemical Analysis of hAHR Expression.
Immunohistochemical analysis was performed as described (26). Lungs were fixed in 0.1 M phosphate buffer containing 4% paraformaldehyde for 24 h and embedded in paraffin. Sections were incubated with anti-AHR antibody in 1:200 dilution, which reacts with both human and mouse AHR (N-19; Santa Cruz Biotechnology). AHR immunoreactivity was visualized with the avidin-biotin-peroxidase system (Vector Laboratories).
TCDD Treatment and Evaluation of Teratogenesis.
TCDD treatment was performed as described (22). On GD12.5, pregnant mice were given TCDD by i.p. administration at a dose of 40 μg/kg body weight (27). On GD18.5, the fetuses were taken out and fixed in 4% paraformaldehyde. The palatal structure was examined by cutting between the upper and lower jaws. The kidneys were sliced longitudinally and stained with hematoxylin/eosin. The presence and severity of hydronephrosis in each kidney was examined under a microscope as previously described (28) by using severity scores ranging from 0 to 3+ (0, normal kidney; 1+, slight decrease in length of papilla; 2+, marked decrease in length of papilla with some loss of renal parenchyma; 3+, complete absence of papilla, shell of kidney remaining with only a small amount of renal parenchyma). For statistical analysis, pairwise comparisons were made by Mann–Whitney U test, by using StatView for Macintosh version 5.0 (SAS Institute, Cary, NC).
Results
Replacement of the Mouse Ahr Gene with hAHR cDNA.
We hypothesize that the specific functional characteristics of the hAHR molecule form the principal basis for the pattern of human responses to xenobiotics that interact with the AHR. To characterize responses mediated by hAHR, we generated a mouse possessing hAHR instead of murine AHR. hAHR cDNA was introduced into the mouse Ahr locus by homologous recombination, thereby disrupting the mouse Ahr gene (Fig. 1A). The cDNA was recombined so that hAHR is expressed under the control of the endogenous mouse Ahr promoter. Sixteen independent G418-resistant ES clones were obtained of 240 by PCR screening, and seven clones were further confirmed as correctly targeted ES cells by genomic DNA blot analysis. EcoRI-digested genomic DNA from the three representative positive clones (nos. 14, 25, and 58) revealed 11.0- and 6.2-kb fragments derived from the intact and targeted alleles, respectively, when hybridized with the 5′-external probe (Fig. 1B).
Figure 1.

Generation of the hAHR knock-in mouse. (A) Strategy for hAHR cDNA knock-in by homologous recombination. E, H, and B are restriction sites for EcoRI, HindIII, and BamHI, respectively. Neo indicates the neomycin-resistance gene, and HSV-tk is the thymidine kinase gene under control of the herpes simplex virus promoter. The 5′-genomic probe used for DNA blot analysis is indicated by the hatched box. The positions of wild-type (pr 3) and mutant allele-specific (pr 2) primers and the common primer (pr 1) used in the genotyping PCR are indicated by arrowheads. The EcoRI restriction fragments detected with the 5′-genomic probe in the wild-type and targeted allele are denoted by horizontal bars. (B) DNA blot analyses of three recombinant ES clones. Genomic DNA was prepared from the ES clones (nos. 14, 25, and 58), and aliquots (10 μg) were digested by EcoRI. EcoRI digestion generated 11.0- and 6.2-kb bands for the wild-type and targeted alleles, respectively, by using the 5′-genomic probe. (C) Genotyping of the Ahr gene by DNA blot analysis. Genomic DNA was extracted from the tails of heterozygous and homozygous hAHR mice and wild-type Ahrb-1/b-1 mice and digested by EcoRI for DNA blot analysis. (D) Genotyping of littermates from the intercrosses of heterozygotes. PCR fragments of wild-type amplified with pr1 and pr3 (Ahrb-1; 280 bp) and mutant allele with pr1 and pr2 (hAHR; 240 bp) as depicted in A. H/H, H/b, and b/b indicate homozygous and heterozygous hAHR mice and wild type (Ahrb-1/b-1), respectively.
These three clones harboring hAHR were used for the generation of chimeric offspring. The male chimeras were mated with C57BL/6J females to obtain heterozygotes of the hAHR allele. They were subsequently bred into a C57BL/6J genetic background through the seventh generation, and the backcrossed heterozygous animals were interbred to yield hAHR homozygous mutant mice. The transmission of the targeted allele to the offspring was confirmed by genomic DNA blot analysis, and the genotype was determined by PCR by using tail DNA as a template (Fig. 1 C and D). Of 124 offspring obtained from heterozygous matings, wild-type (Ahrb-1/b-1), heterozygous, and homozygous hAHR mutant mice numbered 29, 71, and 24, respectively, conforming to the expected Mendelian inheritance ratio. Homozygous hAHR mice were viable, and no abnormalities were observed.
Expression of hAHR in hAHR Knock-in Mice.
The expression of hAHR and mouse Ahr mRNAs was examined by RNA blot analysis by using polyA RNAs isolated from major AHR-expressing organs including liver, lung, kidney, intestine, and thymus (Fig. 2A). A cDNA fragment encoding the PAS domain of C57BL/6 AHR, which shows 83% homology with the corresponding human molecule, was used as a common probe for detecting both mouse Ahr and hAHR mRNAs. The larger band detected in heterozygous hAHR mice and wild-type Ahrb-1/b-1 mice corresponds to the 5.4-kb transcript derived from the endogenous Ahrb-1 gene, and the shorter 5.0-kb transcript observed in heterozygous and homozygous hAHR is derived from the hAHR knock-in allele. This result establishes that, whereas the homozygous hAHR mouse lacks mRNA for murine Ahr, it expresses mRNA for hAHR. Further, the level of expression of hAHR mRNA is comparable to that of endogenous murine Ahr mRNA in the other strains.
Figure 2.

Expression of hAHR in multiple tissues of the hAHR knock-in mouse. (A) RNA blot analysis of polyA RNA (5 μg/lane) extracted from five representative organs of homozygous and heterozygous hAHR mice and Ahrb-1/b-1 mice. Human and mouse Ahr transcripts (hAHR and Ahrb-1, respectively) are indicated (Left). The same membrane was rehybridized with 32P-labeled cDNA of mouse GAPDH. H/H, H/b, and b/b are described in the Fig. 1 legend. (B) RT-PCR analyses of hAHR and murine Ahr mRNA expression in kidney and palate of GD18.5 fetuses. The reverse transcription was conducted either in the presence (+) or absence (−) of reverse transcriptase. PCR products representing the transcripts derived either from hAHR or Ahrb-1 are indicated on the left. (C and D) Immunohistochemical analysis of hAHR protein in the lung of a homozygous hAHR mouse. Immunoreactivity of AHR protein was observed in the alveolar epithelial cells of homozygous hAHR lung (C), whereas no immunoreactivity was observed in the lung of Ahr−/− mouse (D). Original magnifications, ×400 (C and D).
The embryonic expressions of mouse Ahr and hAHR mRNAs were examined by RT-PCR at the stage of GD18.5. As observed in the RNA blot analysis of adult tissues, the hAHR mRNA was expressed in the embryonic palate and kidney of homozygous and heterozygous hAHR mice. The abundance was comparable with that of the mouse Ahr mRNA expressed in Ahrb-1/b-1 and heterozygous hAHR mice (Fig. 2B). These results demonstrate that hAHR mRNA is transcribed under the control of the mouse Ahr promoter in both adult and embryonic hAHR knock-in mice.
To ascertain that hAHR protein is expressed from the knock-in allele, immunohistochemical analysis was performed on lung sections obtained from hAHR knock-in homozygous mouse and the Ahr-null mutant (22). Intense signals were detected in the alveolar epithelial cells of hAHR knock-in animals (Fig. 2C). The signal intensity of Ahr-null mutant lung (Fig. 2D) was as faint as the hAHR knock-in lung without the antibody (data not shown). Thus, hAHR protein is expressed from the knock-in allele.
The hAHR Knock-in Mouse Displays a Distinct Induction Profile of AHR Target Genes to Different AHR Ligands.
The response of the hAHR knock-in mouse to two prototypical AHR ligands, 3-MC and TCDD, was examined. To characterize the distinct properties, if any, of the hAHR, two strains of control mice were used for the analysis. One strain is a wild-type mouse in the C57BL/6J genetic background, which possesses AHR with high affinity for TCDD. The other strain is a congenic mouse, D2N-Ahrd, possessing AHR with low affinity (from DBA/2 mouse) in the C57BL6J genetic background. Because the hAHR knock-in mouse was backcrossed into C57BL/6J, these two strains of mouse enabled us to compare the characteristics of hAHR to those of C57BL/6J and DBA/2 AHR in the same genetic background.
Robust expression of the CYP1A1 and CYP1A2 genes was observed in the liver of Ahrb-1/b-1 mice after administration of 3-MC, whereas the magnitudes of induction in homozygous hAHR and Ahrd/d mice were much weaker and comparable to each other (Fig. 3A). The relative mean band intensities for CYP1A1 were 1.0 and 0.9 and were 1.0 and 1.1 for CYP1A2 in homozygous hAHR and Ahrd/d mice, respectively. After treatment with TCDD, the induction of the two genes was strongest in Ahrb-1/b-1 mice, intermediate in Ahrd/d mice, and weakest in homozygous hAHR mice (Fig. 3B). The fold inductions in homozygous hAHR, Ahrd/d, and Ahrb-1/b-1 mice were 1.0, 4.9, and 14.6 for CYP1A1, and 1.0, 5.7, and 8.4 for CYP1A2, respectively.
Figure 3.

Inducible expression of AHR target genes. Northern blot analysis of AHR-regulated CYP1A1 and CYP1A2 was performed. Six-week-old homozygous hAHR, Ahrd/d, and Ahrb-1/b-1 mice were treated with 80 mg/kg 3-MC (A) or 100 μg/kg TCDD (B). Total hepatic RNA was isolated 24 h after treatment and subjected to Northern analysis (10 μg/lane). Equal loading was confirmed by the abundance of GAPDH transcripts.
When the responses of Ahrb-1/b-1 and Ahrd/d mice were compared, the CYP1A1 expression levels were higher in Ahrb-1/b-1 than in Ahrd/d mice, which is consistent with previous reports (9, 12, 13). It is noteworthy that the responsiveness of homozygous hAHR mice to 3-MC was almost comparable to that of Ahrd/d mice, whereas the responsiveness to TCDD was much weaker. The differential response between Ahrd/d and homozygous hAHR mice was unexpected, because a previous study indicated that hAHR and DBA/2 AHR exhibit similar dissociation constants for TCDD binding as measured in vitro (9, 19). This result suggests that ligand binding does not fully define the integrated function of hAHR.
hAHR Knock-in Mouse Is Relatively Resistant to TCDD-Induced Teratogenicity.
The responses to TCDD mediated by hAHR are weaker than that by DBA/2 and C57BL/6 AHR when measured as inducibility of CYP1A family genes. Teratogenicity is a more integrated and complex toxicological manifestation of TCDD action. The most prominent teratogenic effects of TCDD on mouse fetus are cleft palate and hydronephrosis, both of which depend completely on AHR expression (29). The frequency and severity of these teratogenic effects of TCDD were examined in hAHR knock-in fetuses. Homozygous hAHR knock-in females were mated with males of the same genotype and given a single i.p. dose of 40 μg of TCDD per kg of body weight at GD12.5. Ahrb-1/b-1 and Ahrd/d females were treated in the same way as controls. All dams were weighed to monitor the normal continuation of the pregnancy and killed at GD18.5 to remove fetuses for examination of cleft palate and hydronephrosis.
As reported previously, cleft palate was observed in 100% of the wild-type Ahrb-1/b-1 fetuses exposed to TCDD (Fig. 4 A and C and Table 1) (22). By contrast, none of the treated homozygous hAHR fetuses showed abnormal palatogenesis (Fig. 4 B and D and Table 1). An intermediate frequency (30%) of cleft palate was observed in the Ahrd/d fetuses. Differences in the severity of cleft palate were not apparent in any of the symptomatic fetuses of any genotype. This anomaly was most frequent in Ahrb-1/b-1, intermediate in Ahrd/d, and least frequent in homozygous hAHR mice, in accordance with the transcriptional inducibility of AHR target genes, which was strongest in Ahrb-1/b-1, intermediate in Ahrd/d, and weakest in homozygous hAHR mice. Thus, a strong correlation between the incidence of cleft palate in each strain and the intrinsic transcriptional activity of their respective AHR molecules was observed.
Figure 4.
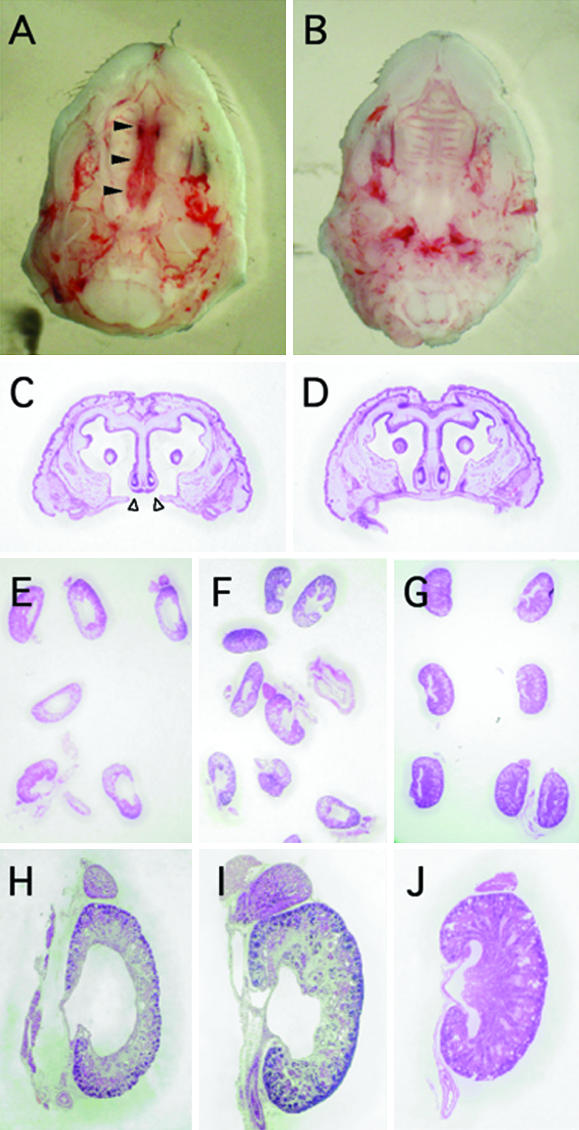
Fetal teratogenesis after maternal administration of TCDD. (A and C) Cleft palate in an Ahrb-1/b-1 fetus is shown. Filled arrowheads in A and open arrowheads in C indicate the failure of palatine shelves to fuse. Note that homozygous hAHR fetuses showed no cleft palates after TCDD treatment (B and D). (E, F, H, and I) Fetal hydronephrosis induced by TCDD. Ahrb-1/b-1 (E and H) and homozygous hAHR (F and I) fetuses are shown. (G and J) Unaffected kidneys from untreated Ahrb-1/b-1 fetuses are shown.
Table 1.
Incidence of anomalies caused by TCDD in homozygous hAHR, Ahrb-1/b-1, and Ahrd/d fetuses
| Genotype of fetuses | TCDD dose, μg/kg | Dams examined, n | Fetuses examined, n | Fetuses with
|
||||
|---|---|---|---|---|---|---|---|---|
| Cleft palate
|
Hydronephrosis
|
|||||||
| n | %* | n | %* | Severity: 0–3.0† | ||||
| Ahrb-1/b-1 | 0 | 2 | 13 | 0 | 0 | 2 | 12.5 | 0.19 ± 0.10‡ |
| Ahrb-1/b-1 | 40 | 5 | 29 | 29 | 100 | 26 | 89.7 | 2.54 ± 0.14§ |
| Ahrd/d | 0 | 2 | 15 | 0 | 0 | 2 | 13.3 | 0.20 ± 0.10‡ |
| Ahrd/d | 40 | 5 | 30 | 9 | 30 | 25 | 81.7 | 1.98 ± 0.14§ |
| Homo-hAHR | 0 | 2 | 16 | 0 | 0 | 1 | 6.3 | 0.03 ± 0.03‡ |
| Homo-hAHR | 40 | 5 | 37 | 0 | 0 | 30 | 81.1 | 1.19 ± 0.01§ |
Percentage of fetuses with each anomaly of all fetuses examined.
The criteria for severity scores are described in Materials and Methods. Data are expressed as mean ± SE.
Significant difference between TCDD-treated and -untreated fetuses of each genotype (P < 0.0001).
Significant difference between TCDD-treated homozygous hAHR fetuses and Ahrb-1/b-1 or Ahrd/d fetuses (P < 0.0001).
Hydronephrosis, another teratogenic effect of TCDD, is characterized by a dilated renal pelvis. The severity of this anomaly in the fetal kidney was scored from 0 (normal) to 3 (severest) according to criteria described previously (28). When kidneys scored at 1, 2, or 3 were counted as hydronephrotic, 89.7% of the Ahrb-1/b-1 offspring suffered from this teratogenic outcome after TCDD treatment (Fig. 4 E and H, and Table 1 for TCDD-treated animals; Fig. 4 G and J, and Table 1 for untreated animals). A similar incidence was observed in a previous study (22). Ahrd/d and homozygous hAHR fetuses also displayed this teratogenic effect with incidences of 81.7% and 81.1%, respectively (Fig. 4 F and I, and Table 1). Thus, there is no substantial difference in the incidence of hydronephrosis among the mice expressing the three distinct Ahr (hAHR) genes. When severity score values were compared among the TCDD-treated fetuses, they averaged 2.54, 1.98, and 1.19 for the Ahrb-1/b-1, Ahrd/d and homozygous hAHR genotypes, respectively (Table 1). Therefore, hydronephrosis observed in the homozygous hAHR fetuses was significantly less severe compared with that in either Ahrb-1/b-1 or Ahrd/d fetuses. Nonetheless, the average score of TCDD-treated homozygous hAHR fetuses (1.19) was still significantly higher than that of untreated homozygous hAHR fetuses (0.03), clearly demonstrating that the TCDD-activated hAHR mediates renal teratogenesis in mice. Although the magnitude of CYP gene induction is dramatically different depending on the Ahr genotype, the incidence of hydronephrosis is surprisingly comparable among the three strains. These results revealed that differences between human and murine AHR allowed for the emergence of discrete biological effects; e.g., hydronephrosis, but not cleft palate in homozygous hAHR mice.
To exclude the possibility that maternal factors affect the teratogenic manifestations on the fetuses, heterozygous hAHR parents were used to obtain homozygous hAHR and Ahrb-1/b-1 fetuses. Heterozygous mothers were treated with TCDD as described above, and fetuses were examined for both cleft palate and hydronephrosis. As described in Table 2, the incidence of cleft palate was 100% and 0% in Ahrb-1/b-1 and homozygous hAHR fetuses, respectively, which is identical to the results presented in Table 1. The incidence and severity (mean score) of hydronephrosis were 100% and 2.47 for Ahrb-1/b-1 and 66.6% and 1.17 for homozygous hAHR fetuses, respectively. Again, a more moderate effect in the homozygous hAHR fetuses is suggested, the severity difference being statistically significant. Therefore, we conclude that the TCDD-induced teratogenic effects are independent of maternal genotypes, and that fetal AHR activity is critical for determining the outcomes.
Table 2.
Incidence of anomalies caused by TCDD in fetuses from heterozygous hAHR parents
| Genotype of fetuses | TCDD dose, μg/kg | Dams examined, n | Fetuses examined, n | Fetuses with
|
||||
|---|---|---|---|---|---|---|---|---|
| Cleft palate
|
Hydronephrosis
|
|||||||
| n | %* | n | %* | Severity: 0–3.0 | ||||
| Ahrb-1/b-1 | 40 | 9 | 9 | 100 | 9 | 100.0 | 2.47 ± 0.14† | |
| Hetero-hAHR | 40 | 7 | 25 | 12 | 48 | 22 | 88.0 | 2.46 ± 0.13 |
| Homo-hAHR | 40 | 12 | 0 | 0 | 8 | 66.6 | 1.17 ± 0.01† | |
Percentage of fetuses with each anomaly out of all fetuses examined.
Significant difference between homozygous hAHR and Ahrb-1/b-1 fetuses (P < 0.0001).
Discussion
One of the central issues in the uncertainty surrounding risk assessments for TCDD and its structural analogs is whether humans are relatively sensitive or resistant to the toxicities of this class of compounds. Because the pleiotropic adverse effects induced by these toxins involve multiple processes, the human response is generated by the summation and integration of the properties inherent to the human components, including expression level, ligand-binding affinity, and transcriptional activity of the AHR, as well as the variety, function and activity of the AHR target genes. Through numerous preceding studies, the primary structure of the AHR protein has been regarded as one of the most critical factors determining the susceptibility and specificity of responses of animals to various PAH/HAHs including dioxin. On the basis of several observations in vitro, polymorphic variation in the Ahr gene is considered the primary basis for differences in sensitivity to TCDD among strains of mice (9–11). In this study, we attempted to establish an in vivo system to evaluate the specific function of the hAHR protein to better evaluate its role in determining possible patterns of human responses to PAH/HAHs.
For this purpose, we adopted a knock-in strategy to introduce hAHR cDNA into the mouse Ahr genomic locus by homologous recombination. This strategy offers an obvious advantage compared with a transgenic method, because the introduced sequence is transcribed under the same regulatory mechanisms of the replaced gene (30). As desired, expression levels of the hAHR transcript were almost the same with those of endogenous mouse Ahr mRNA in multiple AHR-expressing tissues of adult mice and GD18.5 embryos. The hAHR protein was detected by immunostaining in the lungs of homozygous hAHR mice.
A possible explanation of the relative resistance of the hAHR knock-in mouse to TCDD lies in the qualitative difference between the human and mouse AHR molecules. Assuming that the abundance of the hAHR protein is the same as that of the endogenous mouse AHR, our results imply that the hAHR-mediated response to TCDD in vivo is much lower than that of DBA/2 AHR, although previous reports showed that their affinities to TCDD, as measured in vitro, are almost the same (9, 19). Alignment of the primary amino acid sequences of the two molecules indicates the considerable divergence in the C-terminal regions (9) and the deletion analysis differently localized the transcriptional activity within the regions (31). Such structural diversity of the C-terminal region might lead to species-specific interaction behaviors with transcriptional cofactors. TCDD-activated hAHR may not recruit coactivators as efficiently as the DBA/2 counterpart. One possibility must be noted that the incompatibility between TCDD-activated hAHR and the mouse coactivators may cause the reduced response of hAHR knock-in mouse to TCDD.
hAHR was not detectable by immunoblot analysis with the current antiserum, and its abundance relative to the constitutive level of mouse AHR protein could not be determined. Considering this lack of quantitative information, limited protein accumulation might account for the attenuated responsiveness of hAHR knock-in mice to TCDD. hAHR may have an intrinsically shorter life than mouse AHR at physiological expression levels in vivo.
The susceptibility of embryonic kidneys of homozygous hAHR mice to the teratogenic effects of TCDD is noteworthy. The pathogenesis of this renal lesion induced by TCDD involves hyperplasia of the ureteric epithelium, resulting in an occlusion of the ureter and subsequent hydronephrosis (32). Adverse effects on the kidney and urinary tract have also been reported in humans exposed to TCDD (33). However, studies in Ben Tre Province in Vietnam, where defoliant containing dioxin was sprayed extensively, revealed little increase in the prevalence of cleft lip and/or palate compared with that observed in Japan (34), suggesting that hAHR is less potent to mediate the manifestation of cleft palate, and that a higher dose might be required for it. Consistent with these human reports, our analysis showed that hAHR, although expressed in mice, mediated the development of hydronephrosis induced by TCDD, but not cleft palate at our experimental dose. Thus, the knock-in animal seems to mimic some aspects of the human responses to PAH/HAHs.
An intriguing utilization of our knock-in mouse strategy would be as an in vivo system for the qualitative and quantitative assessment of possible human responses to various PAH/HAHs. In this study, D2N-Ahrd mice responded more strongly to TCDD than to 3-MC, whereas the hAHR knock-in mice responded almost equally to these two compounds. These results clearly show that the relative efficacy profiles, examined by TCDD and 3-MC, are different between D2N-Ahrd and our hAHR knock-in mouse. Therefore the efficacy profile specific to hAHR can be displayed by analyses of the responses of the hAHR knock-in mouse to an array of PAH/HAHs. Because environmentally relevant levels of exposure to dioxin and related compounds have garnered much concern in terms of their possible effects on reproductive, neurobehavioral, and immunological functions of humans, our hAHR knock-in mouse will serve as a humanized model mouse, exhibiting the human-specific responses to PAH/HAH congeners. This mouse should help define the range of biological and toxicological effects that could be expected to affect humans and thereby reduce some uncertainty in risk assessments of these persistent environmental contaminants.
Acknowledgments
We thank Drs. H. Ueda and S. Kimura at the Environmental Health Department of the Ministry of the Environment for valuable suggestions and support to initiate this project, and Dr. T. Kensler for the discussion and critical reading of the manuscript. We also thank Dr. N. Morito, Ms. N. Kaneko, and R. Kawai for help. This work was supported by grants from Exploratory Research for Advanced Technology Environmental Response Project (M.Y.), the Ministry of Education, Science, Sports and Culture (H.M. and M.Y.), the Ministry of Health, Labor, and Welfare (H.M., C.T., and M.Y.), Japan Society for the Promotion of Science–Research for the Future Program (M.Y.), Core Research for Evolutional Science and Technology (Y.F.-K., S.O., Y.A., C.T., and M.Y.), Program for Promotion Basic Research Activities for Innovative Biosciences (H.M. and S.T.), and Special Coordination Fund for Promoting Science and Technology (H.M.).
Abbreviations
- AHR
aryl hydrocarbon receptor
- hAHR
human AHR
- hAHR
human AHR knock-in allele
- Ahrd
DBA/2 Ahr allele
- Ahrb-1
C57BL/6 Ahr allele
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- 3-MC
3-methylcholanthrene
- PAH
polycyclic aromatic hydrocarbons
- HAH
halogenated aromatic hydrocarbons
- ES
embryonic stem
- GD
gestation day
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Poland A, Knutson C. Annu Rev Pharmacol Toxicol. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- 2.Whitelock J P., Jr Annu Rev Pharmacol Toxicol. 1990;30:251–277. doi: 10.1146/annurev.pa.30.040190.001343. [DOI] [PubMed] [Google Scholar]
- 3.Landers J P, Bunce N J. Biochem J. 1991;276:273–287. doi: 10.1042/bj2760273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swanson H J, Bradfield C A. Pharmacogenetics. 1993;3:213–230. doi: 10.1097/00008571-199310000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Hoffman E C, Reyes H, Chu F-F, Sander F, Conly L H, Brooks B A, Hankinsin O. Science. 1991;252:954–958. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- 6.Okey A B, Riddick D, Harper P A. Toxicol Lett. 1994;70:1–22. doi: 10.1016/0378-4274(94)90139-2. [DOI] [PubMed] [Google Scholar]
- 7.Rowlands J, Gustafsson J A. CRC Crit Rev Toxicol. 1997;27:109–134. doi: 10.3109/10408449709021615. [DOI] [PubMed] [Google Scholar]
- 8.Poellinger L. Food Addit Contam. 2000;17:261–266. doi: 10.1080/026520300283333. [DOI] [PubMed] [Google Scholar]
- 9.Ema M, Ohe N, Suzuki M, Mimura J, Sogawa K, Ikawa S, Fujii-Kuriyama Y. J Biol Chem. 1994;269:27337–27343. [PubMed] [Google Scholar]
- 10.Poland A, Glover E. Mol Pharmacol. 1990;38:306–312. [PubMed] [Google Scholar]
- 11.Poland A, Palen D, Glover E. Mol Pharmacol. 1994;46:915–921. [PubMed] [Google Scholar]
- 12.Ema M, Matsushita N, Sogawa K, Ariyama T, Inazawa J, Nemoto T, Ota M, Oshimura M, Fujii-Kuriyama Y. J Biochem. 1994;116:845–851. doi: 10.1093/oxfordjournals.jbchem.a124605. [DOI] [PubMed] [Google Scholar]
- 13.Dolwick K M, Schmidt J V, Carver L A, Swanson H I, Bradfield C A. Mol Pharmacol. 1993;44:911–917. [PubMed] [Google Scholar]
- 14.Okey A B, Vella L M, Harper P A. Mol Pharmacol. 1989;35:823–830. [PubMed] [Google Scholar]
- 15.Pohjanvirta R, Unkila M, Tuomisto J. Pharmacol Toxicol. 1993;73:52–56. doi: 10.1111/j.1600-0773.1993.tb01958.x. [DOI] [PubMed] [Google Scholar]
- 16.Pohjanvirta R, Wong J M Y, Li W, Harper P A, Tuomisto J, Okey A B. Mol Pharmacol. 1998;54:86–93. doi: 10.1124/mol.54.1.86. [DOI] [PubMed] [Google Scholar]
- 17.Tuomisto J T, Viluksela M, Pohjanvirta R, Tuomisto J. Toxicol Appl Pharmacol. 1999;155:71–81. doi: 10.1006/taap.1998.8564. [DOI] [PubMed] [Google Scholar]
- 18.Mocarelli P, Gerthoux P M, Ferrari E, Patterson D G, Jr, Kieszak S M, Brambilla P, Vincoli N, Signorini S, Tramacre P, Carreri V, et al. Lancet. 2000;355:1858–1863. doi: 10.1016/S0140-6736(00)02290-X. [DOI] [PubMed] [Google Scholar]
- 19.Micka J, Milatovich A, Menon A, Grabowski G A, Puga A, Nebert D W. Pharmacogenetics. 1997;7:95–101. doi: 10.1097/00008571-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Kohkalainen M, Tuomisto J, Pohjanvirta R. Biochem Biophys Res Commun. 2001;285:1121–1129. doi: 10.1006/bbrc.2001.5317. [DOI] [PubMed] [Google Scholar]
- 21.Abnet C C, Tanguay R L, Heideman W, Peterson R E. Toxicol Appl Pharmacol. 1999;159:41–51. doi: 10.1006/taap.1999.8719. [DOI] [PubMed] [Google Scholar]
- 22.Mimura J, Yamashita K, Nakamura K, Morita M, Takagi T N, Nakao K, Ema M, Sogawa K, Yasuda M, Katsuki M, et al. Genes Cell. 1997;2:645–654. doi: 10.1046/j.1365-2443.1997.1490345.x. [DOI] [PubMed] [Google Scholar]
- 23.Hooper M, Hardy K, Handyside A, Hunter S, Monk M. Nature. 1987;326:292–295. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- 24.Ema M, Sogawa K, Watanabe N, Chujoh Y, Matsushita N, Gotoh O, Funae Y, Fujii-Kuriyama Y. Biochem Biophys Res Commun. 1992;184:246–253. doi: 10.1016/0006-291x(92)91185-s. [DOI] [PubMed] [Google Scholar]
- 25.Fernandez-Salguero P, Pineau T, Hilbert D M, McPhail T, Lee S S T, Kimura S, Nebert D W, Rudikoff S, Ward J M, Gonzalez F J. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- 26.Moriguchi T, Sakurai T, Takahashi S, Goto K, Yamamoto M. J Biol Chem. 2002;277:16985–16992. doi: 10.1074/jbc.M107962200. [DOI] [PubMed] [Google Scholar]
- 27.Yasuda M, Igarashi E, Datu A R, Igawa H. Teratology. 1986;34:454–455. [Google Scholar]
- 28.Birnbaum L S, Harris M W, Barnhart E R, Morrissey R E. Toxicol Appl Pharmacol. 1987;90:206–216. doi: 10.1016/0041-008x(87)90328-0. [DOI] [PubMed] [Google Scholar]
- 29.Birnbaum L S, Abbott B. In: Methods in Developmental Toxicology and Biology. Klug S, Thiel R, editors. London: Blackwell Science; 1997. pp. 51–63. [Google Scholar]
- 30.Tsai F Y, Browne C P B, Orkin S H. Dev Biol. 1998;196:218–227. doi: 10.1006/dbio.1997.8842. [DOI] [PubMed] [Google Scholar]
- 31.Kumar M B, Ramadoss P, Reen R K, Vanden Heuvel J P, Perdew G H. J Biol Chem. 2001;276:42302–42310. doi: 10.1074/jbc.M104798200. [DOI] [PubMed] [Google Scholar]
- 32.Hoffman R E, Stehr-Green P A, Webb K B, Evans R G, Knutson A P, Schramm W F, Staak J L, Gibson B B, Steinberg K K. J Am Med Assoc. 1986;255:2031–2038. [PubMed] [Google Scholar]
- 33.Abbott B D, Birnbaum L S, Pratt R M. Teratology. 1987;35:329–334. doi: 10.1002/tera.1420350307. [DOI] [PubMed] [Google Scholar]
- 34.Natsume N, Kawai T, Le H. Cleft Palate Craniofac J. 1998;35:183–185. doi: 10.1597/1545-1569_1998_035_0183_tte_2.3.co_2. [DOI] [PubMed] [Google Scholar]