

Abstract
The metal ion dependence of the catalytic activity of recombinant Escherichia coli dUTP pyrophosphatase (dUTPase), an essential enzyme preventing incorporation of uracil into DNA, has been investigated by steady-state kinetic, electron paramagnetic resonance, and electron nuclear double resonance methods. Values of kcat and kcat/Km were 4.5 ± 0.1 s−1 and 0.49 ± 0.1 × 106 M−1⋅s−1 in the absence of divalent metal ions, 14.7 ± 2.2 s−1 and 25.1 ± 7.4 × 106 M−1⋅s−1 in the presence of Mg2+ or Mn2+, and 24.2 ± 3.6 s−1 and 2.4 ± 0.7 × 106 M−1⋅s−1 when supported by VO2+ or bis(acetylacetonato)oxovanadium(IV). Binding of VO2+ to the enzyme in the presence of dUDP, a nonhydrolyzable substrate analog, was specific and competitive with Mg2+. Electron paramagnetic resonance spectra of the ternary enzyme–VO2+-chelate–dUDP complex revealed a pattern of 31P superhyperfine coupling specifying two structurally equivalent phosphate groups equatorially coordinated to the VO2+ ion. Proton electron nuclear double resonance spectra revealed an equatorial acetylacetonate ligand, indicating that one of the organic ligands had been displaced. By molecular graphics modeling, we show that the diphosphate group of enzyme-bound dUDP is sterically accessible to a hemi-chelate form of VO2+. We propose a similar location compatible with all kinetic and spectroscopic results to account for the reactivity of VO2+ and the VO2+-chelate in dUTP hydrolysis. In this location the metal ion could promote an ordered conformation of the C-terminal fragment that is obligatory for catalysis but dynamically flexible in the free enzyme.
Contrary to ordinary expectation, the nucleic acid uracil can be incorporated into DNA in the cell because DNA polymerases do not distinguish between thymine and uracil. The most important cellular vehicle for hindering this process is the enzyme dUTP pyrophosphatase (dUTPase) (1, 2). This enzyme hydrolyzes dUTP into dUMP and pyrophosphate, thereby removing dUTP from the DNA biosynthetic pathway. Although several high to medium resolution x-ray studies of dUTPases from human (3), bacterial (4–6), and viral (7, 8) sources have been reported, there is no understanding of the structural basis of the catalytic mechanism. The C-terminal portion of each subunit of this trimeric enzyme is partly disordered in crystals and does not contribute to the electron density map. Furthermore, the binding site of Mg2+ in the active site has not been identified. In the crystal structure of the equine infectious anemia virus (EIAV) enzyme, a Sr2+ ion bound to both the α- and β-phosphate groups of dUDP has been localized (7), but in a position that would sterically prevent nucleophilic attack by a hydrolytic water molecule. Nonetheless, the catalytic importance of the C-terminal residues and the dependence of dUTPase action on Mg2+ have been amply demonstrated by studies of the enzyme in solution (9–12). Because dUTPase has acquired increasing attention as a potential target enzyme in cancer and antiretroviral chemotherapy, determination of the structural basis of its catalytic action is important in the design of chemotherapeutic agents.
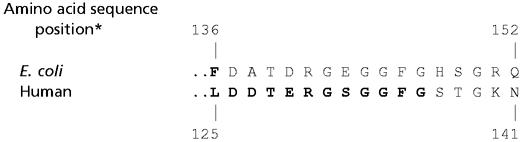
The most thoroughly studied species of dUTPases belong to the Eu(karyotic), Ba(cterial), and R(etroviral), or EuBaR, subgroup in which the protein consisting of 133–168 amino acid residues per subunit forms a homotrimeric complex (3–8, 13). Although sequence identity is as low as 31%, all members of this subgroup exhibit a similar fold with unusual intimacy between neighboring subunits. Each of the three active sites is comprised of residues from two subunits, while the C-terminal region of the third subunit partially covers the entrance. This arrangement of the three polypeptide chains is illustrated in Fig. 1 for the Escherichia coli enzyme. Correspondingly, amino acid sequences of the ordered and disordered segments of the C-terminal portions of the E. coli and human enzymes are compared in Table 1. In the E. coli enzyme the C-terminal region is ordered only on binding 2′-deoxyuridine with a 5′-triphosphate group in the presence of a divalent metal ion (9, 10, 14). Because ordering of the C-terminal region of the polypeptide chain has been shown to be required for catalytic action by the enzyme (9–11), an approach must be adopted for structural analysis that can be applied to macromolecules in solution.
Figure 1.

X-ray determined structure of Escherichia coli dUTPase (4, 5). The subunits of the trimeric enzyme are shown in different colors: A, red; B, yellow; and C, blue. The light-blue ribbon structure illustrates one subunit of the human enzyme (3) superpositioned according to structurally equivalent residues (7). The yellow arrow identifies the ultimate C-terminal residue, Phe-136, that is observed by x-ray in the E. coli enzyme (4–6). Rendering this residue in ball-and-stick form highlights its location in each subunit of the enzyme. The dUDP inhibitor bound to the E. coli enzyme (5) and Sr2+ in the EIAV enzyme (7) in green identify the location of one of the active sites.
Table 1.
Comparison of the C-terminal amino acid residues of E. coli and human dUTPases
 |
In a series of investigations from this laboratory, we have shown that vanadyl (VO2+) is a sensitive paramagnetic probe of Mg2+ and Ca2+ binding sites in nucleotides and proteins for structure analysis by electron paramagnetic resonance (EPR) and electron nuclear double resonance (ENDOR) spectroscopy (15–19). Not only can 31P superhyperfine (shf) structure of coordinating phosphate groups in nucleotide complexes be resolved (16, 17), but the observed EPR intensity derives only from macromolecule-bound VO2+ because free VO2+ near neutral pH polymerizes to form an EPR-silent precipitate (20). Therefore, spectra are not complicated by separate contributions from macromolecule-bound and unbound VO2+.
In steady-state kinetic studies, we have observed that VO2+ supports hydrolysis of dUTP catalyzed by E. coli dUTPase. The catalytic efficiency of the enzyme increased nearly 2-fold in comparison to the turnover rate in the presence of Mg2+, and binding of VO2+ to the enzyme is specific and competitive with Mg2+. Of particular interest is our observation that the enzymatic action of dUTPase is supported with equivalent catalytic efficiency by an organic chelate of VO2+, namely, bis(acetylacetonato)oxovanadium(IV) [VO(acac)2]. This observation, coupled with evidence on the basis of EPR and ENDOR spectroscopy that the organic chelate is bound to the enzyme in the presence of nucleotide, indicates that the Sr2+ site (7) cannot account for dUTP hydrolysis into dUMP and pyrophosphate. These results require complete alteration of all previous suggestions for the catalytic and structural role of Mg2+ in dUTP hydrolysis catalyzed by dUTPase.
Materials and Methods
General.
Q-Sepharose and Sephacryl S-200 were purchased from Amersham Biosciences; dUTP, dUDP, and buffer materials were purchased from Sigma; phenol red indicator was purchased from Merck; Chelex resin (sodium form) was purchased from Bio-Rad; and vanadyl sulfate hydrate and VO(acac)2 were purchased from Sigma-Aldrich. VO(d7-acac)2 was synthesized from perdeuterated starting materials following the procedure described for the synthesis of 13C-enriched VO(acac)2 (21). All other materials were of analytical reagent grade; deionized distilled water was used throughout.
Enzyme Preparation and Assay.
dUTPase from E. coli was purified as described by Persson et al. (22). The purified preparation appeared as a single band on SDS/PAGE [14% (wt/vol) gels; ref. 23]. Protein bands were visualized with Bio-Safe Coomassie stain (Bio-Rad); quantitative analysis was done by densitometry on a Gel Doc densitometer (Bio-Rad). The protein by this method is shown to be of at least 95% purity. Protein concentration was measured spectrophotometrically by using an extinction value for A0.1% of 0.52 at 280 nm (11) and a molecular weight of 49,000 for the trimeric enzyme (24). Enzyme concentrations are reported in this communication with respect to the monomer content of solutions.
Enzyme activity was measured under steady-state conditions with the phenol red indicator method (9, 12). Enzyme concentration was varied over the 5–10 × 10−8 M range and dUTP concentration was varied from 5 to 200 × 10−6 M in 0.001 M N-[tris(hydroxymethyl)methyl]-2-aminoethanesulfonic acid adjusted to pH 7.5, containing 0.15 M KCl and 4.0 × 10−5 M phenol red indicator. Proton release was followed at 559 nm by spectrophotometry on a Jenway 6105 (Dunmow, U.K.) or a Jasco V550 (Easton, MD) with thermostated cuvettes at 25°C. Initial velocity was determined from the slope of the initial part of the progress curve. The integrated Michaelis–Menten equation was also applied to evaluate progress curves (12). The results were in good agreement with initial velocity data.
To avoid adventitious divalent metal ions in assays, buffer and nucleotide solutions were treated first with a 70-ml Chelex column. Divalent cations Mg2+, Mn2+, and VO2+ were added in 1:1 stoichiometric ratio to the dUTP content. For addition of VO2+, all solutions were first freshly purged with N2. Stock solutions of ≈0.001 M VO2+ with dUTP in 1:1 molar ratio were prepared by addition of aliquots of 0.2 M VOSO4 in H2O (pH <2.0) to an appropriate aliquot of 0.1 M dUTP (pH <5.0). The pH was then adjusted to 7.0 with small amounts of 0.2 M NaOH. Preparation of the VO2+-dUTP mixture in this manner was required to keep VO2+ in solution and to prevent pH jumps in the enzyme activity assay mixtures. Alternatively, 0.005 M stock solutions of VO(acac)2 in ethanol/water (50:50 vol/vol) were added to appropriate aliquots of the 0.1 M dUTP, and the pH similarly adjusted. All VO2+-containing solutions were freshly prepared before enzyme activity assays or EPR studies.
Atomic absorption measurements of buffer solutions and of the stock enzyme solution, determined with a Varian Spectra AA220 spectrometer, showed no detectable Mg2+. The lower limit of Mg2+ detection was ≤0.1 ppm.
EPR and ENDOR Spectroscopy.
Concentrated stock solutions of VO2+ were prepared by dissolving VOSO4 in a small volume of H2O under an N2 atmosphere. The desired quantity of the VO2+ ion was then added to dUDP or dUTP solutions as described above for enzyme activity assays. The final concentration of VO2+ was generally ≈0.002 M for nucleotide complexes and ≈0.001 M for dUTPase complexes. All solutions were purged with nitrogen gas before use and stored frozen in EPR sample tubes to prevent oxidation of the metal ion.
EPR and ENDOR spectra were recorded with a Bruker ESP300E (Billerica, MA) spectrometer operating at 9.5 GHz and equipped with an Oxford Instruments ESR910 (Concord, MA) liquid helium cryostat and a Bruker ENDOR accessory, as described (17, 25). Typical experimental conditions for EPR measurements were as follows: sample temperature, 20 K; microwave frequency, 9.45 GHz; incident microwave power, 64 μW; modulation frequency, 12.5 kHz; and modulation amplitude, 0.8 G. Typical experimental conditions for ENDOR were as follows: microwave power, 6.4 mW; rf power, 50 W; rf modulation frequency, 12.5 kHz; and rf modulation depth, 10 kHz. The static laboratory magnetic field was not modulated for ENDOR.
Molecular Modeling.
Molecular modeling of dUTPase was carried out with INSIGHT 95 (Accelrys, San Diego) running on an R4400 Indigo2 (Silicon Graphics, Mountain View, CA) work station with High Impact Graphics. The atomic coordinates were obtained from the Protein Data Bank for E. coli (4, 5) and EIAV (7) enzymes. Coordinates for human UTPase were provided by J. A. Tainer (3). For modeling of the vanadyl cation, the V⩵O bond length was fixed at 1.59 Å (26). Valence angles involving equatorial donor-ligand atoms and equatorial and axial metal-ligand bond lengths were applied as determined for the [VO(CH3OH)5]2+ complex (15).
Results and Discussion
Steady-State Kinetic Analysis of Metal Ion Substitution.
From initial velocity data we have estimated the steady-state kinetic parameters for hydrolysis of dUTP catalyzed by E. coli dUTPase in the presence of three different divalent metal ions. In these studies we observed that Mg2+ can be replaced by either Mn2+ or VO2+ with retention of comparable catalytic activity. The results are summarized in Table 2. Whereas the reaction in the presence of Mn2+ is governed by kinetic parameters identical to those for Mg2+, the value of kcat for hydrolysis supported by VO2+ is 2-fold greater. Nonetheless, because of decreased affinity of the enzyme for the substrate in the presence of VO2+, as reflected in the Km value, the value of kcat/Km indicates that catalytic efficiency is greatest in the presence of Mg2+.
Table 2.
Comparison of the influence of divalent metal ions on steady-state kinetic parameters governing hydrolysis of dUTP catalyzed by recombinant E. coli dUTPase
| Metal ion | kcat (s−1) | kcat/Km (M−1⋅s−1 × 106) |
|---|---|---|
| No metal ion | 4.5 ± 0.1 | 0.49 ± 0.1 |
| Mg2+ | 14.7 ± 2.2 | 25.1 ± 7.4 |
| Mn2+ | 14.5 ± 2.2 | — |
| VO2+ | 27.3 ± 4.1 | 2.8 ± 0.9 |
| VO(acac)2 | 24.2 ± 3.6 | 2.4 ± 0.7 |
—, Not determined.
On the basis of EPR titrations in parallel experiments, we have observed that VO2+ is chelated by dUTP and dUDP near neutral pH, forming a VO2+–deoxyuridine nucleotide (dUXP) complex of limiting 1:2 stoichiometry in the presence of excess nucleotide. This behavior is identical to that observed for binding of adenine and guanine nucleotides by VO2+ (16, 17). The binding affinity of VO2+ for pyrophosphate is ≈103-fold greater than that of Mg2+ (27). Therefore, the VO2+ ion exists either bound to nucleotide or precipitated as an EPR-silent polymeric species near neutral pH (20). To avoid complications arising from either excess VO2+ or excess nucleotide, aliquots of equimolar mixtures of dUTP and VO2+ were added to the buffered enzyme solution to determine kinetic parameters. For direct comparison, steady-state kinetic parameters are also reported in Table 2 for Mg2+- and Mn2+-supported hydrolysis under similar conditions. In separate experiments, we observed that the apparent Km of dUTPase for Mg2+ is 8.0 ± 1.0 × 10−6 M in the presence of saturating concentrations of dUTP. Also, Vmax is increased by 17% when the Mg2+ concentration is raised from 4.0 × 10−5 M to 1.0 × 10−3 M. These results are in accord with the observations of others (12).
The results in Table 2 show that there is significant substrate turnover in the absence of added divalent metal ions. As described in Materials and Methods, we found that the level of adventitious Mg2+ in solutions of the purified enzyme and buffers was below the detection limit of the atomic absorption instrumentation. Therefore, we cannot attribute the observed catalytic activity of ≈4.5 s−1 to contaminant divalent metal ions, and conclude, therefore, that the enzyme catalyzes hydrolysis of dUTP in the absence of added divalent metal ion. On this basis, Mg2+ must be viewed as enhancing the catalytic action of the enzyme rather than as an obligatory cofactor. Comparison of the results in Table 2 indicate that substrate affinity for the enzyme in the absence of added Mg2+ is considerably lower than in its presence, indicating that Mg2+ is involved in binding of dUTP to the enzyme. Larsson et al. (12) also observed low substrate turnover rates in the absence of added metal ion but assumed that this activity was due to low levels of Mg2+ retained through the preparation. No analytical data were presented in confirmation.
EPR and ENDOR of VO2+ in Ternary dUTPase–VO2+–dUXP Complexes.
In frozen solution, the EPR spectrum of VO2+ is characterized by an axially symmetric g matrix and exhibits eight parallel and eight perpendicular absorption lines due to the (I = 7/2) 51V nucleus (28, 29). This pattern is observed in Fig. 2 for VO2+ in the ternary dUTPase–VO2+–dUDP complex (spectrum a) and the ternary complex formed with VO(acac)2 and dUDP (spectrum c). By analysis of the prominent spectral components due to hyperfine coupling of the unpaired electron with the 51V nucleus, we estimated values for A∥ and g∥ of 180.5 × 10−4 cm−1 and 1.936, respectively, for VO2+ in the ternary dUTPase–VO2+–dUDP complex and 179.1 × 10−4 cm−1 and 1.931, respectively, in the [VO(dUDP)2] complex. These values are characteristic of complexes with only oxygen-donor atoms in the equatorial plane (18, 28). As shown in spectrum b, loss of EPR absorption is observed only on addition of at least a 10-fold molar excess of Mg2+. Because no EPR absorption was observed on addition of VO2+ to the enzyme alone or after displacement by Mg2+, we conclude that VO2+ binds specifically to the enzyme and only in the same site as Mg2+.
Figure 2.

Comparison of first-derivative EPR absorption spectra of VO2+. (Left) a, dUTPase–VO2+–dUDP complex; b, identical conditions to those in a but after addition at 5°C of a 10-fold excess of Mg2+; and c, dUTPase–(VO2+-chelate)–dUDP complex. (Right) An expanded comparison of the −3/2⊥ feature of the EPR spectrum (indicated by an arrow in Left) of the VO(acac)2–dUDP mixture in the absence (black spectrum) and presence (red spectrum) of dUTPase. The intensity ratios of 1:2:1, the three hyperfine lines with ap = 7.19 G, and the total line width of 19.78 G are also indicated in red. Molar ratios of enzyme–VO2+–dUDP or of enzyme–VO(acac)2–dUDP were 1.25:1:1, respectively, at a dUTPase concentration of 1.35 × 10−3 M buffered to pH 7 in 0.1 M NaCl with 0.01 M Hepes.
Fig. 2 Right compares the −3/2⊥ feature of the EPR absorption spectra of VO(acac)2 in the presence of an equimolar amount of dUDP before and after addition of dUTPase. The EPR and ENDOR spectra of VO(acac)2 in the presence or absence of dUDP alone were identical. However, comparison of the two spectra in Fig. 2 Right shows that a complex between VO(acac)2 and dUDP is formed only in the presence of dUTPase. In the absence of the enzyme, a weakly resolved doublet underlying the −3/2⊥ spectral feature is observed (in the black spectrum) through an inflection point at ≈3,268 G. [In contrast to frozen aqueous solutions, a doublet is prominently observed for VO(acac)2 in frozen methanol.] The line width is increased, however, from 14.8 to 19.8 G on addition of enzyme together with appearance of a new feature at 3,278 G (red spectrum). We attribute the increase in line width to shf coupling of 31P with the unpaired electron, as observed in other VO2+–nucleotide complexes (16, 17). Whereas the low-field component of the shf pattern is not resolved because of the underlying doublet nature of the −3/2⊥ component, the high-field feature provides a measure of its peak-to-peak amplitude. Together with the increase in line width, this is consistent with a shf pattern of three components of 1:2:1 relative amplitude, as indicated in Fig. 2 Right. Such a pattern expected for two structurally equivalent 31P-containing groups coordinated directly to V4+, and the estimated magnitude of the coupling, 1.85 × 10−4 cm−1, is similar to values observed for other VO2+–nucleotide complexes (16, 17).
Fig. 3 compares proton ENDOR spectra of the ternary enzyme complex formed with VO(acac)2 or VO(d7-acac)2 in the presence of dUDP. Comparison of the spectrum of the ternary enzyme complex to that of free VO(acac)2 shows that the characteristic resonance features attributable to acetylacetonate methyl hydrogens (30) are recognizable but overlap significantly with resonance features of protein residues. The characteristic ENDOR features of the methyl groups are absent in the spectrum of the ternary complex formed with VO(d7-acac)2, confirming their chemical origin. In all three spectra, the A⊥ hyperfine component characteristic of a water molecule bound axially to V4+ (15) is observed. Therefore, the results in Figs. 2 and 3 specify that the equatorial coordination sites of the VO2+ moiety in the ternary complex are occupied by oxygen atoms of two structurally equivalent phosphate groups on one side and two carbonyl oxygens of the acetylacetonate ligand on the other.
Figure 3.

Comparison of first-derivative ENDOR spectra of the ternary dUTPase–(VO2+-chelate)–dUDP complex resulting from addition of dUTPase, VO(acac)2, and dUDP in 1.25:1:1 molar ratio as in spectrum c of Fig. 2. (Upper) ENDOR (black) spectrum of the dUTPase–(VO2+-chelate)–dUDP complex. The red dots represent the ENDOR spectrum of VO(acac)2 in the presence of only dUDP (added in 1:1 stoichiometry). It is seen that the resonance features of the methyl groups of VO(acac)2 free in solution (30), labeled H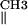 and H
and H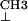 , directly overlap with the prominent features of the spectrum of the ternary enzyme complex. The weaker resonance features belonging to the bridge hydrogens of the acetylcacetonate ligand are identified by arrows. Also, the A⊥ component of a solvent hydroxyl proton, labeled HOH, that is observed for H2O coordinated in the axial position trans to the vanadyl oxygen (15) is shown. (Lower) Proton ENDOR spectrum of the ternary dUTPase–(VO2+-chelate)–dUDP complex formed with VO(d7-acac)2. The loss of the prominent methyl proton resonances seen in Upper confirms their chemical origin. The central Larmor frequency region shows only broad overlapping resonance features of protein residues.
, directly overlap with the prominent features of the spectrum of the ternary enzyme complex. The weaker resonance features belonging to the bridge hydrogens of the acetylcacetonate ligand are identified by arrows. Also, the A⊥ component of a solvent hydroxyl proton, labeled HOH, that is observed for H2O coordinated in the axial position trans to the vanadyl oxygen (15) is shown. (Lower) Proton ENDOR spectrum of the ternary dUTPase–(VO2+-chelate)–dUDP complex formed with VO(d7-acac)2. The loss of the prominent methyl proton resonances seen in Upper confirms their chemical origin. The central Larmor frequency region shows only broad overlapping resonance features of protein residues.
Modeling VO2+–Nucleotide and VO2+–Protein Interactions in dUTPase.
Fig. 4 illustrates the coordination geometry of [VO(acac)+] with dUDP bound in the active site of dUTPase that accounts for 31P shf coupling to V4+ observed by EPR and the VO2+-bound acetylacetonate ligand detected by ENDOR. Although the uracil base and the deoxyribose moiety are positioned so as to preserve their interactions with the β5–β6 hairpin turn in the protein, which is important for nucleotide recognition (3–8), the two phosphate groups remain sterically accessible for binding of [VO(acac)+]. Both the phosphate oxygens and the organic ligand are coordinated in the equatorial plane of the metal ion, perpendicular to the V⩵O bond, as required by our spectroscopic observations. The only well defined metal ion binding site in dUTPase is that of Sr2+ in the ternary dUTPase–Sr2+–dUDP complex of the EIAV enzyme (7). The hemi-chelate form of VO2+ revealed by ENDOR cannot be sterically accommodated into the Sr2+ site, and hydrolysis of dUDP catalyzed by dUTPase has not been observed. Moreover, dUDP in the presence of divalent metal ions does not elicit the ordered conformation of C-terminal residues detected by circular dichroism that is obligatory for enzyme-catalyzed substrate turnover (9–11).
Figure 4.

Stereoview of the coordination geometry of [VO(acac)+] in the active site of E. coli dUTPase with dUDP proposed on the basis of EPR and ENDOR results. The uracil and deoxyribose moieties were superpositioned onto their counterparts of dUDP in EIAV dUTPase (7). Atoms of the acetylacetonate ligand, color-coded according to element and labeled “acac,” are shown on the front side of the metal ion with the α- and β-phosphate oxygens of dUDP coordinated to the metal ion from the back side. Both supply equatorial oxygen-donor atoms to the vanadium (rendered as a mauve sphere). Subunit A residues are red, subunit C residues are blue, and residues corresponding to the structurally disordered C-terminal fragment of the B subunit in the E. coli enzyme and modeled according to the conformation of ordered C-terminal residues of human dUTPase (ref. 3; cf. Table 1) are cyan. Also shown is the 5′-triphosphate moiety of dUTP (light purple and labeled dUTP) coordinated to the V4+ ion (orange sphere) to illustrate a possible mode of metal ion binding in the catalytically competent ternary complex and its interaction with C-terminal residues. The Sr2+ ion, as coordinated to the diphosphate moiety of dUDP in the homologous EIAV enzyme (7), is yellow. The light-blue crosses indicate the corresponding locations of three hydrogen-bonded water molecules in the active site of the human enzyme (3) nearest to the α-phosphorus of the 5′-triphosphate group. It is seen that these water molecules overlap significantly with the Sr2+ ion. The Lee–Richards solvent-accessible surface (31) of active site residues is color-coded according to subunit. Active-site residues are labeled for purposes of orientation and are color-coded according to subunit origin.
Because the VO2+-chelate supports substrate hydrolysis with kinetic efficiency comparable to that of Mg2+ and VO2+ alone and because the Sr2+ site cannot sterically accommodate the hemi-chelate form of VO2+, we have inspected the active site of the enzyme by molecular graphics to identify the most likely position for binding a divalent metal ion in its catalytically active form. In our graphics analysis we reasoned that a catalytically competent metal ion binding site must account for the following experimental observations:
(i) enhancement of the rate of substrate hydrolysis on addition of Mg2+ or other catalytically facilitatory divalent metal ions;
(ii) enhancement of the rate of substrate hydrolysis in the presence of VO2+ compared with that in the presence of Mg2+;
(iii) nucleophilic attack by the hydrolytic water molecule at the α-phosphorus position of the dUTP substrate as demonstrated with 18O-enriched water (12);
(iv) compatibility of the metal ion binding site to accommodate VO2+ chelated by two phosphate groups of the nucleotide in structurally equivalent equatorial positions on one side with the carbonyl oxygens of the acetylacetonate ligand on the other side; and
(v) metal ion-assisted ordering of the C-terminal fragment (9–11) in a manner compatible with kinetic enhancement of substrate hydrolysis.
None of these experimental observations can be accounted for by the x-ray-defined Sr2+ binding site in the ternary dUTPase–Sr2+–dUDP complex (7).
As seen in Fig. 4, we find that the [VO(acac)+] species can occupy a similar position in the presence of dUTP to that shown for dUDP. In the ternary complex with dUTP, however, coordination of the metal ion to oxygen atoms of the β- and γ-phosphate groups of the substrate positions it closer to C-terminal residues of the neighboring subunit so as to be able to accept the carbonyl oxygen of Gly-147 as an axial ligand. This coordination geometry could facilitate stabilization of an ordered conformation of other C-terminal residues through the side-chain interactions identified by graphics modeling. The most important of these interactions in the ordered C-terminal conformation are those of His-148 with the terminal phosphate group of the substrate and Ser-149 with Lys-77. The latter two residues are conserved in all species. Also, Arg-151 can readily interact with the side chain of Asp-107. Modeling of the dUTP substrate by superpositioning uracil and deoxyribose moieties onto their counterparts of the dUDP molecule (7) showed that the phosphate groups of the substrate can interact favorably with residues Arg-141, -116, or -71. Thus, while the substrate is firmly anchored into the active site at one end through these interactions, binding of the metal ion to the β- and γ-phosphate groups at the other allows it to serve in a bridging capacity to mediate stabilization of the C-terminal region of the third subunit comprising the active site. Conformational ordering of the C-terminal fragment is obligatory for catalytic activity (9–11).
In identifying the divalent metal ion binding site to account for observed catalytic properties detailed above, of importance is our observation that catalysis in the presence of VO2+ is associated with a 2-fold increase in kcat over that for Mg2+ (cf. Table 2). As pointed out above, the affinity of pyrophosphate for VO2+ is ≈103-fold higher than for Mg2+ (27). The higher rate of substrate turnover and higher affinity of pyrophosphate for VO2+ are directly compatible with binding to the β- and γ-phosphate groups because product stabilization would be more favorably facilitated by VO2+ through increased covalent metal–ligand interactions. Moreover, through enhanced product stabilization, the proposed metal binding site can similarly account for enhancement of the catalytic rate constant in the presence of divalent metal ions over that observed in their absence.
The x-ray structure of human dUTPase, having dUDP in the active site, shows a cluster of water molecules hydrogen bonded to each other and to the side chains of Asp-32 and Gln-119 (3). On the basis of homology and the similar protein folds of the two enzymes, a comparable cluster of hydrogen-bonded water molecules is likely to be found also in the E. coli enzyme. As seen in Fig. 4, when the human and E. coli structures are superpositioned according to structurally homologous residues (3–7), Wat-277 is poised for inline nucleophilic attack on the α-phosphorus of the substrate. The geometry of nucleophilic attack could occur within a trigonal bipyramidal configuration, in which the bridging oxygen to the β-phosphorus becomes the leaving group. Nucleophilic attack at the α-phosphorus by a solvent molecule is required on the basis of isotope studies (12). The cluster of water molecules hydrogen bonded to each other and to the side chain of Asp-32 is consistent with a general-base catalyzed mechanism activating the nucleophilic water. The x-ray-defined Sr2+ site (7) cannot account for these catalytic properties. Moreover, binding of Sr2+, as shown in Fig. 4, would lead to displacement of the three clustered water molecules closest to the Sr2+ ion.
General Conclusions.
Based on steady-state kinetic studies, we have concluded that dUTPase can catalyze hydrolysis of dUTP in the absence of divalent metal ions. The divalent metal ions used in this study, Mg2+, Mn2+, or VO2+, only enhanced the catalytic action of the enzyme and, therefore, do not act as obligatory cofactors. The most interesting result in Table 2, showing that the organic chelate VO(acac)2 supports hydrolysis comparable to that by VO2+, was unusual and unanticipated. EPR and ENDOR results demonstrated that VO2+ was bound to the phosphate groups of dUDP together with a chelating acetylacetonate ligand. Because the VO2+-chelate supports substrate hydrolysis essentially as efficiently as VO2+, Mg2+, or Mn2+, we conclude that these metal ions all bind in the same site of the catalytically competent enzyme–substrate complex and that this site is shared by the VO2+ moiety of the hemi-chelate.
We have rationalized the enhancement of the rate of hydrolysis of dUTP catalyzed by dUTPase on addition of divalent metal ion as a general base catalyzed process in which a cluster of hydrogen-bonded water molecules in the active site provides the attacking nucleophilic water at the α-phosphorus atom. Nucleophilic attack is accompanied by binding of the divalent metal ion to the terminal β- and γ-phosphate groups promoting product stabilization. Through molecular modeling we have identified a metal ion binding site that is compatible with previously reported kinetic (12) and structural (9, 10) observations and is sterically compatible with binding of a [VO(acac)+] ligand. These requirements cannot be satisfied by the x-ray-defined position of the Sr2+ site in the EIAV enzyme (7). These results thus provide new insight into the role of divalent metal ions in the mechanism of action of dUTPase.
Acknowledgments
We thank Dr. R. Persson for the pET3a plasmid containing the E. coli dUTPase gene, and Dr. E. V. Galtseva for synthesis of VO(d7-acac)2. This work was supported by grants to M.W.M. from the National Institutes of Health (DK57599) and National Science Foundation (MCB-0092524) and grants to B.G.V. from the Hungarian National Science Foundation (OTKA T034120 and TS044730) and the Howard Hughes Medical Institute (55000342).
Abbreviations
- dUTPase
dUTP pyrophosphatase
- EIAV
equine infectious anemia virus
- ENDOR
electron nuclear double resonance
- EPR
electron paramagnetic resonance
- shf
superhyperfine
- VO(acac)2
bis(acetylacetonato)oxovanadium(IV)
References
- 1.Pearl L H, Savva R. Nat Struct Biol. 1996;3:485–487. doi: 10.1038/nsb0696-485. [DOI] [PubMed] [Google Scholar]
- 2.Ingraham H A, Dickey L, Goulian M. Biochemistry. 1986;25:3225–3230. doi: 10.1021/bi00359a022. [DOI] [PubMed] [Google Scholar]
- 3.Mol C D, Harris J M, McIntosh E M, Tainer J A. Structure (London) 1996;4:1077–1092. doi: 10.1016/s0969-2126(96)00114-1. [DOI] [PubMed] [Google Scholar]
- 4.Cedergren-Zeppezauer E S, Larsson G, Nyman P O, Dauter Z, Wilson K S. Nature. 1992;355:740–743. doi: 10.1038/355740a0. [DOI] [PubMed] [Google Scholar]
- 5.Larsson G, Svensson L A, Nyman P O. Nat Struct Biol. 1996;3:532–538. doi: 10.1038/nsb0696-532. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez A, Larsson G, Persson R, Cedergren-Zeppezauer E. Acta Crystallogr D. 2001;57:767–774. doi: 10.1107/s0907444901004255. [DOI] [PubMed] [Google Scholar]
- 7.Dauter Z, Persson R, Rosengren A M, Nyman P O, Wilson K S, Cedergren-Zeppezauer E S. J Mol Biol. 1999;285:655–673. doi: 10.1006/jmbi.1998.2332. [DOI] [PubMed] [Google Scholar]
- 8.Prasad G S, Stura E A, McRee D E, Laco G S, Hasselkus-Light C, Elder J H, Stout C D. Protein Sci. 1996;5:2429–2437. doi: 10.1002/pro.5560051205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vertessy B G. Proteins Struct Funct Genet. 1997;28:568–579. [PubMed] [Google Scholar]
- 10.Vertessy B G, Larsson G, Persson T, Bergman A C, Persson R, Nyman P O. FEBS Lett. 1998;421:83–88. doi: 10.1016/s0014-5793(97)01545-7. [DOI] [PubMed] [Google Scholar]
- 11.Vertessy B G, Persson R, Rosengren A M, Zeppezauer M, Nyman P O. Biochem Biophys Res Commun. 1996;219:294–300. doi: 10.1006/bbrc.1996.0226. [DOI] [PubMed] [Google Scholar]
- 12.Larsson G, Nyman P O, Kvassman J O. J Biol Chem. 1996;271:24010–24016. doi: 10.1074/jbc.271.39.24010. [DOI] [PubMed] [Google Scholar]
- 13.McGeoch D J. Nucleic Acids Res. 1990;18:4105–4110. doi: 10.1093/nar/18.14.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nord J, Nyman P, Larsson G, Drakenberg T. FEBS Lett. 2001;492:228–232. doi: 10.1016/s0014-5793(01)02257-8. [DOI] [PubMed] [Google Scholar]
- 15.Mustafi D, Makinen M W. Inorg Chem. 1988;27:3360–3368. [Google Scholar]
- 16.Mustafi D, Telser J, Makinen M W. J Am Chem Soc. 1992;114:6219–6226. [Google Scholar]
- 17.Jiang F, Makinen M W. Inorg Chem. 1995;34:1736–1744. [Google Scholar]
- 18.Makinen M W, Mustafi D. Metal Ions Biol Syst. 1995;31:89–127. [PubMed] [Google Scholar]
- 19.Mustafi D, Nakagawa Y, Makinen M W. Cell Mol Biol. 2000;46:1345–1360. [PubMed] [Google Scholar]
- 20.Chasteen N D. Struct Bonding (Berlin) 1983;53:105–138. [Google Scholar]
- 21.Junge H, Musso H, Zahorszky U I. Chem Ber. 1968;101:793–800. [Google Scholar]
- 22.Persson R, Nord J, Roth R, Nyman P O. Prep Biochem Biotechnol. 2002;32:157–172. doi: 10.1081/PB-120004128. [DOI] [PubMed] [Google Scholar]
- 23.Cleveland D W, Fischer S G, Kirschner M W, Laemmli U K. J Biol Chem. 1977;252:1102–1106. [PubMed] [Google Scholar]
- 24.Lundberg L G, Thoresson H O, Karlström O H, Nyman P O. EMBO J. 1983;2:967–971. doi: 10.1002/j.1460-2075.1983.tb01529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mustafi D, Nakagawa Y. Proc Natl Acad Sci USA. 1994;91:11323–11327. doi: 10.1073/pnas.91.24.11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ballhausen C J, Djurinskij B F, Watson K J. J Am Chem Soc. 1968;90:3305–3309. [Google Scholar]
- 27.Martell A E, Smith R M. Critical Stability Constants. VI. New York: Plenum; 1988. p. 447. [Google Scholar]
- 28.Chasteen N D. Biol Magn Res. 1981;3:53–119. [Google Scholar]
- 29.Kivelson D, Lee S K. J Chem Phys. 1964;41:1896–1903. [Google Scholar]
- 30.Makinen M W, Brady M J. J Biol Chem. 2002;277:12215–12220. doi: 10.1074/jbc.M110798200. [DOI] [PubMed] [Google Scholar]
- 31.Lee B, Richards F M. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]