

Abstract
The structures of Saccharomyces cerevisiae, Dictyostelium, and Caenorhabditis elegans actin bound to gelsolin segment-1 have been solved and refined at resolutions between 1.9 and 1.75 Å. These structures reveal several features relevant to the ATP hydrolytic mechanism, including identification of the nucleophilic water and the roles of Gln-137 and His-161 in positioning and activating the catalytic water, respectively. The involvement of these residues in the catalytic mechanism is consistent with yeast genetics studies. This work highlights both structural and mechanistic similarities with the small and trimeric G proteins and restricts the types of mechanisms responsible for the considerable enhancement of ATP hydrolysis associated with actin polymerization. The conservation of functionalities involved in nucleotide binding and catalysis also provide insights into the mechanistic features of members of the family of actin-related proteins.
Actin is a ubiquitous 43-kDa adenine nucleotide-binding protein involved in a wide range of fundamental processes in eukaryotic cells, including cell motility (1), cytokinesis (2), vesicle transport (3, 4), and the establishment and maintenance of cell morphology (5, 6). Actin is also an essential component of numerous specialized structures in higher eukaryotes, such as the intestinal brush border (7, 8) and stereocilia (9, 10). The ability of actin to participate in these diverse processes is a consequence of the dynamic and polymorphic nature of the filamentous actin (F-actin) assemblies that are formed in vivo.
Central to understanding cytoskeletal regulation is the contribution of the adenine nucleotide to actin structure, function, and dynamics. Bound nucleotide is required to stabilize the actin monomer under physiological conditions, because nucleotide-free actin denatures at a rate of 0.2 s−1 (11). ATP hydrolysis occurs subsequent to monomer addition to the growing filament, thus “marking” the oldest part of the filament (i.e., ADP-actin), and may serve as a timing mechanism for filament disassembly, analogous to the role of GTP hydrolysis in G proteins (12, 13). Furthermore, several regulatory proteins (e.g., profilin, cofilin, and thymosin β-4) show specificity for either ATP- or ADP-actin, which is thought to be an important element of cytoskeletal regulation (12). Actin dynamics also depends on the identity of the bound metal–nucleotide complex (14–21).
The structure of the actin monomer has been known for over a decade, and the structures of mammalian Ca2+-ATP actins in complex with DNase I (22), profilin (23), gelsolin segment-1 (24), and gelsolin segments-4–6 (25) are available. An orthorhombic crystal form of the Ca2+-ATP Dictyostelium actin/gelsolin segment-1 structure has also been reported (26). These structures highlighted specific regulatory interactions and revealed the remarkable architectural similarity between actin and a wide range of proteins (27), including the heat shock cognate ATPases (e.g., HSC70) and sugar kinases (e.g., hexokinase) (28–30). This structural homology likely reflects functional and mechanistic similarities, because these proteins couple changes in nucleotide state to conformation rearrangements required for activity. Because these structures did not contain the physiologically relevant Mg2+-ATP metal nucleotide, a number of essential issues related to actin's ATPase activity remain to be addressed. We report the monoclinic crystal structures of Saccharomyces cerevisiae, Dictyostelium, and Caenorhabditis elegans actin in complex with gelsolin segment-1 at resolutions that range from 1.9 to 1.75 Å. The metal nucleotide complexes represented in these structures include Ca2+-, Li+-, and the physiologically relevant Mg2+-ATP species. The relatively high resolution of these structures highlights mechanistic similarities to the small and trimeric G proteins, allows for the identification of the nucleophilic water and catalytic base, and suggests a structural explanation for the reduced ability of calcium to support nucleotide hydrolysis. These structures, in conjunction with the Holmes model of the actin filament (31), also limit the types of mechanisms that may be responsible for the greater than three orders of magnitude enhancement in hydrolytic activity associated with the monomer to filament transition (32). Furthermore, the present work is directly relevant to a mechanistic understanding of members of the family of actin-related proteins that share considerable sequence and structural similarity with actin (33, 34).
Methods
Preparation and Crystallization of Nonvertebrate Actin–Gelsolin Complexes.
Standard methods were used for the preparation of S. cerevisiae, Dictyostelium discoiteum (35), and C. elegans (36) actin. Human gelsolin segment-1 was expressed in Escherichia coli BL21(DE3) and purified from inclusion bodies as described (37) with minor modifications.
The yeast Mg2+-ATP actin-gelsolin segment-1 complex was prepared by mixing yeast actin with gelsolin (1:1.25 stoichiometry) in 2 mM Tris⋅HCl/2 mM ATP/4 mM MgCl2/0.5 mM EGTA/1 mM NaN3, pH 8.0. Crystals were grown by hanging drop vapor diffusion using precipitant composed of 1.4–1.7 M ammonium sulfate in 100 mM Hepes (pH 7.5), containing 2 mM Mg-ATP. Diffraction from these crystals is consistent with the monoclinic space group C2 (a = 176.66 Å, b = 68.09 Å, b = 54.59 Å, β = 102.55°), with a single actin-segment-1 complex per asymmetric unit. The Dictyostelium Mg2+-ATP actin complex was crystallized from 1.5 M Li2SO4/2 mM Mg2+-ATP/0.5 mM EGTA/0.1 M Hepes, pH 7.5. Replacement of Mg2+ with Ca2+ was accomplished by soaking the Mg2+-ATP crystals in 1.6 M Li2SO4/10 mM CaCl2/2 mM Na+-ATP/0.1 M Hepes, pH 7.5 for 4 months. Crystals of the Li+-ATP Dictyostelium actin complex were grown from 1.7 M Li2SO4/2 mM Mg2+-ATP/0.5 mM EGTA/0.1 M Tris, pH 7.9. Crystals of the C. elegans Mg2+-ATP actin complex were grown from 1.55 M Li2SO4/2 mM Mg2+-ATP/0.5 mM EGTA/0.1 M Hepes, pH 7.5.
Data Collection and Structure Solution.
Data were collected at −180°C on beam line X9B at the National Synchrotron Light Source, Brookhaven National Laboratory, at a wavelength of 0.98 Å by using a MAR345 image plate detector. Before flash freezing, the crystals were serially transferred to mother liquor containing between 10% and 20% (vol/vol) glycerol. Data were processed with the program denzo and scaled and merged with scalepack (38).
The structure of the yeast Mg2+-ATP actin complex was solved by molecular replacement with amore (39) using the rabbit skeletal muscle actin/gelsolin segment-1 complex as the search model (coordinates kindly provided by P. McLaughlin, University of Edinburgh, Edinburgh). Refinement with program x-plor (40) and manual rebuilding, using 2Fo − Fc and Fo − Fc, difference Fourier syntheses, resulted in a final R factor of 19.3% (Rfree is 23.1%) at 1.9-Å resolution (Table 1, which is published as supporting information on the PNAS web site, www.pnas.org). Solvent molecules were built into 3.5-σ peaks in difference Fourier syntheses, and Mg2+, Ca2+, and SO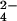 ions were subsequently assigned on the basis of observed electron density, coordination number and geometry, and refined B factors. The final model consists of 3,882 protein atoms (2,890 from actin, 992 from gelsolin segment-1), a single ATP, one Mg2+, one Ca2+, one SO
ions were subsequently assigned on the basis of observed electron density, coordination number and geometry, and refined B factors. The final model consists of 3,882 protein atoms (2,890 from actin, 992 from gelsolin segment-1), a single ATP, one Mg2+, one Ca2+, one SO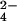 , and 427 water molecules (Fig. 1). The Ramachandran plot, computed with the program procheck (41), showed that 93.7% of the residues reside in the most favorable region of conformational space, whereas 6.3% lie in additional allowed regions. The first four residues of yeast actin were not observed in electron density maps and were not included in the final model. Residues 40–41 in the DNase I loop were modeled as alanine because of weak electron density. The structures of the Dictyostelium and C. elegans complexes were solved by difference Fourier methods and refined as described above, with Rfree values that ranged from 23.2% to 25.8% (Table 1).
, and 427 water molecules (Fig. 1). The Ramachandran plot, computed with the program procheck (41), showed that 93.7% of the residues reside in the most favorable region of conformational space, whereas 6.3% lie in additional allowed regions. The first four residues of yeast actin were not observed in electron density maps and were not included in the final model. Residues 40–41 in the DNase I loop were modeled as alanine because of weak electron density. The structures of the Dictyostelium and C. elegans complexes were solved by difference Fourier methods and refined as described above, with Rfree values that ranged from 23.2% to 25.8% (Table 1).
Figure 1.

Overall structure of the yeast actin/gelsolin segment-1 complex. Gelsolin (purple) binds to subdomains 1 and 3 of actin (yellow). Sixteen water molecules (aqua) contribute to the actin-gelsolin binding interface. Eight water molecules (red) form a network across the actin nucleotide binding cleft, contacting residues in subdomains 1, 3, and 4 (red backbone). The gelsolin-associated Ca2+ ion is represented as pink. The adenine nucleotide is also shown, with the Mg2+ ion in black.
Genetics.
To attempt to make the H161A actin, the appropriately mutated actin sequence was placed on the centromeric plasmid pRS314 (42) marked with the TRP1 gene. This plasmid was used to transform a trp1, ura3-52 S. cerevisiae haploid strain in which the chromosomal ACT1 gene had been disrupted by replacement of the coding sequence with the LEU2 gene; wild-type actin was expressed in these recipient cells from another centromeric plasmid containing the URA3 gene. After transformation with the mutant plasmid and selection on tryptophan-deficient medium, surviving cells were obtained in quantities comparable to that achieved in a wild-type control. We attempted to eliminate the wild-type actin plasmid by growing the cells repeatedly in trp−/ura+ medium, and after replica plating colonies from 60 surviving cells on trp−/ura− medium, only three ura− colonies were identified. Plasmid rescue and DNA sequencing for each of these colonies revealed the plasmid contained a wild-type actin sequence. For a mutant actin that has little or no impact on cell viability, this procedure gives us 50–55 ura−/trp− colonies, all of which will contain the mutant sequence. Similar results were obtained when the cells were selected on 5-fluoroacetic acid plates to force the elimination of the URA3 plasmid.
To attempt to generate the Q137 E mutant, we used the procedure of Cook et al. (43) to construct a URA3-marked centromeric plasmid, pCENQ137E. The plasmid was introduced into a homozygous ura3–52, leu2 diploid strain of yeast in which one of the two chromosomal actin genes had been disrupted by the LEU2 gene. Transformed diploids able to grow on ura−/leu− medium were readily obtained. After sporulation of these cells, tetrad dissection was performed in an attempt to identify leu−/ura+ colonies. Repeated dissections produced no such surviving haploid cells.
Results and Discussion
Nonvertebrate Actin Structures.
Like the mammalian actins, nonvertebrate actins are composed of two major domains that form a deep cleft representing the metal–nucleotide binding site (Fig. 1). These two domains can be further divided into smaller subdomains; the first consists of subdomain 1 (residues 1–32, 70–144, and 338–375) and subdomain 2 (residues 33–69), and the second domain is composed of subdomain 3 (residues 145–180 and 270–337) and subdomain 4 (residues 181–269). Consistent with the high level of sequence conservation (>85%), the overall structural similarity of the vertebrate and nonvertebrate actins is highlighted by the small deviations between these independent structures. The most variable region of the actin molecule appears to be the DNase I loop (residues 35–53 in subdomain 2), as this segment is disordered in several of the structures and is present in a different conformation in each of the structures where there is observable electron density (for example; ref. 44). As demonstrated by the recent ADP-actin structure (45), the plasticity of the DNase I loop likely reflects the potential of this segment to alter conformation in response to nucleotide state and polymerization. The five nonvertebrate actin structures described here exhibit rms deviations that range from 0.47 to 0.77 Å (excluding the DNase I loop), and these deviations extend to 1.3 Å when mammalian α- and β-actins are considered.
In the nonvertebrate actin/gelsolin segment-1 crystal structures, electron density maps (calculated with either 2Fo − Fc or Fo − Fc coefficients) clearly identified a sulfate ion located on a crystallographic 2-fold axis. This sulfate makes hydrogen bonds with the backbone amide nitrogens of residues 62, 203, and 204 from subdomains 2 and 4 of two symmetry related actin molecules and likely contributes to the formation and maintenance of the crystal lattice. Two water molecules complete the coordination sphere, resulting in a total of eight hydrogen bonds with a geometry frequently observed for anion-binding sites in proteins (46). A number of additional bound water molecules are also observed in the present structures. Of particular note is a network of eight water molecules that runs across the nucleotide-binding cleft. This network is stabilized by interactions with Gly-15, Asp-157, Arg-210, Glu-214, Lys-336, and Tyr-337, connecting subdomains 1, 3, and 4, and may thus contribute to the overall dynamic and function of the actin monomer (Fig. 2). This network is partially conserved in all of the nonvertebrate actin structures, and appears to be fully conserved in the mammalian ADP-actin structure.
Figure 2.

Details of the water-mediated hydrogen bonding network across the nucleotide-binding cleft. Eight solvent molecules (black) run across the nucleotide binding cleft and may play a role in transducing changes in the nucleotide state into structural and dynamic changes relevant to polymerization and interactions with regulatory proteins.
Nucleotide–Metal Binding Site.
ATP binds at the bottom of the cleft formed by the two major lobes of the actin molecule and contacts all four subdomains, thus providing a mechanism for nucleotide dependent conformational changes. The ATP moiety in the yeast actin structure displays 2′-endo sugar pucker, the glycosidic torsion angle (χglyc = O4′-C1′-N9-C4) adopts the anticonformation (χglyc = −113°), and the polyphosphate tail assumes a relatively extended conformation. As a member of the nucleoside triphosphate hydrolase superfamily, actin is characterized by the so-called P loops, P1, P2, and P3, that are responsible for recognizing the polyphosphate tail, the divalent (monovalent) cation, and the purine base, respectively (47) (Fig. 4, which is published as supporting information on the PNAS web site). In the nonvertebrate actins, these loops correspond to residues 11–22, 150–159, and 295–306, contributed from subdomains 1, 3, and 4, respectively (Table 2, which is published as supporting information on the PNAS web site).
Based on the coordination number and geometry, the ATP-bound cation in the present yeast actin structure can be confidently identified as a Mg2+ ion. The ion exhibits nearly ideal octahedral coordination (48), making a bidentate interaction with nonbridging β- and γ-phosphoryl oxygens of ATP, with the remainder of the coordination shell being completed by four water molecules (Fig. 3, and Figs. 4 and 5, which are published as supporting information on the PNAS web site). The four coordinating water molecules make hydrogen bonds with carboxylate side chain functionalities of Asp-11 (P1 loop) and Asp-154 (P2 loop), and with the side chain of Gln-137. The Mg2+ ion does not make direct contacts with actin, with the nearest protein atom being the side chain oxygen of Gln-137 (4.16 Å).
Figure 3.

Proposed mechanism for ATP hydrolysis. (A–C) The final model of the Dictyostelium Ca2+-ATP, Mg2+-ATP, and Li+-ATP actin complexes, respectively. The polyphosphate tail of ATP, the metal, coordinating water molecules, putative water nucleophile (WAT709), His-161, and Gln-137 are highlighted. The final refined electron density calculated with 2Fo − Fc coefficients is displayed at 1.5 σ (blue). The placement of the lithium ion was determined by difference Fourier, by using Fo,Mg − Fo,Li as coefficients and phases from the refined Li+-ATP structure; density is displayed at 3.5 σ (red). WAT709 is well positioned for direct in-line nucleophilic attack of the γ-phosphate center, and in the Ca2+- and Mg2+-ATP structures forms a water-mediated hydrogen bond with the ND of His-161. In the Li+-ATP structure, His-161 adopts a different side chain rotamer conformation that supports a direct hydrogen bond between NE and WAT709. We propose that Gln-137 directs that placement of this water, which is activated by His-161 through either direct or indirect proton abstraction. (D–F) The geometry of WAT709 and the γ-phosphoryl in the different metal nucleotide complexes. As the series moves from Ca2+- to Mg2+- to Li+-ATP, WAT709 moves closer to the γ-phosphoryl and the β-γ bridging oxygen-Pγ-WAT709 angle moves closer to linearity. Although speculative, these structures may provide a model of the actin-associated ATPase reaction coordinate prior to reaching the transition state.
In addition to the interaction with bound metal, the polyphosphate tail makes numerous contacts with elements of the P1 loop. The nonbridging γ-phosphoryl oxygens interact with the backbone amide nitrogen and the side chain hydroxyl of Ser-14, as well as with the backbone amide nitrogen of Asp-157 contributed from the P2 loop. The β,γ- bridging oxygen also forms a potential hydrogen bond with the backbone amide of Asp-157. One of the nonbridging β-phosphoryl oxygens hydrogen bonds to the backbone amides of Gly-15 and Met-16, whereas one of the α-phosphoryl oxygens interacts with the backbone amide of Gly-302 from the P3 loop. Lys-18 is the sole basic functionality interacting with the nucleotide, forming a bidentate interaction with nonbridging α- and β-phosphoryl oxygens. Of particular interest is a water molecule (WAT709) that ranges from 3.90 to 4.15 Å from the γ-phosphoryl depending on the identity of the nucleotide-bound metal (Fig. 3 and supporting information). The role of this water as the putative nucleophile in the hydrolytic mechanism is described below.
In the nonvertebrate Mg2+-ATP actins, the adenosine ring occupies a hydrophobic pocket formed by the side chains of Met-305 and Phe-306 from the P3 loop, as well as the methylene carbons of the side chains from Lys-213 and Glu-214. The endocyclic and exocyclic nitrogens of the purine ring form water-mediated hydrogen bonds with NZ of Lys-213 and Lys-336, and with the main chain carbonyls of residues 213, 214, and 305. The exocyclic ribose oxygens also participate in specific polar contacts, with O2′ interacting with the side chain functionalities of Lys-213 and Glu-214, and O3′ interacting with the side chains of Asp-157 and Lys-213. The interactions involved in binding adenine nucleotide are conserved in the Dictyostelium and C. elegans actins.
The substitution of Li+-ATP for Mg2+-ATP in Dictyostelium actin appears to cause only minor alterations, as the metal maintains hexacoordination with the same ligands and only very modest alterations in bond lengths. The Ca2+-ATP Dictyostelium actin structure shows heptacoordination around the calcium, with distances that are systematically longer than the Mg2+-ATP structure, as expected. The equivalent of WAT709 also exhibits a slight change in position relative to the γ-phosphoryl.
The Mechanism of ATP Hydrolysis in the Actin Monomer.
The present structures, in particular the isomorphous series of metal-ATP Dictyostelium actin structures, provide a framework for examining the kinetic and mechanistic features of the actin catalyzed ATP hydrolytic reaction (Fig. 3). In all of the nonvertebrate structures, the equivalent of WAT709 appears to be positioned through its interaction with Gln-137 to act as the nucleophile for direct in-line attack at the γ-phosphoryl of ATP. In the Dictyostelium Mg2+-ATP structure, WAT709 is positioned 4.01 Å from the γ-phosphoryl of ATP and the β-γ bridging oxygen-Pγ-WAT709 angle is 167°. A comparable organization is present in all of the nonvertebrate actin structures, and of particular note, the same relative geometry of the Pγ, water nucleophile, and the equivalent of Gln-137 (Gln-61 in ras and Gln-204 in the Giα1 structure) is present in the trimeric (49, 50) and small (51) G proteins, supporting the assignment of WAT709 as the catalytic water.
In the nonvertebrate Mg2+-ATP actin structures the ND of His-161 participates in a water-mediated hydrogen bond with WAT709, suggesting that His-161 may activate WAT709 by functioning as a general base catalyst. The assignment of His-161 as the catalytic base is supported not only by its proximity to the putative water nucleophile and γ-phosphoryl, but also the lack of additional, appropriately positioned functional groups in the nucleotide-binding cavity. Three closest appropriate side chains, Asp-11, Asp-154, and His-73, are at least 5, 7, and 8 Å distant from the putative water nucleophile, respectively, with the two carboxylates participating in water-mediated interactions with the nucleotide-associated metal. Mutagenesis experiments involving these residues, as well as others in the vicinity of the active site, have identified a number of functionalities critical for catalytic activity and viability (reviewed in ref. 52). However, in the case of the aspartic acidic residues, the observed activities and phenotypes are likely the consequence of improper metal coordination, as there exists no evidence supporting their repositioning to allow for direct activation of the nucleophile, either in actin or any other member of the superfamily. The negative charge that accumulates in the transition state is stabilized by the divalent cation that coordinates the β- and γ-phosphates, a series of hydrogen bonds involving main chain and side chain polar groups and a single ionic interaction involving Lys-18. In the Dictyostelium Ca2+-ATP structure the distance between WAT709 and the γ-phosphoryl increases to 4.15 Å, whereas the β-γ bridging oxygen-Pγ-WAT709 angle decreases to 157°. This increase in distance and excursion from linearity suggests a partial explanation for the reduced ability of Ca2+ to support ATP hydrolysis, as even very small changes in relative geometry can have significant effects on reactivity. Of note, the water-mediated hydrogen bond between WAT709 and the δ nitrogen of His-161 is preserved in the Ca2+-ATP structure.
Of particular mechanistic interest is the Li+-ATP Dictyostelium actin structure, which exhibits the opposite trend observed in the Ca2+-ATP structure. In the Li+-ATP structure, the separation between WAT709 and the γ-phosphoryl decreases to 3.90 Å and the β-γ bridging oxygen-Pγ-WAT709 angle approaches linearity at 174°. Furthermore, in the model of the Li+-ATP structure, His-161 displays an altered side chain conformation (χ1 = −56°, χ2 = −77°) compared with the Mg2+-ATP (χ1 = −82°, χ2 = 117°) structure. Notably, in this conformation the NE of His-161 participates in a direct hydrogen bond with WAT709 (3.30 Å) (Fig. 3). Although His-161 reproducibly refines to this position, careful examination of the electron density suggests that there may be partial occupancy of the bridging water molecule, which would necessitate partial occupancy of a side chain rotamer similar to that observed in the Mg2+-ATP structure. These observations highlight the mobile properties of His-161, which might be expected of a residue involved in catalysis. Thus, the isomorphous series of Dictyostelium metal–ATP structures exhibits a continuum of WAT709 positions that range from the least likely to support phosphoryl transfer in the Ca2+-ATP structure (i.e., greatest distance from γ-phosphoryl and least in line) to the most favorable in the Li+-ATP structure (i.e., shortest distance from γ-phosphoryl and the most linear approach). The unambiguous identification of ATP in the active site is noteworthy, as it suggests that turnover in the crystalline state is significantly reduced relative to solution, likely as a consequence of crystal contacts (and/or binding of gelsolin S1) that hinder local/global conformational alterations required for hydrolysis (see below).
The current structural analysis strongly implicates Gln-137 and His-161 as primary participants in the actin-associated ATPase reaction. These assignments are supported by genetic studies suggesting that the presence of Gln and His at residues 137 and 161, respectively, is required for viability in yeast. Numerous attempts were made to alter Gln-137 to a glutamic acid, a substitution that occurs naturally in other members of the actin superfamily, as well as to mutate His-161 to alanine. These studies involved two basic approaches. The first was the introduction of the mutant carried on a centromeric plasmid into a diploid strain of yeast that already harbored a single disrupted chromosomal actin gene, followed by tetrad dissection to isolate viable cells producing only the mutant actin (53). The second approach involved a plasmid shuffling protocol using haploid cells carrying a wild-type actin coding sequence on a URA3-marked plasmid. After introduction of the plasmid containing the mutant actin coding sequence, elimination of the WT plasmid was attempted both in the absence and in the presence of 5-fluoroorotic acid, which provides a more stringent selection (54). In no case were viable cells obtained that expressed only the mutant actins. In all cases parallel controls with mutant constructs at other positions that were known to produce viable cells were successful.
The metal-dependent structural changes observed in the actin monomer are consistent with a recent reevaluation of the role of bound divalent cation. Blanchoin and Pollard (55) used quenched-flow methods to demonstrate that the rate of ATP hydrolysis by Mg2+-F-actin (0.30 s−1) is 6-fold higher than that of Ca2+-F-actin (0.05 s−1) (limitations in instrumental deadtime precluded the examination of shorter reaction times that may have distinguished Li+- and Mg2+-ATP). Although absolute magnitudes are difficult to predict, these kinetic results support our proposal that magnesium promotes an arrangement in which the putative water nucleophile is positioned closer to the ideal “in-line” geometry. It is important to note that although the placement of water molecules can be subjective, especially at the resolution of these studies, the systematic trend observed for WAT709 is consistent with the available kinetic data and suggests that these structures may represent movements along the reaction coordinate before achieving the transition state (Fig. 3).
Comparison with Other NTPases.
Monomeric actin exhibits an extremely slow hydrolytic activity toward ATP (apparent first order rate constant of 10−5 s−1) (32), which is consistent with the relative sparseness of functional groups that interact with the polyphosphate tail (Fig. 3 and supporting information). A particularly meaningful manner in which to view these hydrolytic rates is to examine the catalytic power, which is defined as the ratio of the maximal turnover number for the enzyme catalyzed reaction (i.e., kcat) compared with the pseudofirst order rate constant for the same reaction under the same solvent conditions (ksoln) (56). Thus, although the hydrolytic rate for G-actin is slow, it is still well over three orders of magnitude faster than the solvent catalyzed reaction, the equivalent of a catalytic power of ≈103.6 (32, 56). For comparison, the catalytic power of typical ATPases, such as the mitochondrial F1 ATPase and myosin are ≈1010.7 (56). These large differences in catalytic power can be attributed to specific protein functionalities that contribute to transition state stabilization and activation of the water nucleophile. In the F1 ATPase, two arginines and one lysine interact with the nonbridging oxygens of the γ-phosphoryl of ATP, whereas in the nonvertebrate Mg2+-ATP actin structures, the sole protein derived basic functionality in the vicinity of the polyphosphate tail is Lys-18. Monomeric actin is more similar to the small G proteins (i.e., ras) and trimeric G proteins (i.e., Giα1), which display relatively low catalytic powers of 105.7 and 107.1, respectively (56). The G proteins, like actin, have a reduced number of basic functionalities interacting with the polyphosphate tail as compared with the F1 ATPase, and are thus less effective at stabilizing the transition state. The G proteins also appear to lack a general base, consistent with the lowered hydrolytic rates that are observed. Although the equivalent of Gln-137 in actin has been proposed to serve as a general base in the G protein catalyzed reactions (51, 57), the pKa of the carboxamide side chain argues against such a role, and this residue is likely to effect catalysis through positioning the water nucleophile, as well as providing some degree of stabilization to the developing charge as the transition state is approached (58, 59). It is anticipated that Gln-137 may play a similar role in actin catalyzed ATP hydrolysis.
Implications for the F-actin ATPase Activity.
Although our current structural analysis is based solely on the actin monomer, this model may provide some insights into the mechanistic features required for the greatly enhanced rate of ATP hydrolysis associated with actin polymerization. In the case of the small G proteins, the rates of GTP hydrolysis are enhanced several orders of magnitude by the binding of GTPase activating proteins (GAPs), which deliver additional functional groups to the active site to provide transition state stabilization (60). By analogy, an attractive mechanism accounting for the enhanced ATPase rate associated with actin polymerization would be that neighboring subunits in the filament provide additional functional groups that facilitate ATP hydrolysis. However, examination of the Holmes model for the actin filament shows that the closest approach of an appropriate functionality from a neighboring subunit to the γ-phosphoryl is at a distance of >17 Å, indicating that a very different mechanism for activation must be operating. Considerable structural work has indicated that the trimeric G proteins exist in an autoinhibited state that exhibits inefficient turnover. In contrast to the small G protein GAPs, the regulators of G protein signaling (RGSs) responsible for activation of the trimeric G proteins do not make any direct contacts with GTP in the active site. Instead, the RGSs induce a small rearrangement of active site residues that promotes the formation of a catalytically active conformation, resulting in rate enhancements of up to several orders of magnitude (49, 61) (reviewed in ref. 62). In the case of the actin filament, we propose a modification of this mechanism in which the lateral and longitudinal contacts formed during polymerization result in a conformational rearrangement of catalytic residues, especially His-161 and perhaps Gln-137, which accounts for the enhanced hydrolytic activity of the actin filament. Several recent demonstrations of structural plasticity within the monomer (44, 63) and the filament (64–67) are consistent with this notion. Furthermore, given the great paucity of appropriate functionalities in the vicinity of the γ-phosphoryl, we speculate that the conformational alterations associated with polymerization may also contribute to the enhanced ATPase activity of the filament through ground state destabilization. This proposal is analogous to that recently suggested for ras, in which Mg2+-GTP begins to acquire transition-state-like features (i.e., geometry and bond order) in the enzyme-bound ground state (68).
Implications for the Actin-Related Proteins (Arps).
Actin also provides a framework for examining the mechanistic considerations relevant to members of the family of Arps. The P loops in all of the Arps show significant conservation with actin, indicating that all Arps may have the potential to bind nucleotide (Table 2, which is published as supporting information on the PNAS web site) (28–30, 33). Of particular note is the Arp2/3 complex that is responsible for de novo nucleation of actin filaments. ATP hydrolysis appears to be required for Arp2/3 function, and structural (34, 69) and biochemical (70) data suggest that ATP hydrolysis is coupled with a conformational reorganization within the Arp2/3 complex, which allows the Arp2 and Arp3 subunits to adopt an arrangement that mimics the growing barbed end of an actin filament and promotes the nucleation of new filaments. Like actin, both Arp2 and Arp3 possess the equivalent of Gln-137 and His-161, suggesting mechanistic similarities for nucleotide hydrolysis. Importantly, the Arp2/3 complex does not exhibit measurable ATPase activity, either alone or in the presence of its activators N-WASP and F-actin (70). As the majority of Arp2/3 activators contain a G-actin binding domain (termed the WH2 module) in close proximity to the Arp2/3 binding domain, it has been suggested that the role of the WH2 module is to promote an interaction between a filament-associated Arp2/3 complex and an actin monomer, resulting in stimulation of the Arp2/3 ATPase activity (70). This hypothesis shares considerable similarity with our proposed mechanism for the rate enhancement of the actin ATPase associated with actin polymerization. Finally, the recent Ca2+-ADP-G-actin structure (45), and a host biochemical studies (52), implicate Ser-14, Ser-33 Tyr-69, Asp-157, and Arg-183 as key components of the mechanism that directs the nucleotide-dependent conformational properties of the actin monomer (45). Identical or highly conservative residues are present at these positions in both Arp2 and Arp3, suggesting that these molecules couple nucleotide state and conformation via a mechanism similar to that used by actin.
Supplementary Material
Acknowledgments
We thank Anne Bresnick for comments on the manuscript. P.A.R., D.G.D., C.F., J.C., and S.C.A. acknowledge support from the National Institutes of Health. S.O. acknowledges support from the American Heart Association and the National Science Foundation.
Abbreviation
- F-actin
filamentous actin
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1YAG, 1NM1, 1NLV, 1NMD, and 1D4X).
References
- 1.Mitchison T J, Cramer L P. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- 2.Glotzer M. Annu Rev Cell Dev Biol. 2001;17:351–386. doi: 10.1146/annurev.cellbio.17.1.351. [DOI] [PubMed] [Google Scholar]
- 3.Goode B L, Drubin D G, Barnes G. Curr Opin Cell Biol. 2000;12:63–71. doi: 10.1016/s0955-0674(99)00058-7. [DOI] [PubMed] [Google Scholar]
- 4.Gavin R H. Int Rev Cytol. 1997;173:207–242. doi: 10.1016/s0074-7696(08)62478-x. [DOI] [PubMed] [Google Scholar]
- 5.Matsudaira P. Semin Cell Biol. 1994;5:165–174. doi: 10.1006/scel.1994.1021. [DOI] [PubMed] [Google Scholar]
- 6.Matsudaira P. Trends Biochem Sci. 1991;16:87–92. doi: 10.1016/0968-0004(91)90039-x. [DOI] [PubMed] [Google Scholar]
- 7.Mooseker M S. Annu Rev Cell Biol. 1985;1:209–241. doi: 10.1146/annurev.cb.01.110185.001233. [DOI] [PubMed] [Google Scholar]
- 8.Heintzelman M B, Mooseker M S. Curr Topics Dev Biol. 1992;26:93–122. doi: 10.1016/s0070-2153(08)60442-1. [DOI] [PubMed] [Google Scholar]
- 9.Cotanche D A, Lee K H. Curr Opin Neurobiol. 1994;4:509–514. doi: 10.1016/0959-4388(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 10.Hofer D, Ness W, Drenckhahn D. J Cell Sci. 1997;110:765–770. doi: 10.1242/jcs.110.6.765. [DOI] [PubMed] [Google Scholar]
- 11.De La Cruz E M, Pollard T D. Biochemistry. 1995;34:5452–5461. doi: 10.1021/bi00016a016. [DOI] [PubMed] [Google Scholar]
- 12.Pollard T D, Blanchoin L, Mullins R D. Annu Rev Biophys Biomol Struct. 2000;29:545–576. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- 13.Blanchoin L, Pollard T D. J Biol Chem. 1999;274:15538–15546. doi: 10.1074/jbc.274.22.15538. [DOI] [PubMed] [Google Scholar]
- 14.Gaszner B, Nyitrai M, Hartvig N, Koszegi T, Somogyi B, Belagyi J. Biochemistry. 1999;38:12885–12892. doi: 10.1021/bi990748y. [DOI] [PubMed] [Google Scholar]
- 15.Kim E, Motoki M, Seguro K, Muhlrad A, Reisler E. Biophys J. 1995;69:2024–2032. doi: 10.1016/S0006-3495(95)80072-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moraczewska J, Strzelecka-Golaszewska H, Moens P D, dos Remedios C G. Biochem J. 1996;317:605–611. doi: 10.1042/bj3170605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moraczewska J, Wawro B, Seguro K, Strzelecka-Golaszewska H. Biophys J. 1999;77:373–385. doi: 10.1016/S0006-3495(99)76896-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nyitrai M, Hild G, Belagyi J, Somogyi B. Biophys J. 1997;73:2023–2032. doi: 10.1016/S0006-3495(97)78232-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nyitrai M, Hild G, Lakos Z, Somogyi B. Biophys J. 1998;74:2474–2481. doi: 10.1016/S0006-3495(98)77955-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frieden C, Patane K. Biochemistry. 1985;24:4192–4196. doi: 10.1021/bi00336a056. [DOI] [PubMed] [Google Scholar]
- 21.Barden J A, dos Remedios C G. Eur J Biochem. 1985;146:5–8. doi: 10.1111/j.1432-1033.1985.tb08612.x. [DOI] [PubMed] [Google Scholar]
- 22.Kabsch W, Mannherz H G, Suck D, Pai E F, Holmes K C. Nature. 1990;347:37–44. doi: 10.1038/347037a0. [DOI] [PubMed] [Google Scholar]
- 23.Schutt C E, Myslik J C, Rozycki M D, Goonesekere N C, Lindberg U. Nature. 1993;365:810–816. doi: 10.1038/365810a0. [DOI] [PubMed] [Google Scholar]
- 24.McLaughlin P J, Gooch J T, Mannherz H G, Weeds A G. Nature. 1993;364:685–692. doi: 10.1038/364685a0. [DOI] [PubMed] [Google Scholar]
- 25.Robinson R C, Mejillano M, Le V P, Burtnick L D, Yin H L, Choe S. Science. 1999;286:1939–1942. doi: 10.1126/science.286.5446.1939. [DOI] [PubMed] [Google Scholar]
- 26.Matsuura Y, Stewart M, Kawamoto M, Kamiya N, Saeki K, Yasunaga T, Wakabayashi T. J Mol Biol. 2000;296:579–595. doi: 10.1006/jmbi.1999.3467. [DOI] [PubMed] [Google Scholar]
- 27.Lo Conte L, Brenner S E, Hubbard T J, Chothia C, Murzin A G. Nucleic Acids Res. 2002;30:264–267. doi: 10.1093/nar/30.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W, Holmes K C. FASEB J. 1995;9:167–174. doi: 10.1096/fasebj.9.2.7781919. [DOI] [PubMed] [Google Scholar]
- 29.Flaherty K M, McKay D B, Kabsch W, Holmes K C. Proc Natl Acad Sci USA. 1991;88:5041–5045. doi: 10.1073/pnas.88.11.5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bork P, Sander C, Valencia A. Proc Natl Acad Sci USA. 1992;89:7290–7294. doi: 10.1073/pnas.89.16.7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holmes K C, Popp D, Gebhard W, Kabsch W. Nature. 1990;347:44–49. doi: 10.1038/347044a0. [DOI] [PubMed] [Google Scholar]
- 32.Pollard T D, Weeds A G. FEBS Lett. 1984;170:94–98. doi: 10.1016/0014-5793(84)81376-9. [DOI] [PubMed] [Google Scholar]
- 33.Kelleher J F, Atkinson S J, Pollard T D. J Cell Biol. 1995;131:385–397. doi: 10.1083/jcb.131.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robinson R C, Turbedsky K, Kaiser D A, Marchand J B, Higgs H N, Choe S, Pollard T D. Science. 2001;294:1679–1684. doi: 10.1126/science.1066333. [DOI] [PubMed] [Google Scholar]
- 35.Bresnick A R, Condeelis J. Methods Enzymol. 1991;196:70–83. doi: 10.1016/0076-6879(91)96009-g. [DOI] [PubMed] [Google Scholar]
- 36.Ono S. Cell Motil Cytoskel. 2001;43:128–136. doi: 10.1002/(SICI)1097-0169(1999)43:2<128::AID-CM4>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 37.Way M, Pope B, Weeds A G. J Cell Biol. 1992;116:1135–1143. doi: 10.1083/jcb.116.5.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 39.Navaza J. Acta Crystallogr D. 2001;57:1367–1372. doi: 10.1107/s0907444901012422. [DOI] [PubMed] [Google Scholar]
- 40.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1994;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 41.Laskowski R A, MacArthur M W, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 42.Chen X, Cook R K, Rubenstein P A. J Cell Biol. 1993;123:1185–1195. doi: 10.1083/jcb.123.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cook R K, Blake W T, Rubenstein P A. J Biol Chem. 1992;267:9430–9436. [PubMed] [Google Scholar]
- 44.Chik J K, Lindberg U, Schutt C E. J Mol Biol. 1996;263:607–623. doi: 10.1006/jmbi.1996.0602. [DOI] [PubMed] [Google Scholar]
- 45.Otterbein L R, Graceffa P, Dominguez R. Science. 2001;293:708–711. doi: 10.1126/science.1059700. [DOI] [PubMed] [Google Scholar]
- 46.Chakrabarti P. J Mol Biol. 1993;234:463–482. doi: 10.1006/jmbi.1993.1599. [DOI] [PubMed] [Google Scholar]
- 47.Walker J E, Saraste M, Runswick J, Gay N J. EMBO J. 1982;1:945–516. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glusker J P. Adv Protein Chem. 1991;42:1–76. doi: 10.1016/s0065-3233(08)60534-3. [DOI] [PubMed] [Google Scholar]
- 49.Coleman D E, Sprang S R. J Biol Chem. 1999;274:16669–16672. doi: 10.1074/jbc.274.24.16669. [DOI] [PubMed] [Google Scholar]
- 50.Coleman D E, Berghuis A M, Lee E, Linder M E, Gilman A G, Sprang S R. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- 51.Pai E F, Krengel U, Petsko G A, Goody R S, Kabsch W, Wittinghofer A. EMBO J. 1990;9:2351–2359. doi: 10.1002/j.1460-2075.1990.tb07409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schuler H. Biochim Biophys Acta. 2001;1549:137–147. doi: 10.1016/s0167-4838(01)00255-2. [DOI] [PubMed] [Google Scholar]
- 53.Cook R K, Root D, Miller C, Reisler E, Rubenstein P A. J Biol Chem. 1993;268:2410–2415. [PubMed] [Google Scholar]
- 54.Chen X, Rubenstein P A. J Biol Chem. 1995;270:11406–11414. doi: 10.1074/jbc.270.19.11406. [DOI] [PubMed] [Google Scholar]
- 55.Blanchoin L, Pollard T D. Biochemistry. 2002;41:597–602. doi: 10.1021/bi011214b. [DOI] [PubMed] [Google Scholar]
- 56.Mildvan A S. Proteins. 1997;29:401–416. [PubMed] [Google Scholar]
- 57.Goody R S, Pai E F, Schlichting I, Rensland H, Scheidig A, Franken S, Wittinghofer A. Philos Trans R Soc London Ser B. 1992;336:3–10. doi: 10.1098/rstb.1992.0037. ; discussion 10–11. [DOI] [PubMed] [Google Scholar]
- 58.Maegley K A, Admiraal S J, Herschlag D. Proc Natl Acad Sci USA. 1996;93:8160–8166. doi: 10.1073/pnas.93.16.8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chung H H, Benson D R, Schultz P G. Science. 1993;259:806–809. doi: 10.1126/science.8430333. [DOI] [PubMed] [Google Scholar]
- 60.Scheffzek K, Ahmadian M R, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, Wittinghofer A. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 61.Tesmer J J, Berman D M, Gilman A G, Sprang S R. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 62.Ross E M, Wilkie T M. Annu Rev Biochem. 2000;69:795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 63.Page R, Lindberg U, Schutt C E. J Mol Biol. 1998;280:463–474. doi: 10.1006/jmbi.1998.1879. [DOI] [PubMed] [Google Scholar]
- 64.Galkin V E, VanLoock M S, Orlova A, Egelman E H. Curr Biol. 2002;12:570–575. doi: 10.1016/s0960-9822(02)00742-x. [DOI] [PubMed] [Google Scholar]
- 65.Galkin V E, Orlova A, Lukoyanova N, Wriggers W, Egelman E H. J Cell Biol. 2001;153:75–86. doi: 10.1083/jcb.153.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Belmont L D, Orlova A, Drubin D G, Egelman E H. Proc Natl Acad Sci USA. 1999;96:29–34. doi: 10.1073/pnas.96.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McGough A, Pope B, Chiu W, Weeds A. J Cell Biol. 1997;138:771–781. doi: 10.1083/jcb.138.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cheng H, Sukal S, Deng H, Leyh T S, Callender R. Biochemistry. 2001;40:4035–4043. doi: 10.1021/bi0021131. [DOI] [PubMed] [Google Scholar]
- 69.Volkmann N, Amann K J, Stoilova-McPhie S, Egile C, Winter D C, Hazelwood L, Heuser J E, Li R, Pollard T D, Hanein D. Science. 2001;293:2456–2459. doi: 10.1126/science.1063025. [DOI] [PubMed] [Google Scholar]
- 70.Dayel M J, Holleran E A, Mullins R D. Proc Natl Acad Sci USA. 2001;98:14871–14876. doi: 10.1073/pnas.261419298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.