

Abstract
Mycobacterium tuberculosis, the causative agent of human tuberculosis, is unique among bacterial pathogens in that it displays a wide array of complex lipids and lipoglycans on its cell surface. One of the more remarkable lipids is a sulfated glycolipid, termed sulfolipid-1 (SL-1), which is thought to mediate specific host-pathogen interactions during infection. However, a direct role for SL-1 in M. tuberculosis virulence has not been established. Here we show that MmpL8, a member of a large family of predicted lipid transporters in M. tuberculosis, is required for SL-1 production. The accumulation of an SL-1 precursor, termed SL1278, in mmpL8 mutant cells indicates that MmpL8 is necessary for an intermediate step in the SL-1 biosynthesis pathway. We use a novel fractionation procedure to demonstrate that SL-1 is present on the cell surface, whereas SL1278 is found exclusively in more internal layers. Importantly, we show that mmpL8 mutants are attenuated for growth in a mouse model of tuberculosis. However, SL-1 per se is not required for establishing infection as pks2 mutants, which are defective in an earlier step in SL-1 biosynthesis, have no obvious growth defect. Thus, we hypothesize that either MmpL8 transports molecules in addition to SL-1 that mediate host-pathogen interactions or the accumulation of SL1278 in mmpL8 mutant cells interferes with other pathways required for growth during the early stages of infection.
Mycobacterium tuberculosis is an intracellular pathogen that is highly adapted to infect and persist within mammalian tissues (1, 2). Although little is known about the molecular mechanisms of M. tuberculosis virulence, an emerging paradigm is that cell wall lipids play important roles in the pathogenesis of this organism (3). The cell wall of M. tuberculosis is a complex structure that is responsible for many of the unique properties of the organism (4). One of the distinguishing features of the mycobacterial cell wall is the extremely long chain fatty acids, termed mycolic acids, which are esterified to the arabinogalactan core. In addition, a highly diverse set of surface-exposed lipids and glycolipids are noncovalently associated with the mycolic acids, creating an asymmetric bilayer structure (ref. 5, see Fig. 6). Together, these features of the cell wall create a protective barrier that is vital for the survival of the organism within the host.
Figure 6.

Model of the role of MmpL8 in the SL-1 biosynthetic pathway. See Discussion for details of the model. HPA, hydroxyphthioceranic acid; ML, mycolate layer; AG, arabinogalactan layer; PG, peptidoglycan layer; CM, cytoplasmic membrane.
A rich literature exists that also implicates mycobacterial surface-exposed lipids in modulating specific host-pathogen interactions. Early work established a correlation between levels of two major cell wall lipids, sulfolipid-1 (SL-1, the most abundant sulfatide) and phthiocerol dimycocerosate (PDIM), and the virulence of M. tuberculosis (6, 7). Indeed, purified SL-1, as well as other mycobacterial lipids such as trehalose 6,6′ dimycolate and lipoarabinomannan, have interesting immunomodulatory activity when administered to animals or cultured immune cells (8–14). PGL-1, a glycosylated version of PDIM found in Mycobacterium leprae, can suppress lymphocyte responses as well as facilitate Schwann cell invasion (15, 16). Finally, the increased pathology of a M. tuberculosis clinical isolate has been attributed to differences in its surface-exposed lipids (17).
Further evidence for the importance of surface-exposed lipids in the pathogenesis of M. tuberculosis came from two genetic screens that uncovered genes involved in lipid synthesis and secretion that are required for virulence (18, 19). The ppsA-E and fadD28 genes are involved in synthesizing PDIM, and mutants lacking these genes exhibit a specific growth defect in the mouse lung. Although PDIM may be required for cell wall integrity (20), the precise function of this molecule during infection is unknown. Interestingly, mmpL7 mutants fail to secrete PDIM and exhibit the same in vivo phenotype as PDIM-deficient mutants (18–20).
MmpL7 is a member of a family of 13 predicted lipid transporters encoded by the M. tuberculosis genome. Many of these mmpL genes are adjacent to polyketide synthase genes in the chromosome, prompting the speculation that individual MmpL proteins transport the lipids produced by the neighboring synthases (21). Indeed, this hypothesis is supported by the fact that mmpL7, ppsA-E, and fadD28 are clustered together in the genome. In this study, we describe the identification of another mmpL family member, mmpL8, and show that this gene, like the neighboring polyketide synthase pks2 (22), is essential for SL-1 biosynthesis. Furthermore, we show that although SL-1 is dispensable during the acute stage of infection, mmpL8 is absolutely required for normal M. tuberculosis growth in vivo.
Materials and Methods
Media and Strains.
M. tuberculosis cells (Erdman strain) were cultured as described (18). For MS analysis, cultures were grown in a modified Sauton media (23). The mmpL8∷Tn5367–1, mmpL8∷Tn5367–2, ΔmmpL8, Δpks2, Δpks2 + ppks2, and ΔmmpL8 + pmmpL8 strains are named jcm130, jcm131, jcm108, jcm119, scm6, and scm8, respectively.
Signature-Tagged Mutagenesis.
Signature-tagged mutagenesis, mutant isolation, tag amplification, and insertion determination were performed exactly as described (18).
Construction of M. tuberculosis Knockout Strains and Complementation Constructs.
The ΔmmpL8 (jcm108) and Δpks2 (jcm119) mutant strains were created by homologous recombination using specialized transducing phages phjsc311 and phjsc377, respectively (24). These deletions replaced 3254 bp of mmpL8 (aa 5–1089) and 6305 bp of pks2 (aa 14–2116) with a hygromycin resistance cassette. Correct replacement of both genes was confirmed by Southern blot. For Δpks2, genomic DNA was digested with AatII, and the blot was probed with a 610 bp 5′ flank, revealing a 2,871-bp fragment for wild-type and a 3,918-bp fragment for the mutant. For ΔmmpL8, genomic DNA was digested with SmaI, and the blot was probed with a 709-bp 3′ flank, revealing a 2,638-bp fragment for wild-type and a 3,832-bp fragment for the mutant.
The pks2 (pSEC7) and mmpL8 (pSEC16) complementation vectors each contained the entire ORF under the control of the endogenous promoter and were created by subcloning from a cosmid, cERD4F2, into the integrating vector pMV306.kan.
Biochemical Analysis of PDIM and SL-1.
Actively growing cultures were diluted to OD600 = 0.6 in a final volume of 50 ml. 14C-propionate (7.5 μCi, 1 Ci = 37 GBq) was added and incubation continued for 16 h at 37°C. Cell pellets were extracted essentially as described (18). Lipids were separated on 10 cm × 10 cm HPTLC plates (Alltech Associates) by using either hexanes/ether (9:1) to resolve PDIM, or chloroform/methanol/water (60:30:6) to resolve SL-1. Radioactive sulfate experiments were performed as above, except 100 μCi of 35S-SO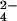 was incubated with 10 ml of cells for 48 h.
was incubated with 10 ml of cells for 48 h.
For pulse–chase experiments, the cells were labeled for 30 min, pelleted, washed with media, and resuspended in 100 ml of radioactive-free 7H9 media. Ten-milliliter aliquots were removed at different time points and the cells were harvested for lipid extraction.
To analyze surface-exposed lipids from M. tuberculosis, 7.5 μCi of 14C-propionate was added and incubated for 2 h to strongly label both SL-1 and SL1278. Cell pellets were resuspended in 1-ml hexanes/0.05% decylamine and sonicated by using a Branson Sonifier 250 with a 2-in cup horn chamber (Branson) at 25% for 90 s. The supernatant and cell pellet were recovered by centrifugation, and decylamine was removed by extracting 3× with 0.1 N HCl. Lipids were extracted and analyzed as described above.
MS.
Lipid 2 from ΔmmpL8 mutant cells was partially purified by using preparative TLC plates and eluted with CHCl3/MeOH (1:2). The recovered lipids were dried under nitrogen and dissolved in CHCl3/MeOH (2:1). Spectra were obtained by using a Bruker Apex II Fourier transform ion cyclotron resonance MS (Bruker, Billerica, MA) equipped with a 7T actively shielded magnet as described (23). Spectra were acquired in the negative ion mode using electrospray ionization and were composed of 512,000 data points. Authentic SL-1 was obtained from Colorado State University (National Institutes of Health, National Institute of Allergy and Infectious Disease Contract N01 AI-75320).
Infection of Individual M. tuberculosis Mutants.
Actively growing wild-type, jcm108, and jcm119 strains were washed, resuspended in 1× PBS and 0.1% Tween, and sonicated. C57BL/6 mice were infected by tail injection with 1–2 × 106 colony-forming units (cfu) of each strain by using an OD600 = 3 × 108 cfu⋅ml−1. Organs from infected mice were homogenized and plated as described (18).
Results
Isolation of mmpL8 Transposon Mutants from a Screen for Attenuated Strains.
In an extension of a previous screen to identify genes involved in virulence of M. tuberculosis, we identified two mutants with transposon insertions in the same gene, mmpL8 (Fig. 1A). Because each transposon contained a unique sequence tag, the representation of these mutants within pools of 48 random mutants could be quantified by amplification of all of the tags, followed by labeling and hybridization to filter arrays spotted with each individual tag. As shown in Fig. 1B, both of these mutants were equally represented in inoculum pools, yet markedly underrepresented after 3 weeks of growth in the lungs of infected mice. These results suggested that mmpL8 is required for growth during the early stages of infection (see below).
Figure 1.

Two mmpL8 mutants of M. tuberculosis identified by signature-tagged mutagenesis. (A) Diagram of mmpL8 and surrounding genes denoting the transposon insertion sites (1,660 and 2,238 bp with respect to the start codon) in each mutant. (B) Signature DNA tags from mutant mycobacteria harvested from the inoculum and the lungs of two mice after 3 weeks of growth were amplified, radiolabelled, and hybridized to tag array filters (18). Tags from the two underrepresented mmpL8 mutants are shown next to control tags that did not vary throughout the experiment. (C) ΔmmpL8 and Δpks2 strains were generated by specialized transduction, and Southern blot revealed the expected bands indicating the replacement of the genomic sequence with the hygromycin cassette. For details, see Materials and Methods.
Inspection of the genomic location of mmpL8 revealed that it is near pks2, which encodes a polyketide synthase required for synthesizing phthioceranic and hydroxyphthioceranic acids, the methyl-branched acyl constituents of sulfolipid-1 (SL-1) (ref. 22, Fig. 1A). To study mmpL8 and pks2, we removed the entire ORFs from a wild-type M. tuberculosis strain and confirmed these deletions by Southern blot (Fig. 1C). Importantly, deletion of either gene did not affect growth in culture as the doubling time of ΔmmpL8, Δpks2, and wild-type cells were 20.2 ± 0.3, 20.2 ± 0.2, and 20.2 ± 0.3 h, respectively.
Initial Characterization of mmpL8 Role in Mycobacterial Lipid Biosynthesis.
To test the hypothesis that MmpL8 functions in the SL-1 biogenesis pathway, we monitored SL-1 synthesis by labeling cells with 14C-propionate and separating total lipids by TLC (Fig. 2A). Because both PDIM and SL-1 contain methyl-branched fatty acids, these lipids (as well as other methyl-branched lipids in M. tuberculosis) are labeled by 14C-propionate, yet they are distinguishable by their mobility on TLC plates. As expected, SL-1, a major species in wild-type cells, is completely absent in extracts prepared from the Δpks2 mutants (Fig. 2A, lane 2). Reintroduction of the pks2 gene restored the ability of the strain to synthesize SL-1 (lane 3). Interestingly, we noted a minor species (lipid 2) that was present in the complemented Δpks2 strain as well as the wild-type, but was absent in Δpks2 cells.
Figure 2.

TLC analysis of lipids from mmpL8 and pks2 M. tuberculosis mutants is shown. (A) Cells were labeled with 14C-propionate, and lipids were extracted and separated under two TLC solvent conditions. The first (Upper) resolves SL-1, whereas the second (Lower) resolves PDIM. Equal amounts of lipids were spotted onto both TLCs. The position of SL-1, lipid 2, and the two forms of PDIM are noted. (B) Cells were labeled with 35S-SO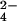 , and lipids were extracted and separated by TLC, as in A Upper. (C) Proposed structure of SL-1 as determined by Mougous et al. (23) and Goren et al. (25, 26).
, and lipids were extracted and separated by TLC, as in A Upper. (C) Proposed structure of SL-1 as determined by Mougous et al. (23) and Goren et al. (25, 26).
Like Δpks2 mutants, each of the mmpL8 transposon mutants and the ΔmmpL8 strain failed to synthesize SL-1 (Fig. 2A, lanes 4–6). This surprising result was confirmed by MS, which demonstrated that SL-1 was absent in extracts from both ΔmmpL8 and Δpks2 mutant cells (data not shown). In contrast to the Δpks2 mutant, however, mmpL8 mutants accumulated a lipid species that co-migrated with the lipid 2 species detected in wild-type cells. This defect was due specifically to disruption of mmpL8 as a single copy of the intact gene fully restored SL-1 synthesis (lane 7). Notably, PDIM synthesis was not perturbed in either mmpL8 or pks2 mutants, suggesting that these genes function specifically in SL-1 and lipid 2 biosynthesis.
Next, we sought to determine whether lipid 2 and SL-1 share common structural features (see Fig. 2C). The observation that lipid 2 is labeled with 14C-propionate suggests that, like SL-1, this lipid also contains methyl-branched fatty acids (Fig. 2A). To determine whether lipid 2 contains a sulfur atom, we labeled both wild-type and ΔmmpL8 cells with 35S-SO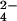 and separated total lipids by TLC. As shown in Fig. 2B (lane 1), SL-1 from wild-type cells is labeled strongly with 35S-sulfur, and over-exposure of the TLC revealed that lipid 2 also contains 35S-sulfur (data not shown). Importantly, the lipid 2 that accumulates in ΔmmpL8 cells also contains 35S-sulfur (lane 2). As expected, Δpks2 cells lack both SL-1 and lipid 2 (lane 3). Taken together, these results confirm that lipid 2 shares significant structural features with SL-1.
and separated total lipids by TLC. As shown in Fig. 2B (lane 1), SL-1 from wild-type cells is labeled strongly with 35S-sulfur, and over-exposure of the TLC revealed that lipid 2 also contains 35S-sulfur (data not shown). Importantly, the lipid 2 that accumulates in ΔmmpL8 cells also contains 35S-sulfur (lane 2). As expected, Δpks2 cells lack both SL-1 and lipid 2 (lane 3). Taken together, these results confirm that lipid 2 shares significant structural features with SL-1.
Mass Spectrometric Analysis of a Novel Lipid in ΔmmpL8 Mutant Cells.
To further characterize the structure of lipid 2, we sought to determine its mass by using FT-ICR MS. Spectra from partially purified lipid 2 were complex, and included numerous lipid-like molecules (data not shown). Lipids can be identified based on a characteristic 14 Da spacing, which reflects heterogeneity in the acyl chain lengths that vary by (CH2)n units. To distinguish the peaks corresponding to lipid 2 from contaminants, we used a novel approach that combines 34S heavy isotopic labeling with high-resolution MS, to identify sulfur-containing compounds from complex mixtures (23). In short, sulfur-containing molecules isolated from cells grown in the presence of 34SO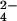 will shift by 2 Da as compared with those grown in the presence of 32SO
will shift by 2 Da as compared with those grown in the presence of 32SO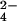 . By using this method on partially purified lipid 2 from mmpL8 mutant cells, a group of lipoforms within the m/z range of 1270–1450 shifted by 2 Da in the 34S-labeled sample indicating the presence of a sulfur atom in the underlying structure (Fig. 3 A and B).
. By using this method on partially purified lipid 2 from mmpL8 mutant cells, a group of lipoforms within the m/z range of 1270–1450 shifted by 2 Da in the 34S-labeled sample indicating the presence of a sulfur atom in the underlying structure (Fig. 3 A and B).
Figure 3.

Structural characterization of lipid 2 by MS is shown. (A) Cells were labeled with 32S-SO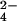 or 34S-SO
or 34S-SO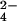 , lipid 2 was partially purified by preparative TLC, and both lipid samples were analyzed by Fourier transform ion cyclotron resonance MS. Comparison of both spectra revealed a single group of species (m/z values denoted above each peak) that shifted by 2 mass units, indicating the presence of one sulfur atom in each molecule. An unrelated, sulfur-free compound within the spectra is marked with an asterisk. (B) A magnified view of the spectra from A that shows two peaks that shifted when cells were labeled with 34S-SO
, lipid 2 was partially purified by preparative TLC, and both lipid samples were analyzed by Fourier transform ion cyclotron resonance MS. Comparison of both spectra revealed a single group of species (m/z values denoted above each peak) that shifted by 2 mass units, indicating the presence of one sulfur atom in each molecule. An unrelated, sulfur-free compound within the spectra is marked with an asterisk. (B) A magnified view of the spectra from A that shows two peaks that shifted when cells were labeled with 34S-SO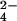 . (C) Proposed structure of the SL-related lipid 2 species, SL1278, as determined by exact mass analysis. The mass measured by MS differs from the calculated mass of the proposed structure by 11 ppm.
. (C) Proposed structure of the SL-related lipid 2 species, SL1278, as determined by exact mass analysis. The mass measured by MS differs from the calculated mass of the proposed structure by 11 ppm.
In an effort to assign an elemental composition to lipid 2, we performed exact mass measurements on the smallest lipoform (m/z 1278, SL1278). The measured mass of SL1278 was determined to be m/z 1,277.91 and is within 11 ppm of the exact mass of a partially acylated sulfated trehalose (Fig. 3C). This structure is consistent with the previously elucidated structure of fully acylated sulfolipid-1 (Fig. 2C). The exact position of the hydroxyphthioceranic acid could not be determined by this approach, but it is likely to be present on either the 6- or 6′-position of trehalose. SL1278 was recently identified as a minor sulfated species found in M. tuberculosis (23), consistent with our finding that SL1278 is present in minor quantities in wild-type cells (Fig. 2A, lane 1).
Biochemical Analysis of Cell Wall Lipids in M. tuberculosis mmpL Mutants.
To determine whether SL1278 is a true biosynthetic precursor of SL-1, we performed pulse–chase experiments. Wild-type and mmpL8 mutant cells were labeled briefly with 14C-propionate, and total lipids were extracted at various time points and analyzed by TLC (Fig. 4A). At early time points, both wild-type and ΔmmpL8 mutant cells were capable of synthesizing significant amounts of SL1278. In wild-type cells, radiolabel appeared in SL-1 at later time points, whereas in mmpL8 mutant cells, radiolabeled SL1278 persisted throughout the time course. Taken together these results strongly suggest that SL1278 is a naturally occurring SL-1 precursor and that MmpL8 is required for SL-1 maturation.
Figure 4.

SL1278 is a biosynthetic precursor of SL-1 and is not exposed on the mycobacterial cell surface. (A) Wild-type (Upper) and ΔmmpL8 mutant (Lower) cells were pulsed labeled with 14C-propionate for 30 min, washed to remove unincorporated label, and grown in liquid media. At the times indicated during the chase, cells were harvested, and lipids were extracted and analyzed by TLC. The ΔmmpL8 sample at t = 60 is underloaded, not deficient in SL1278. (B) Cells were labeled with 14C-propionate for 2 h, and surface-exposed lipids (S) were extracted by using hexanes/decylamine and gentle sonication. Cell pellets (P) containing the lipids not extracted by this procedure were harvested by centrifugation and lipids from both fractions were extracted and analyzed by TLC.
Because MmpL8 is a member of a family of lipid transporters thought to export lipids from the cytoplasmic membrane to the cell wall of M. tuberculosis, we sought to determine the subcellular localization of SL-1 and SL1278. To this end, we developed a gentle fractionation procedure in which surface-exposed lipids were extracted from the outer leaflet of the cell wall, whereas lipids that reside in deeper layers remained cell associated. In brief, radioactively labeled cells were suspended in a mixture of mild organic solvents, gently sonicated, and centrifuged. Comparison of solvent-exposed lipids (supernatant, S) to the total lipid sample (pellet, P) allowed us to identify lipids present on the surface of the cell wall.
Previous work has demonstrated that PDIM is normally located on the cell surface in wild-type cells but present in the cytoplasmic membrane of mmpL7 mutant cells (18, 20). Therefore, to validate our new procedure, we determined the subcellular localization of PDIM in both wild-type and mmpL7 mutant cells (Fig. 4B). As expected, large amounts of PDIM were detected in the supernatant fraction from wild-type cells (Fig. 4B Lower, lanes 1 and 2). The presence of PDIM in the pellet fraction is likely a consequence of the gentle extraction procedure, resulting in incomplete extraction of surface-exposed lipids. Importantly, PDIM was found exclusively in the cell pellet fraction of mmpL7 mutant cells, in agreement with MmpL7's proposed function as a PDIM transporter (Fig. 4B Lower, lanes 3 and 4). This result demonstrates the utility of this assay for probing the exterior portions of the cell wall for the presence of lipids.
Next, we sought to determine the subcellular localization of SL-1 and SL1278 in wild-type and ΔmmpL8 mutant cells. Based on our pulse–chase experiments, we labeled cells for 2 h to visualize both species by TLC. In wild-type cells, SL-1 is found in the supernatant fraction, demonstrating that this lipid is present on the cell surface, whereas SL1278 is never detected on the cell surface (Fig. 4B, lanes 1 and 2). Likewise, in ΔmmpL8 cells, SL1278 is found exclusively in the cell pellet fraction, suggesting that it resides within interior portions of the cell (Fig. 4B, lanes 5 and 6). SL-1 secretion is fully restored in the mmpL8 complemented strain (Fig. 4B, lanes 7 and 8). To ensure that SL1278 is soluble in the extraction solvent, we performed harsher, lytic extraction procedures and found abundant levels of SL1278 in the supernatant fractions from wild-type and ΔmmpL8 mutant cells (data not shown). Importantly, PDIM secretion is normal in ΔmmpL8 mutant cells, whereas SL-1 synthesis and secretion is normal in ΔmmpL7 mutant cells, demonstrating that these two MmpL family members act on distinct lipid substrates.
Role of MmpL8 and SL-1 in M. tuberculosis Infection.
The identification of mmpL8 from our transposon screen suggested that SL-1 is important for M. tuberculosis pathogenesis. To test this notion, we infected mice by the i.v. route with wild-type, Δpks2, and ΔmmpL8 mutant strains and monitored their growth in vivo. Surprisingly, Δpks2 mutant cells grew at rates indistinguishable from that of wild-type cells in the spleen, liver, and lung, indicating that SL-1 is not necessary for M. tuberculosis growth during the innate phase of immunity (Fig. 5 A–C). In marked contrast, ΔmmpL8 mutant cells were attenuated in all of these organs. In the spleen and liver, ΔmmpL8 mutant cells grew initially but, unlike wild-type cells, were rapidly eliminated after the 7-day time point. ΔmmpL8 mutants also showed a subtle yet statistically significant growth defect in the lung that resulted in threefold fewer bacteria by 6 weeks after infection compared with wild-type cells (Fig. 5C). Taken together, these results demonstrate that, although SL-1 is dispensable for M. tuberculosis replication during infection, MmpL8 is required for sustained bacterial growth and persistence.
Figure 5.

mmpL8, but not pks2, is required for growth in a mouse model of tuberculosis. C57/B6 mice were infected by tail-vein injection with 1–2 × 106 cells of wild-type (filled circles), ΔmmpL8 (open circles), or Δpks2 (filled triangles) mutant M. tuberculosis cells. Growth in the spleen (A), liver (B), and lung (C) was monitored by harvesting organs at 1, 7, 14, 21, and 42 days after infection, and bacillary loads were determined by plating dilutions on solid media. Each data point represents the average of colony-forming units from five infected mice, and error bars indicate the standard error of the means. The data shown here is from one experiment and a duplicative experiment gave nearly identical results. *, P value of 0.003.
Discussion
In this study, we identified mmpL8 in an unbiased screen for genes required for M. tuberculosis virulence. Using this same methodology, we had previously identified a homologous lipid transporter, mmpL7, which is required for PDIM secretion and M. tuberculosis virulence (18). Surprisingly, mmpL8 mutants show a dramatic SL-1 synthesis defect, whereas mmpL7 mutants can fully synthesize PDIM and only fail to export it to the cell wall. Furthermore, we have shown that mmpL8 functions in a step downstream of pks2 in the SL-1 biosynthetic pathway, as a partially acylated precursor, termed SL1278, accumulated intracellularly in mmpL8 mutants. Using a powerful new technique to identify sulfated compounds, we were able to assign a molecular structure to this intermediate that is consistent with previous structural analysis of SL-1 (25, 26). Finally, pulse–chase analysis revealed that this is a natural intermediate in wild-type cells, rather than an aberrant by-product of mmpL8 mutation.
These results have allowed us to draw a model of SL-1 biosynthesis and the role of mmpL8 in distal steps of this pathway (Fig. 6). Because pks2 mutants synthesize neither SL-1 nor SL1278, and because SL1278 contains one hydroxyphthioceranic acid, it is likely that Pks2 works in a more proximal step in the pathway. Additional enzymes must also function upstream of MmpL8, as the trehalose head-group of SL1278 has undergone sulfation and two acyltransferase reactions. The enzymes that catalyze these reactions and the order in which they function is unclear. Likely candidates for the sulfation step are one or more of the three sulfotransferases encoded by the M. tuberculosis genome (27). It is also tempting to speculate that the other genes in the mmpL8 genomic region, papA1 or fadD23 (Fig. 1A), may encode the acyltranferases required for the assembly of SL-1.
Why might MmpL8, a predicted lipid transporter, be required for SL-1 biosynthesis? One possibility is that MmpL8, unlike MmpL7, may have some unknown catalytic activity and directly modifies SL-1 during its production. Perhaps more likely is that MmpL8 plays an indirect role in SL-1 biogenesis, consistent with its proposed lipid transport function. As shown in Fig. 6, MmpL8 may flip SL1278 across the plane of the cell membrane, allowing it to be further modified into mature SL-1 before delivery to the cell wall. The enzymes that catalyze these later steps are unknown.
Delivery of lipid intermediates to extracytoplasmic synthases may be a common feature among some members of the MmpL family and homologues in related bacterial species. For example, Mycobacterium smegmatis mutants deficient in TmtpC, a mmpL family member, are unable to synthesize glycopeptidolipid (28). Likewise, when the mmpL homologue, actII-ORF3 is disrupted in Streptomyces coelicolor, actinorhodin synthesis is altered (29). Thus, mmpL7 mutants may still synthesize mature PDIM because the entire synthesis machinery is localized to the cytoplasmic side of the cell membrane.
In addition to MmpL8's role in SL-1 biosynthesis, we have also shown that MmpL8 is required to establish a high-level infection in mice. We initially isolated mmpL8 mutants based on their competitiveness in a pooled infection and subsequently demonstrated that these mutants are defective for growth in all organs tested when infected as a clonal population. Interestingly, pks2 mutants, which are also defective for SL-1 biosynthesis, are not attenuated during mouse infection. Thus, despite previous evidence implicating SL-1 in pathogenesis, our results clearly demonstrate that SL-1 per se is not required for growth of M. tuberculosis during the acute phase of infection in mice. We cannot rule out that SL-1 may be required during chronic infection of mice or perhaps plays a role in human M. tuberculosis infection that is not apparent in the mouse model of tuberculosis. However, the clear defect of mmpL8 mutants suggests that other MmpL8-dependent processes, besides SL-1 synthesis, are important for M. tuberculosis to thrive during innate immune responses.
There are several models that could explain the paradoxical in vivo growth of pks2 and mmpL8 mutants. The simplest model is that MmpL8 may transport other substrates in addition to SL-1, and it is these molecules that are important for pathogenesis. Although we have no direct evidence that argues against this model, using numerous experimental approaches we have not identified additional lipids whose synthesis or secretion is blocked in mmpL8 mutants. A second model is that the accumulation of SL1278 may be generally toxic to the mmpL8 mutant cells. However, this effect would have to be manifest only during infection, as mmpL8 mutants grow with wild-type kinetics in liquid culture. Alternatively, the accumulation of SL1278 in mmpL8 mutant cells may cause a specific inhibition of other pathways required to avert host defenses. For example, the accumulation of the sulfated lipid may inhibit a sulfotransferase, leading to a perturbation in sulfate homeostasis and, as a result, an alteration of host–pathogen interactions. Whether any of these or perhaps other models can explain the phenotypic differences between mmpL8 and pks2 mutants remains unknown.
MmpL8 is the second member of the MmpL lipid tranporter family demonstrated to be required for virulence. It is likely that other members of this family of transporters perform critical functions during M. tuberculosis virulence. Whether the lipids transported by MmpL proteins play only a structural role in the cell wall or perhaps mediate specific host–pathogen interactions remains to be elucidated. In either case, the MmpL family is an attractive target for the design for new antituberculosis drugs.
Acknowledgments
We thank W. R. Jacobs, Jr., for the transposon and phages, L.-M. Ting and L. Quadri for assistance with making the pks2 knockout, S. Stanley and L. Woo for expert assistance with mouse experiments, all members of the Cox laboratory for helpful discussions, A. Sil for critical reading of the manuscript, and all members of the Sil laboratory for helpful discussions. S.E.C. is supported by a National Science Foundation Graduate Research Fellowship. J.S.C. gratefully acknowledges the support of the Pew Scholars Program in the Biomedical Sciences and the Sandler Family Supporting Foundation. This work was supported by National Institutes of Health Grant AI68540.
Abbreviations
- SL-1
sulfolipid-1
- PDIM
phthiocerol dimycocerosate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Russell D G. Nat Rev Mol Cell Biol. 2001;2:569–577. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 2.McKinney J D, Jacobs J, W R, Bloom B R. In: Emerging Infections. Fauci A, Krause R, editors. London: Academic; 1998. pp. 51–146. [Google Scholar]
- 3.Glickman M S, Jacobs W R., Jr Cell. 2001;104:477–485. doi: 10.1016/s0092-8674(01)00236-7. [DOI] [PubMed] [Google Scholar]
- 4.Brennan P J, Nikaido H. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 5.Nikaido H, Kim S H, Rosenberg E Y. Mol Microbiol. 1993;8:1025–1030. doi: 10.1111/j.1365-2958.1993.tb01647.x. [DOI] [PubMed] [Google Scholar]
- 6.Goren M B, Brokl O, Schaefer W B. Infect Immun. 1974;9:150–158. doi: 10.1128/iai.9.1.150-158.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goren M B, Brokl O, Schaefer W B. Infect Immun. 1974;9:142–149. doi: 10.1128/iai.9.1.142-149.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goren M B, D'Arcy Hart P, Young M R, Armstrong J A. Proc Natl Acad Sci USA. 1976;73:2510–2514. doi: 10.1073/pnas.73.7.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pabst M J, Gross J M, Brozna J P, Goren M B. J Immunol. 1988;140:634–640. [PubMed] [Google Scholar]
- 10.Zhang L, English D, Andersen B R. J Immunol. 1991;146:2730–2736. [PubMed] [Google Scholar]
- 11.Zhang L, Goren M B, Holzer T J, Andersen B R. Infect Immun. 1988;56:2876–2883. doi: 10.1128/iai.56.11.2876-2883.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brozna J P, Horan M, Rademacher J M, Pabst K M, Pabst M J. Infect Immun. 1991;59:2542–2548. doi: 10.1128/iai.59.8.2542-2548.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lima V M, Bonato V L, Lima K M, Dos Santos S A, Dos Santos R R, Goncalves E D, Faccioli L H, Brandao I T, Rodrigues-Junior J M, Silva C L. Infect Immun. 2001;69:5305–5312. doi: 10.1128/IAI.69.9.5305-5312.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strohmeier G R, Fenton M J. Microbes Infect. 1999;1:709–717. doi: 10.1016/s1286-4579(99)80072-0. [DOI] [PubMed] [Google Scholar]
- 15.Neill M A, Klebanoff S J. J Exp Med. 1988;167:30–42. doi: 10.1084/jem.167.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng V, Zanazzi G, Timpl R, Talts J F, Salzer J L, Brennan P J, Rambukkana A. Cell. 2000;103:511–524. doi: 10.1016/s0092-8674(00)00142-2. [DOI] [PubMed] [Google Scholar]
- 17.Manca C, Tsenova L, Barry C E, III, Bergtold A, Freeman S, Haslett P A, Musser J M, Freedman V H, Kaplan G. J Immunol. 1999;162:6740–6746. [PubMed] [Google Scholar]
- 18.Cox J S, Chen B, McNeil M, Jacobs W R., Jr Nature. 1999;402:79–83. doi: 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- 19.Camacho L R, Ensergueix D, Perez E, Gicquel B, Guilhot C. Mol Microbiol. 1999;34:257–267. doi: 10.1046/j.1365-2958.1999.01593.x. [DOI] [PubMed] [Google Scholar]
- 20.Camacho L R, Constant P, Raynaud C, Laneelle M A, Triccas J A, Gicquel B, Daffe M, Guilhot C. J Biol Chem. 2001;276:19845–19854. doi: 10.1074/jbc.M100662200. [DOI] [PubMed] [Google Scholar]
- 21.Tekaia F, Gordon S V, Garnier T, Brosch R, Barrell B G, Cole S T. Tuber Lung Dis. 1999;79:329–342. doi: 10.1054/tuld.1999.0220. [DOI] [PubMed] [Google Scholar]
- 22.Sirakova T D, Thirumala A K, Dubey V S, Sprecher H, Kolattukudy P E. J Biol Chem. 2001;276:16833–16839. doi: 10.1074/jbc.M011468200. [DOI] [PubMed] [Google Scholar]
- 23.Mougous J D, Leavell M D, Senaratne R H, Leigh C D, Williams S J, Riley L W, Leary J A, Bertozzi C R. Proc Natl Acad Sci USA. 2002;99:17037–17042. doi: 10.1073/pnas.252514899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glickman M S, Cox J S, Jacobs W R., Jr Mol Cell. 2000;5:717–727. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- 25.Goren M B. Biochim Biophys Acta. 1970;210:127–138. doi: 10.1016/0005-2760(70)90068-8. [DOI] [PubMed] [Google Scholar]
- 26.Goren M B, Brokl O, Das B C, Lederer E. Biochemistry. 1971;10:72–81. doi: 10.1021/bi00777a012. [DOI] [PubMed] [Google Scholar]
- 27.Mougous J D, Green R E, Williams S J, Brenner S E, Bertozzi C R. Chem Biol. 2002;9:767–776. doi: 10.1016/s1074-5521(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 28.Recht J, Martinez A, Torello S, Kolter R. J Bacteriol. 2000;182:4348–4351. doi: 10.1128/jb.182.15.4348-4351.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bystrykh L V, Fernandez-Moreno M A, Herrema J K, Malpartida F, Hopwood D A, Dijkhuizen L. J Bacteriol. 1996;178:2238–2244. doi: 10.1128/jb.178.8.2238-2244.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]