

Abstract
The oxidative folding of the Amaranthus α-amylase inhibitor, a 32-residue cystine-knot protein with three disulfide bridges, was studied in vitro in terms of the disulfide content of the intermediate species. A nonnative vicinal disulfide bridge between cysteine residues 17 and 18 was found in three of five fully oxidized intermediates. One of these, the most abundant folding intermediate (MFI), was further analyzed by 1H NMR spectroscopy and photochemically induced dynamic nuclear polarization, which revealed that it has a compact structure comprising slowly interconverting conformations in which some of the amino acid side chains are ordered. NMR pulsed-field gradient diffusion experiments confirmed that its hydrodynamic radius is indistinguishable from that of the native protein. Molecular modeling suggested that the eight-membered ring of the vicinal disulfide bridge in MFI may be located in a loop region very similar to those found in experimentally determined 3D structures of other proteins. We suggest that the structural constraints imposed on the folding intermediates by the nonnative disulfides, including the vicinal bridge, may play a role in directing the folding process by creating a compact fold and bringing the cysteine residues into close proximity, thus facilitating reshuffling to native disulfide bridges.
The cystine knot, or the knottin fold, is a compact structure consisting of a short triple-stranded antiparallel β-sheet reinforced by three disulfide bridges that form a topological knot [see Amaranthus α-amylase inhibitor (AAI) structure, Fig. 1A Inset]. It is found both in small peptides and as a domain of larger proteins (1) and in addition, it is a recurrent substructure of larger cysteine-rich motifs such as the well known conotoxin fold (2). Despite their small size, cystine knots can fulfill a large variety of functions ranging from ion-channel blocking to enzyme inhibition, which makes them ideal protein engineering scaffolds for industrial applications (3, 4). Because cystine knots occur in many unrelated species (fungi, plants, snails, and spiders, etc.), it is presumed that this fold has emerged via convergent evolution (2). The compactness and versatility of this fold are best illustrated by the fact that both the shortest peptide inhibitor of α-amylases, AAI [32 amino acids (Fig. 1A)] (5–8) and the shortest lectin molecule known so far (9), are members of this family.
Figure 1.

Oxidative folding of AAI at pH 8.5, 25°C/100 mg of AAI per liter–1 of refolding buffer (100 mM NH4OAc/1M GdnHCl/1 mM cysteine/0.05 mM cystine/2 mM EDTA). (A) Time-resolved RP-HPLC chromatograms. (Inset) The fold and disulfide connectivity of native AAI. (B) Species abundances are calculated from the peak intensities of the detector readings at 214 nm. (Insets) The disulfide connectivity of the native state and main folding intermediate (MFI), the latter being deduced from the data in Table 1.
Cystine knot peptides are known to form readily in vitro under the conditions of oxidative folding (4). In this work, we describe an analysis of the oxidative folding intermediates of AAI, which shows that the native protein (N) forms via several fully oxidized intermediates with nonnative disulfide bonds, including the vicinal disulfide 17–18. NMR spectroscopy revealed that the main folding intermediate has a structure as compact as the completely folded peptide, comprising a number of slowly interconverting backbone conformations. On the basis of the experimental data, we suggest that the compactness, a prerequisite for efficient folding, may be the consequence of the geometric constraints imposed on the peptide backbone by the vicinal disulfide bridge.
Materials and Methods
Chemicals.
Trypsin and α-chymotrypsin, both sequencing grade, were obtained from Promega and Biochemika (Fluka), respectively. Flavin mononucleotide, obtained from Sigma, was used as the photosensitizer in photochemically induced dynamic nuclear polarization (photo-CIDNP) experiments. All other reagents were of the highest-purity grade available.
Peptide Synthesis.
AAI peptide was produced manually on a 1-mmol scale by solid-phase peptide synthesis using a fluorenyl-9-methyloxycarbonyl methodology. The crude peptide was isolated with yield >90% and purified to homogeneity (>98%, RP-HPLC). The procedure was essentially as described (6), with the exception of cysteine residues, which were introduced with a triphenylmethyl side chain protection (10).
Oxidative Folding.
Oxidative folding was carried out at pH 8.5, 25°C, initiated by placing purified reduced AAI at a concentration of 100 mg⋅liter–1 into the refolding buffer (100 mM ammonium acetate/1 M guanidinium hydrochloride/1 mM cysteine/0.05 mM cystine/2 mM EDTA). At these conditions, the refolding was complete within 20 h. The same distribution of intermediates was observed when the folding was carried out at different pH values (7.0, 7.5, 8.0, and 8.5) and in the absence of guanidinium hydrochloride; the highest pH value and 1 M guanidinium hydrochloride were selected because of the highest yield of N.
Disulfide Mapping.
The five fully oxidized intermediate species were isolated by quenching the oxidative folding reaction after 30, 35, 40, or 45 min by acidification to pH ≈2 [4% trifluoroacetic acid (TFA)] and separated by RP-HPLC. The disulfide bridge connectivities were determined via trypsin/α-chymotrypsin digestion. Each species was purified via RP-HPLC to obtain >98% purity and then incubated at 37°C in 0.1 M ammonium acetate solution at pH 5.2 for 44 h, with a trypsin/α-chymotrypsin/protein ratio of 1:1:20. When digestion was complete, the reaction was quenched by adding 4% trifluoroacetic acid, and the proteolytic fragments were analyzed with a liquid chromatography (LC)–MS system.
RP-HPLC and Electrospray Ionization–MS (ESI-MS).
Analytical and preparative RP-HPLC were carried out on a Gilson system, configured with an 805 manometric module, an 811C dynamic mixer with two model 306 pumps, a 118 UV/VIS detector, computer-controlled Gilson 506 C system module, and Unipoint system software. Analytical separation was done on a Zorbax 300 SB-5C18 (150 × 4.6 mm, Agilent) column at flow rates of 0.8–1.0 ml⋅min−1. Preparative RP-HPLC was done on a Waters Prep LC Universal Base Module with a Prep-Pac cartridge Delta Pac 300 15RC18 (100 × 40 mm, Waters) column at a flow rate of 15 ml⋅min−1. Samples were eluted with linear gradients of solvents A (H2O/0.1% TFA) and B (CH3CN/0.1% TFA). The peptide species in Fig. 1A were isolated with the analytic column and a gradient of 0–20% B (5 min) followed by 20–27% B (30 min) at a flow rate of 0.8 ml⋅min–1. ESI-MS experiments were performed on a Perkin–Elmer API 150 EX electrospray single-quadrupole mass spectrometer, operated at a resolution of 3,000. The samples were analyzed in positive mode either by direct syringe infusion or via a LC-MS system by using two Waters model 510 pumps and a micro bore rapid resolution column (Agilent Zorbax 300 SB-C18, 1.0 × 150 mm, 3.5 μm) and applying a gradient of 0–60% B (60 min) at a flow rate of 40 μl⋅min−1. The molecular weights of the peptides and the masses for theoretical digestion of the intermediates were calculated by using Applied Biosystems software (biomultiview and promac).
NMR and Photo-CIDNP Experiments.
All NMR spectra were recorded on home-built 600- and 750-MHz NMR spectrometers (Oxford Centre for Molecular Sciences) with operating frequencies of 600.20 and 750.02 MHz, respectively. The spectrometers are equipped with Oxford Instruments magnets, Omega software (Bruker, Billerica, MA), and digital control equipment (Bruker), home-built triple resonance probe heads, and home-built linear amplifiers (11). For photo-CIDNP experiments, the light of a 4-W continuous-wave argon ion laser (Spectra-Physics Stabilite 2016–05) with light pulses of 100 ms was introduced into the NMR tube via an optical fiber (diameter 1 mm), inside a coaxial Pyrex insert [Wilmad (Buena, NY) WGS 5BL]. NMR and photo-CIDNP spectra were recorded with 512 and 16 scans, respectively. All NMR data were processed by using felix 2.3 software from Biosym Technologies (San Diego). Pulsed-field gradient diffusion measurements were performed with a pulsed gradient stimulated echo longitudinal encode–decode pulse sequence (12, 13). The length of pulses and delays was held constant, and 20 spectra were acquired with the strength of the diffusion gradient varying between 5% and 100% of its maximum value; each measurement was repeated six times to improve the signal-to-noise ratio and to allow the measurement error to be estimated. The lengths of the diffusion gradient and the stimulated echo were optimized for each sample to give a total decay in the protein signal of between 80% and 90%.
Molecular Modeling.
The three nonnative forms of AAI were modeled with the Biopolymer and Discover modules of InsightII (Biosym Technologies). Ten thousand steps of the steepest descendant method and followed by 1,000 steps of the conjugate gradient technique were performed for energy minimization. The final rms deviation was <0.001 Å. Five random shakes of the starting atom coordinates resulted in identical final models. The rms distances between the Cα atoms of the cysteine residues were calculated by optimal superposition of each intermediate's modeled structure with the native AAI.
Results
Oxidative Folding.
The oxidative folding of AAI proceeds via a number of one-, two-, and three-disulfide intermediate species, revealed by time-resolved RP-HPLC chromatography (Fig. 1A). A total of 12 intermediate species were isolated and characterized by electrospray ionization (ESI)–MS, of which seven had at least one open disulfide bridge (either in the SH form or attached to an exogenous cysteine; data not shown). Interestingly, the five most abundant intermediates (indicated by arrows and denoted MFI, I-1, I-4, I-5, and I-6 in Fig. 1A) were fully oxidized three-disulfide species. Their disulfide topologies were determined by combining trypsin/α-chymotrypsin digestion and ESI-MS, and it was found that three of the five have a vicinal disulfide bond between the adjacent cysteine residues 17 and 18 (Table 1). It has to be noted that, because the sequence contains two adjacent cysteines, some of the disulfide topologies cannot be unequivocally determined; in this case there are two alternatives for species I-1 and I-4 (Table 1). One of the intermediates, termed MFI, is the most abundant species, which accumulated up to 40% after 30 min of oxidative refolding. In contrast to the cystine-knot motif of N (disulfides: 1–18, 8–23, and 17–31), MFI contains bonds between the cysteines that are closest to one another in the sequence (“bead-like” disulfides: 1–8, 17–18, and 23–31), including the vicinal disulfide bridge.
Table 1.
The disulfide structure of the five three-disulfide isomers observed during the in vitro oxidative folding of AAI
 |
Molecular weights and disulfide bonds of peptide fragments were obtained by trypsin/α-chymotrypsin digestion of the five fully oxidized species. The disulfide bonds that occur in the native species are highlighted in red, and those occurring in MFI are shown in green. The box in the final column indicates the vicinal disulfide bridge.
Fig. 1B shows that the fully reduced protein with six thiol groups disappears within minutes of initiating the oxidative folding reaction, which is then followed by the appearance of a large number of intermediate species (Fig. 1A). After about 30 min a well defined collection of intermediate species has formed, showing the predominance of MFI. N accumulates to 90% within 360 min, and the overall yield after 20 h was >95%.
NMR Spectroscopy.
The conformation of MFI was compared with that of N by using 1H NMR spectroscopy combined with photo-CIDNP (14–16). Photo-CIDNP is a powerful tool for the elucidation of residue-specific surface accessibilities of aromatic side-chains, which uses a laser-initiated, reversible chemical reaction between the protein and a flavin photosensitizer dye to generate nuclear spin polarizations that result in enhancements in the intensities of the corresponding NMR signals. In the studies of AAI, photo-CIDNP was used to assign the aromatic signals of MFI and to give further information about its conformation through the solvent accessibility of aromatic residues. AAI contains four aromatic residues (Trp-5, Tyr-21, Tyr-27, and Tyr-28), whose NMR and photo-CIDNP spectra at pH 7 are compared in Fig. 2 for N and the intermediate MFI species. The N spectra are well resolved, with chemical shifts characteristic of a single well defined tertiary structure (7, 8). The pattern of photo-CIDNP effects indicates that Trp-5, Tyr-27, and Tyr-28 are exposed, but that of Tyr-21, which is not polarized and showed no photo-CIDNP signal, exists in a more crowded environment. This result is consistent with the static accessibilities calculated from the 3D structure of AAI (8, 17).
Figure 2.

1H NMR (600 MHz) (A) and photo-CIDNP (B) spectra of the purified native state (N) and of the MFI (0.3 mM protein/0.2 mM flavin mononucleotide, pH* 7, in 2H2O at 25°C). NMR and photo-CIDNP spectra were recorded with 512 and 16 scans, respectively. The assignments of the N-state tyrosine resonances are taken from Lu et al. (8). The tyrosine resonances of the MFI species, labeled YA, YB, and YC, have not been assigned to specific residues. Attempts to measure the spectra of the fully reduced protein at pH 7 were thwarted by the rapid formation of disulfide species (Fig. 1A). (pH* is the measured pH of a 2H2O solution, uncorrected for the deuterium isotope effect.)
The NMR spectrum of the purified MFI species (Fig. 2A) resembles that of the N state: the chemical shifts are well dispersed, and some of the lines are sharp, but the MFI spectrum is clearly more complex. In addition to the major resonances, many of which have obvious counterparts in the spectrum of the N state, there are several weaker and broader signals close to the principal tyrosine and tryptophan lines (denoted by asterisks in the resolution-enhanced Fig. 3 Inset), arising from minor conformations. This is more evident in the 2D 750-MHz 1H COSY spectrum (Fig. 3), from which a partial assignment of the tyrosine signals of MFI can be obtained, as indicated in Figs. 2 and 3. The signal from the H (2, 6) protons of one of the tyrosine residues (YA) has moved ≈0.3 ppm from its position in the spectrum of the native state (7.0–7.2 ppm), to overlap with the H (3, 5) resonances at ≈6.8 ppm. This shift, which gradually increased when the temperature was lowered to 4°C, may arise from the stacking of aromatic side-chains permitted by the changed backbone conformation(s) in the MFI state. Close inspection of this portion of the spectrum (6.7–6.9 ppm) revealed at least two weak doublets (denoted by asterisks in Fig. 3 Inset) in addition to the three H (3, 5) signals and the shifted H (2, 6) peak. The corresponding CIDNP spectrum of MFI (Fig. 2B) has enhanced signals from Trp-5 and all three tyrosines. The changes to the backbone fold(s) imposed by the nonnative disulfides apparently expose the side chain of Tyr-21, itself in close proximity to the vicinal disulfide. Additionally, some of the subsidiary lines are also polarized, suggesting that the minor conformation(s) have similar exposures of the aromatic residues.
Figure 3.

2D 1H 750 MHz COSY of the purified MFI species. This section of the aromatic region shows mainly (H2, H6) and (H3, H5) protons of the three tyrosine residues, YA,YB, YC, and some protons from Trp-5. The spectral width was 6,666 Hz, with 1,024 complex points in both direct and indirect dimensions. (Inset) 1D 1H 750 MHz NMR spectrum of the purified MFI species in which some weaker signals (suggesting the presence of minor conformations in addition to the major conformation) are indicated by asterisks.
Diffusion Measurements.
The molecular dimensions of N and MFI species were compared by performing NMR pulsed-field gradient diffusion measurements (12, 13, 18, 19). The approach focused on the estimation of the effective hydrodynamic radii of both species with a pulsed-gradient-stimulated echo longitudinal encode–decode pulse sequence (12, 18) in which the protein NMR signal decay is measured as a function of gradient field strength. The signal decay rate is proportional to the diffusion coefficient D, which is itself inversely proportional to the effective hydrodynamic radius, RH. By adding a small molecule, in our case dioxane, as an internal radius standard (12, 13), absolute values for protein radii can be estimated as indicated in Eq. 1.
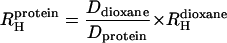 |
1 |
Although such absolute sizes must be interpreted with care (13), measurements of the relative sizes of similar species measured under similar conditions are much more robust. The ratio of effective hydrodynamic radii for N and MFI species was 0.91 ± 0.08, and so the radii of the two species are indistinguishable within the experimental error.
Database Searches and Modeling.
The vicinal disulfide bridge is an eight-membered ring, composed of the main and side chain atoms of the two adjacent cysteine residues. The first conformational analysis of the oxidized Cys–Cys dipeptide suggested that the ring might lead to a turn when found in the middle of a peptide chain (20–25). The analysis of model peptides confirmed that the formation of the vicinal disulfide bond is accompanied by the appearance of a tight β-type turn, and this phenomenon is also used in rational peptide design (26–29).
We have searched the Protein Data Bank for experimentally determined structures, and it was observed that the vicinal disulfide bridge is a relatively rare structural element. It occurs in quite unrelated proteins such as the J-Atracotoxin from Hadronyche versuta (26), methanol dehydrogenase from Methylobacterium extorquens (27–29), the ethanol dehydrogenase from Pseudomonas aeruginosa (30), acetylcholine-binding protein from Lymnaea stagnalis (31), thioesterase I from Bos taurus (32), carboxypeptidase T from Thermoacinomyces vulgaris (33), and antimicrobial peptide hepcidin-25 from Homo sapiens (34) (Fig. 4 A–G). Further, the topology of the turn is much less conserved than in the case of model peptides. The vicinal disulfide bridge is also found in small irregular loops between different secondary structural elements, not only in tight β turns (Fig. 4 A–G). On the other hand, a comparison of these structures (Fig. 4 A–G) reveals that the eight-membered ring of the vicinal disulfide bond is in fact invariably situated within a turn of the main chain, in such a way that the main chain bends away from the ring.
Figure 4.

Schematic view of the vicinal disulfide bonds, which have been structurally characterized. (A) 1dlo: J-Atracotoxin (H. versuta), C13-C14 (26); (B) 1eh5: palmitoyl protein thioesterase I (B. taurus), C45-C46 (32); (C) 1flg: ethanol dehydrogenase (P. aeruginosa), C105-C106 (30); (D) 1i9b acetylcholine-binding protein (L. stagnalis), C187-C188 (31); (E) 1obr: carboxypeptidase T (T. vulgaris), C155-C156 (34); (F) 4aah: methanol dehydrogenase (M. extorquens), C103-C104 (28); (G) 1m4f: hepcidin-25 (H. sapiens), C13-C14 (33); (H) Computer model of MFI, C17-C18 (see text).
A closer examination of the native AAI structure reveals that the six cysteine residues are near enough to each other that, in principle, all of the 15 theoretically possible disulfide topologies can form via minor displacements of the main chain. We built 3D models for three intermediates, MFI, I-1, and I-4, and have found that the rms deviation distances calculated for cysteine Cα atoms with respect to the native structure are 1.72, 1.67, and 1.31 Å, respectively (see Fig. 5, which is published as supporting information on the PNAS web site, www.pnas.org). All three models are globular compact structures resembling the native state. It should be emphasized that modeling of this sort shows that stereochemically allowed structures with nonnative disulfide bridges can be generated from the native tertiary scaffold of AAI protein, but not that these structures are necessarily observed. Energy minimization (and molecular dynamics) is known to be unreliable at predicting loop structures. The region of the vicinal disulfide bridge in the model of MFI (Fig. 4H) features a turn reminiscent of those found in the protein structures (Fig. 4 A–G).
Characterization of the Structure of MFI.
Attempts were made to determine the atomic resolution structure of MFI by using 2D 1H NMR spectroscopy via 1H-1H nuclear Overhauser effects (NOE), which reveal pairs of protons separated by <≈5 Å. Six hundred megahertz of 2D NOE NMR spectroscopy spectra (not shown) of the native and MFI states show similar chemical shift dispersion, as noted for the 1D spectra in Fig. 2, and many NOE cross peaks. Compared with the native state, however, the MFI spectrum was much less well resolved, showing a large number of overlapping resonances, indicating the presence of slowly interconverting conformations with different side chain environments. Rapid interconversion would tend to average chemical shift differences and lead to weak NOEs. Given the poor spectral resolution resulting from multiple conformations, a structure determination was not feasible.
Additionally, CD spectra (not shown) of the native, fully reduced and MFI states were compared. In the far-UV (190–260 nm), the CD spectra of the reduced and intermediate forms of the protein were almost identical and distinct from that of the native state. By contrast, in the near-UV (260–320 nm), the reduced state showed very little CD intensity, whereas the native and intermediate forms had quite similar spectra. These results indicate that MFI possesses a disordered backbone but that at least some of the aromatic side chains are ordered.
Discussion
The in vitro oxidative folding pathways of proteins are quite varied (35–37), and although fully oxidized intermediates with nonnative disulfides are known to occur (35, 38–40), the intermediates studied here (MFI, I-5, and I-6) appear to be the first such folding intermediates possessing a vicinal disulfide bond. The predominance of the vicinal disulfide bridge is conspicuous because of the 15 theoretically possible fully oxidized intermediates of AAI, the three that contain a vicinal bridge were all detected experimentally (Table 1). MFI accumulates in large quantities early in the folding process, so it can be envisaged as an important element of the folding equilibrium.
Although it proved impossible to determine the detailed structure of MFI, a consistent picture nevertheless emerges from the experiments described above. The structure is as compact as the native state, as indicated by the NMR diffusion measurements. Although there is little evidence from far-UV CD for secondary structure, at least some of the side chains are ordered, as evidenced by the near-UV CD, the number of NOEs, the chemical shift dispersion, and the large change in the chemical shift of the H (3, 5) resonances of one of the tyrosine residues from its position in the native state. The additional lines visible in the NMR spectra, responsible for the poor resolution of the 2D NOE NMR spectroscopy cross peaks, point strongly toward slowly interconverting multiple conformations of the polypeptide backbone. Modeling suggests that the nonnative disulphides in MFI do not constrain the backbone to the extent that native-like conformations of the backbone and native-like side chain interactions cannot exist. Nevertheless, the side chain exposures revealed by CIDNP and the differences in chemical shifts show that there are clear differences between the tertiary structures of the native and MFI states. It is clear that the MFI species differs substantially from the fully reduced form, which has no tertiary structure, limited chemical shift dispersion and few NOEs.
It is known that the rate of formation of disulfides in an unstructured chain decreases with the number of residues between the two cysteines (41). On the basis of this observation, the vicinal disulfides should exhibit the highest rate of formation, and bead-like structures, in which disulphide bridges are formed from the cysteines nearest to each other within the sequence, should have a high rate of formation. Of the three intermediates with a vicinal bridge, which would be favored per se, the most abundant, MFI, is indeed the one with the bead-like structure. It is also well known that single domain proteins featuring primarily sequence-local contacts (high “contact order”) tend to fold more rapidly and with lower energy barriers than those characterized by more nonlocal interactions (42).
One could envisage that a vicinal S—S bond (and the associated eight-membered ring) would have a similar structural effect to a proline residue (a five-membered ring) in constraining the movement of the main chain in such a way that a turn is formed, increasing the rigidity of the structure relative to the fully reduced state. In the context of the MFI structure, the bend imposed on the backbone may help to bring various parts of the molecule into close proximity, which could assist the formation of the final, fully oxidized protein. The vicinal disulfide bridge of MFI (Fig. 4H), modeled on the native AAI structure, has the eight-membered ring in a position analogous to those found in the experimentally determined structures (Fig. 4 A–G). One can conclude that the formation of the vicinal disulfide bridge is thus favored both by thermodynamic and kinetic factors.
We speculate that the steric constraints caused by the ring structure may contribute, e.g., via a decrease in entropy, to the formation of a compact structure where both nonnative and native contacts may be important (43). The high contact order conferred on the MFI state of AAI by its bead-like disulfide pattern may well be responsible for its rapid formation in the early stages of oxidative folding. However, it is difficult to say whether this fact or the structural constraints imposed on the folding intermediates by the nonnative disulfides can be considered more important in directing the folding process by creating a compact fold. The compact intermediate, possessing some native-like side chain contacts, could then slowly rearrange via disulfide reshuffling to the equally compact, organized, but more stable native state. Nonnative interactions in protein folding have been suggested to play a potentially significant role even for small proteins with simple topologies (44).
Supplementary Material
Acknowledgments
We are indebted to C. Redfield, J. Boyd, C. Guarnaccia, Zs. Májer, and A. Perczel for expert advice on the NMR, MS, and CD experiments. J.A.J. is a Royal Society University Research Fellow. M.Č. is the recipient of an International Center for Genetic Engineering and Biotechnology predoctoral fellowship. This work was supported in part by the Engineering and Physical Sciences Research Council, the Biotechnology and Biological Sciences Research Council, and the Medical Research Council through the Oxford Centre for Molecular Sciences.
Abbreviations
- AAI
Amaranthus α-amylase inhibitor
- N
native protein
- MFI
main folding intermediate
- NOE
nuclear Overhauser effect
- photo-CIDNP
photochemically induced dynamic nuclear polarization
References
- 1.Narasimhan L, Singh J, Humblet C, Guruprasad K, Blundell T. Nat Struct Biol. 1994;1:850–852. doi: 10.1038/nsb1294-850. [DOI] [PubMed] [Google Scholar]
- 2.Carugo O, Lu S, Luo J, Gu X, Liang S, Strobl S, Pongor S. Protein Eng. 2001;14:639–646. doi: 10.1093/protein/14.9.639. [DOI] [PubMed] [Google Scholar]
- 3.Smith G P, Patel S U, Windass J D, Thornton J M, Winter G, Griffiths A D. J Mol Biol. 1998;277:317–332. doi: 10.1006/jmbi.1997.1621. [DOI] [PubMed] [Google Scholar]
- 4.Craik D J, Daly N L, Waine C. Toxicon. 2001;39:43–60. doi: 10.1016/s0041-0101(00)00160-4. [DOI] [PubMed] [Google Scholar]
- 5.Chagolla-Lopez A, Blanco-Labra A, Patthy A, Sanchez R, Pongor S. J Biol Chem. 1994;269:23675–23680. [PubMed] [Google Scholar]
- 6.Lozanov V, Guarnaccia C, Patthy A, Foti S, Pongor S. J Pept Res. 1997;50:65–72. doi: 10.1111/j.1399-3011.1997.tb00621.x. [DOI] [PubMed] [Google Scholar]
- 7.Pereira P J, Lozanov V, Patthy A, Huber R, Bode W, Pongor S, Strobl S. Structure Fold Des. 1999;7:1079–1088. doi: 10.1016/s0969-2126(99)80175-0. [DOI] [PubMed] [Google Scholar]
- 8.Lu S, Deng P, Liu X, Luo J, Han R, Gu X, Liang S, Wang X, Li F, Lozanov V, et al. J Biol Chem. 1999;274:20473–20478. doi: 10.1074/jbc.274.29.20473. [DOI] [PubMed] [Google Scholar]
- 9.Lu S, Liang S, Gu X. J Protein Chem. 1999;18:609–617. doi: 10.1023/a:1020663619657. [DOI] [PubMed] [Google Scholar]
- 10.Han Y, Albercio F, Barany G. J Org Chem. 1997;62:4307–4312. doi: 10.1021/jo9622744. [DOI] [PubMed] [Google Scholar]
- 11.Soffe N, Boyd J, Leonard M. J Magn Reson A. 1995;116:117–121. [Google Scholar]
- 12.Jones J A, Wilkins D K, Smith L J, Dobson C M. J Biomol NMR. 1997;10:199–203. [Google Scholar]
- 13.Wilkins D K, Grimshaw S B, Receveur V, Dobson C M, Jones J A, Smith L J. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 14.Kaptein R, Dijkstra K, Nicolay K. Nature. 1978;274:293–294. doi: 10.1038/274293a0. [DOI] [PubMed] [Google Scholar]
- 15.Hore P J, Winder S L, Roberts C H, Dobson C M. J Am Chem Soc. 1997;119:5049–5050. [Google Scholar]
- 16.Maeda K, Lyon C E, Lopez J J, Čemažar M, Dobson C M, Hore P J. J Biomol NMR. 2000;16:235–244. doi: 10.1023/a:1008351128089. [DOI] [PubMed] [Google Scholar]
- 17.Hubbard S J, Thornton J M. naccess (University College, London) 1996. [Google Scholar]
- 18.Gibbs S J, Johnson C S. J Magn Reson. 1991;93:395–402. [Google Scholar]
- 19.Stejskal E O, Tanner J E. J Chem Phys. 1965;42:288–292. [Google Scholar]
- 20.Capasso S, Mattia C, Mazzarella L, Puliti R. Acta Crystallogr B. 1977;33:2080–2083. [Google Scholar]
- 21.Hata Y, Matsuura Y, Tanaka N, Ashida T, Kakudo M. Acta Crystallogr B. 1977;33:3561–3564. [Google Scholar]
- 22.Capasso S, Garzillo A M, Marino G, Mazzarella L, Pucci P, Sannia G. FEBS Lett. 1979;101:351–354. doi: 10.1016/0014-5793(79)81042-x. [DOI] [PubMed] [Google Scholar]
- 23.Capasso S, Mazzarella L, Sica F, Zagari A. Int J Pept Protein Res. 1984;24:588–596. doi: 10.1111/j.1399-3011.1984.tb03164.x. [DOI] [PubMed] [Google Scholar]
- 24.Sukumaran D K, Prorok M, Lawrence D S. J Am Chem Soc. 1991;113:706–707. [Google Scholar]
- 25.Avizonis D Z, Farr-Jones S, Kosen P A, Basus V J. J Am Chem Soc. 1996;118:13031–13039. [Google Scholar]
- 26.Wang X, Connor M, Smith R, Maciejewski M W, Howden M E, Nicholson G M, Christie M J, King G F. Nat Struct Biol. 2000;7:505–513. doi: 10.1038/75921. [DOI] [PubMed] [Google Scholar]
- 27.Blake C C, Ghosh M, Harlos K, Avezoux A, Anthony C. Nat Struct Biol. 1994;1:102–105. doi: 10.1038/nsb0294-102. [DOI] [PubMed] [Google Scholar]
- 28.Xia Z, Dai W, Zhang Y, White S A, Boyd G D, Mathews F S. J Mol Biol. 1996;259:480–501. doi: 10.1006/jmbi.1996.0334. [DOI] [PubMed] [Google Scholar]
- 29.Xia Z X, He Y N, Dai W W, White S A, Boyd G D, Mathews F S. Biochemistry. 1999;38:1214–1220. doi: 10.1021/bi9822574. [DOI] [PubMed] [Google Scholar]
- 30.Keitel T, Diehl A, Knaute T, Stezowski J J, Hohne W, Gorisch H. J Mol Biol. 2000;297:961–974. doi: 10.1006/jmbi.2000.3603. [DOI] [PubMed] [Google Scholar]
- 31.Brejc K, van Dijk W J, Klaassen R V, Schuurmans M, van Der Oost J, Smit A B, Sixma T K. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 32.Bellizzi J J, III, Widom J, Kemp C, Lu J Y, Das A K, Hofmann S L, Clardy J. Proc Natl Acad Sci USA. 2000;97:4573–4578. doi: 10.1073/pnas.080508097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teplyakov A, Polyakov K, Obmolova G, Strokopytov B, Kuranova I, Osterman A, Grishin N, Smulevitch S, Zagnitko O, Galperina O. Eur J Biochem. 1992;208:281–288. doi: 10.1111/j.1432-1033.1992.tb17184.x. [DOI] [PubMed] [Google Scholar]
- 34.Hunter H N, Fulton D B, Ganz T, Vogel H J. J Biol Chem. 2002;277:37597–37603. doi: 10.1074/jbc.M205305200. [DOI] [PubMed] [Google Scholar]
- 35.Chang J Y, Li L, Bulychev A. J Biol Chem. 2000;275:8287–8289. doi: 10.1074/jbc.275.12.8287. [DOI] [PubMed] [Google Scholar]
- 36.Narayan M, Welker E, Wedemeyer W J, Scheraga H A. Acc Chem Res. 2000;33:805–812. doi: 10.1021/ar000063m. [DOI] [PubMed] [Google Scholar]
- 37.Wedemeyer W J, Welker E, Narayan M, Scheraga H A. Biochemistry. 2000;39:4207–4216. doi: 10.1021/bi992922o. [DOI] [PubMed] [Google Scholar]
- 38.Creighton T. J Mol Biol. 1974;87:579–602. doi: 10.1016/0022-2836(74)90105-3. [DOI] [PubMed] [Google Scholar]
- 39.Hua Q X, Gozani S N, Chance R E, Hoffmann J A, Frank B H, Weiss M A. Nat Struct Biol. 1995;2:129–138. doi: 10.1038/nsb0295-129. [DOI] [PubMed] [Google Scholar]
- 40.Chang J Y, Li L, Lai P H. J Biol Chem. 2001;276:4845–4852. doi: 10.1074/jbc.M005160200. [DOI] [PubMed] [Google Scholar]
- 41.Wedemeyer W J, Xu X, Welker E, Scheraga H A. Biochemistry. 2002;41:1483–1491. doi: 10.1021/bi011893q. [DOI] [PubMed] [Google Scholar]
- 42.Plaxco K W, Simons K T, Baker D. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 43.Dobson C M, Sali A, Karplus M. Angew Chem Int Ed Engl. 1998;37:868–893. doi: 10.1002/(SICI)1521-3773(19980420)37:7<868::AID-ANIE868>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 44.Capaldi A P, Kleanthous C, Radford S E. Nat Struct Biol. 2002;9:209–216. doi: 10.1038/nsb757. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}