

Abstract
Factors that elevate amyloid-β (Aβ) peptide levels are associated with an increased risk for Alzheimer's disease. Insulysin has been identified as one of several proteases potentially involved in Aβ degradation based on its hydrolysis of Aβ peptides in vitro. In this study, in vivo levels of brain Aβ40 and Aβ42 peptides were found to be increased significantly (1.6- and 1.4-fold, respectively) in an insulysin-deficient gene-trap mouse model. A 6-fold increase in the level of the γ-secretase-generated C-terminal fragment of the Aβ precursor protein in the insulysin-deficient mouse also was found. In mice heterozygous for the insulysin gene trap, in which insulysin activity levels were decreased ≈50%, brain Aβ peptides were increased to levels intermediate between those in wild-type mice and homozygous insulysin gene-trap mice that had no detectable insulysin activity. These findings indicate that there is an inverse correlation between in vivo insulysin activity levels and brain Aβ peptide levels and suggest that modulation of insulysin activity may alter the risk for Alzheimer's disease.
Amyloid-β (Aβ) peptide-containing senile plaques are a prominent feature of the pathology of Alzheimer's disease (AD) and occur consistently in AD of all etiology, from early-onset, familial-linked AD to late-onset AD of indeterminate origin (1). Aβ is formed from the amyloid precursor protein (APP) by sequential enzymatic processing. A β-secretase cleavage first yields the 99-aa C-terminal fragment (CTF) of APP, CTFβ, which then is cleaved by γ-secretase to release Aβ peptides, predominately Aβ40 and Aβ42, and the CTFγ peptides of 49–51 residues (2).
The proteolysis of APP to yield Aβ peptides is a normal physiologic process observed in multiple cell types, although the endogenous function of APP processing and its products is still not well defined (3). To date, all of the genetic mutations linked to AD result in increased Aβ accumulation, albeit by distinct mechanisms.
Although considerable effort has been directed toward generating specific inhibitors of the β- and γ-secretases as a means of preventing Aβ formation (4), the mechanism of Aβ clearance also is of considerable interest because the steady-state concentrations of Aβ peptides are dependent on both their rates of synthesis and their rates of clearance. Recent studies suggest that an important route of Aβ clearance is through hydrolytic cleavage by proteases and peptidases (recently reviewed in refs. 36–38). The peptidase insulysin (EC 3.4.24.56) is one of the enzymes that has been suggested as a candidate Aβ-degrading enzyme primarily based on its ability to degrade Aβ peptides in vitro (5–7).
Insulysin is a zinc metalloprotease, originally identified as an insulin-degrading enzyme (8), that migrates with reported molecular masses of 110–115 kDa on SDS/polyacrylamide gels and has no demonstrated posttranslational modifications. The observed molecular mass of insulysin is consistent with the use of the second of its two potential translation initiation sites, although N-terminal sequencing of authentic insulysin has not been reported. A limited number of peptides in addition to insulin and Aβ also have been identified as insulysin substrates in vitro (9), including β-endorphin (10). To gain insight into the contribution of insulysin to Aβ degradation in vivo, we have examined the steady-state levels of endogenous Aβ peptides in a mouse model in which insulysin gene expression has been disrupted by a gene-trap insertion.
Materials and Methods
Mice.
Omnibank B6.129 mice containing a gene-trap vector VICTR (viral construct for trapping; ref. 11) inserted in intron 1 of the gene encoding insulysin were obtained from Lexicon Genetics (The Woodlands, TX). The standard designation “IDE” that is used to refer to the insulysin-encoding gene is based on the original name for insulysin, insulin-degrading enzyme. Mice heterozygous for the gene trap were bred to produce “knockouts” (IDE−/−) containing the gene trap in both alleles as well as littermate wild-type controls and heterozygotes. The insulysin-deficient mice are healthy and exhibit no overt phenotype in behavior or development, or upon gross examination during necropsy, for at least 10 weeks. Blood glucose and insulin levels in IDE−/− mice determined 4 h after refeeding a standard diet to overnight-fasted animals are not different from those in wild-type littermates, suggesting that insulin degradation and activity are not severely perturbed by the deficiency. Male offspring between 7 and 10 weeks of age were used unless otherwise noted. The mouse genotypes were determined by PCR amplification of DNA that was purified by standard techniques from tail snips with primers to a portion of the VICTR gene-trap sequence (BTK18F, 5′-CCATGGCTCCGGTAGGTCCAG, and BTK84R, 5′-TAATGCAGGTAGCTCCCAGA; 1 min at 94°C followed by 1 min at 60°C for 35 cycles) to distinguish wild-type (IDE+/+) littermates from those carrying either one or two copies of the gene-trap vector. IDE−/+ and IDE−/− mice were distinguished by determining VICTR gene-trap copy number vs. IDE gene copy number by quantitative Southern slot blot of the purified DNA. Serial DNA dilutions (2.0, 1.0, and 0.5 μg) from mice to be genotyped, as well as DNA standards from mice whose genomes contained either two or no copies of the gene trap, were slotted onto duplicate nitrocellulose filters by using a Minifold Slot Blotter II (Schleicher & Schuell). The filters were incubated with hybridization probes to either the neomycin region of the gene-trap vector (corresponding to nucleotides 2996–3271 of pPGKneo-II, GenBank accession no. AF335420) or to IDE (corresponding to nucleotides 1256–1781 of the IDE cDNA sequence, GenBank accession no. BC041675). The probes were synthesized by PCR amplification of cloned sequences, incorporating digoxigenin (DIG)-labeled nucleotides for immunodetection. After washing to remove unbound probe, the blots were incubated sequentially with an alkaline phosphatase-labeled anti-DIG antibody and the chemiluminescent substrate CSPD [disodium 3-(4-methoxyspiro{1,2-dioxetane-3,2′-(5′-chloro)tricyclo[3.3.1.13,7]decan}-4-yl)phenyl phosphate] and exposed to autoradiographic film. All reagents for preparing DIG-labeled probes and for DIG detection were from Roche Molecular Biochemicals. Probe synthesis, hybridization, and detection were performed according to the manufacturer's protocols. Densitometric quantitation of the scanned images was performed with imagequant software (Molecular Dynamics). Confirmation of the genotype by semiquantitative RT-PCR and/or Western blot analysis to measure insulysin mRNA or protein expression in at least one tissue was performed for each mouse by using the techniques described below.
RT-PCR.
Both standard and real-time RT-PCR techniques were used to examine insulysin mRNA levels. For both, total RNA purified by acid/chloroform/phenol extraction (12) was reverse-transcribed with Superscript II (GIBCO/BRL) by using oligo(dT) for priming and following the manufacturer's suggested protocol for RNA messages with a high content of guanine and cytosine residues. Control samples (-RT) were treated identically except that the reverse transcriptase enzyme was not added to the reactions. For semiquantitative comparison within the linear phase of amplification, PCR with the intron-spanning IDE primers 12F, 5′-GGAAGCGTTCGCCGAGATCGCA, and 614R, 5′-TCTGAATCGACAGCGTTCAC, was performed by using a program of 94°C, 55°C, and 72°C for 45 sec each over a range of cycles with a technique we have described previously (13). Dimethyl sulfoxide (0.1%) was added to the amplification reactions to reduce DNA secondary structure. For detection, the PCR products were separated by agarose gel electrophoresis, blotted to nylon membranes, and hybridized with a DIG-labeled probe corresponding to nucleotides 12–614 of the IDE cDNA sequence. PCR of actin from the same reverse-transcribed samples was performed similarly over a range of cycles with DIG nucleotides incorporated during PCR for direct detection as we have described previously (13). PCR reagents were all from Perkin–Elmer. To determine relative insulysin mRNA levels by real-time PCR with the comparative cycle threshold method, the IDE-specific primers ex1/2-F, 5′-CACCTTGCGCTCCATCCT, and ex1/2-R, 5′-GCCGGATTACTCATTGTGCTGTA were used. The PCR products were detected in real time with an IDE-specific Taqman minor groove binder probe, 5′-TGGGAATCCACACAGTC. Insulysin mRNA levels were normalized to 18S rRNA levels that were determined in the same reverse-transcribed samples by using a commercially available 18S rRNA TaqMan assay system (Applied Biosystems) that specifically detects 18S cDNA and not genomic sequences.
Western Blot Analysis of Insulysin.
Two different antibodies to insulysin were elicited by standard techniques in rabbits. The rIDE4020 antibody was elicited by immunization with nickel agarose-purified recombinant rat insulysin with an N-terminal six-histidine tag, which was expressed by a baculovirus system (PharMingen) in Sf9 insect cells. The hIDE7–24 antibody was elicited by immunization with a synthetic peptide that corresponds to amino acids 7–24 (relative to the second ATG) of human insulysin that was coupled to keyhole limpet hemocyanin by a C-terminal cysteine residue (Pierce). Tissue homogenates were prepared in the 10 mM Tris⋅HCl, pH 7.8, lysis buffer that we have described previously (13), which contains protease inhibitors but no detergent, and centrifuged for 10 min at 10,000 × g to remove debris. Protein content of the resulting supernatant fractions was determined by the bicinchoninic acid method (Sigma), using BSA as the standard. Proteins in the supernatant fractions were resolved by electrophoresis in 10% Tris⋅HCl Ready gels (Bio-Rad), using standard sample and electrophoresis buffers containing SDS, and blotted to nitrocellulose (SS Protran; Schleicher & Schuell) electrophoretically. Immunodetection was performed by using the primary antibodies indicated in the figure legends, horseradish peroxidase-conjugated anti-rabbit Ig as the secondary antibody, and the ECL (enhanced chemiluminescence) Western Blotting Analysis System from Amersham Pharmacia. Benchmark prestained protein standards (GIBCO/BRL) were used for molecular weight estimation.
Insulysin Activity Measurements.
Insulysin activity in 10,000 × g supernatant fractions of tissue homogenates prepared in 10 mM potassium phosphate buffer, pH 7.4, containing 0.2 M sucrose was measured by the generation of γ-endorphin from β-endorphin (10). Reaction mixtures of 100 μl containing 30 μM β-endorphin and ≈20 μg of liver extract in 100 mM potassium phosphate buffer, pH 7.4, were incubated for 10 min at 37°C. The reaction was stopped by the addition of 10 μl of 5% trifluoroacetic acid, and the reaction products then were separated by reverse-phase HPLC on a Vydac C4 column (Hesperia, CA), using a linear gradient from 0.1% trifluoroacetic acid in 95% water/5% acetonitrile to 0.1% trifluoroacetic acid in 50% water/50% acetonitrile. Peptides were detected by their absorbance at 214 nm and quantified by peak area.
ELISA Quantitation of Brain Aβ Peptides.
Brains were removed immediately after euthanasia of the mice by CO2 narcosis, frozen in liquid nitrogen, and stored frozen until extraction. Brains were extracted in 0.2% diethylamine in 50 mM NaCl and centrifuged at 20,000 × g for 1 h at 4°C to remove insoluble material. Supernatant fractions were neutralized by the addition of 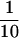 volume of Tris⋅HCl, pH 6.8, and analyzed by sandwich ELISA for Aβ40 and Aβ42 essentially as described previously (14, 15) by using the BNT77/BA27 and BNT77/BC05 antibody systems to detect Aβ40 and Aβ42, respectively. We are grateful to Takeda (Osaka) for their generous gifts of BNT77, BA27, and BC05.
volume of Tris⋅HCl, pH 6.8, and analyzed by sandwich ELISA for Aβ40 and Aβ42 essentially as described previously (14, 15) by using the BNT77/BA27 and BNT77/BC05 antibody systems to detect Aβ40 and Aβ42, respectively. We are grateful to Takeda (Osaka) for their generous gifts of BNT77, BA27, and BC05.
Western Blot Analysis of Brain APP and C-Terminal Fragments.
Equal amounts of protein in neutralized diethylamine brain extracts were resolved by SDS/PAGE on 10–20% Tris-Tricine peptide gels (Bio-Rad) and electroblotted. The O443 antibody against the C-terminal 20 residues of APP (16) was used for immunodetection of APP, CTFβ, and CTFγ by using the chemiluminescent SuperSignal Western blotting system from Pierce. The relative levels of APP and CTFγ were determined by densitometry of the scanned images by using imagequant software (Molecular Dynamics).
Statistical Methods.
The Aβ40 and Aβ42 peptide determinations were analyzed by employing a two-way ANOVA, with IDE allele type and age group as the two factors. The post hoc Student–Newman–Keuls test, with a type I error of 0.05, was applied to allele type to further assess statistically significant results from the ANOVA. The testing was implemented in the SAS (Cary, NC) statistical software package.
Results
To investigate the contribution of insulysin to Aβ degradation in vivo, we examined the steady-state level of endogenous Aβ peptides in mice deficient in insulysin expression. A mouse model in which the IDE gene was disrupted by insertion of a VICTR gene trap into intron 1 was used for these studies. To confirm that insulysin expression was eliminated by this disruption, we initially examined insulysin mRNA and immunoreactive protein levels. This question was of particular importance because intron 1 is upstream of the second of two potential protein translation initiation sites in the IDE gene (17, 18). In mice containing two copies of the gene trap (IDE−/−), no insulysin mRNA was detected in liver, a tissue with a normally high level of insulysin expression, whereas an intermediate decrease was found in heterozygote (IDE+/−) mice (Fig. 1A). An intermediate decrease in immunoreactive insulysin protein also was found in liver from the heterozygote mice, whereas no insulysin protein was detected in the IDE−/− mice (Fig. 1B). In addition to the 113-kDa insulysin protein, the insulysin antibodies also detect a prominent protein band of ≈120 kDa and a less prominent doublet of ≈75 kDa in some tissues in which insulysin is expressed. These latter proteins are not products of the insulysin gene because their presence is not correlated with IDE genotype or with insulysin mRNA or enzyme activity levels.
Figure 1.

Liver insulysin expression in IDE gene-trap mice. (A) Equal amounts of total RNA from sex-matched littermates carrying one (IDE−/+), no (IDE−/−), or two (IDE+/+) copies of the wild-type IDE allele were reverse-transcribed (+RT) or treated identically but without addition of reverse transcriptase (−RT). Portions of the same reverse-transcribed RNA were subjected to PCR with insulysin-specific primers (Top) and β-actin-specific primers (Middle) over a range of cycles (22, 24, and 26 cycles for the +RT insulysin samples and 26 cycles for the −RT insulysin samples; 12, 14, and 16 for the +RT actin and 16 cycles for the −RT actin sample). The insulysin PCR products were displayed by hybridization with a DIG nucleotide-labeled insulysin probe and chemiluminescent detection as described in Materials and Methods. Densitometric quantitation of the insulysin products is displayed in Bottom. (B) Equal amounts of liver protein (10 μg per lane) were resolved by SDS/PAGE and analyzed by Western blotting with an antibody elicited with recombinant rat insulysin. The location of the 113-kDa insulysin protein is indicated.
To confirm that insulysin activity also was absent from the IDE−/− mice, we examined the insulysin-mediated cleavage of β-endorphin to γ-endorphin (10). Insulin was not used as a substrate because insulysin is only one of several enzymes that can degrade insulin, whereas insulysin is the only enzyme known to metabolize β-endorphin to γ-endorphin. As shown in Fig. 2, no activity was detected in the IDE−/− mice, whereas heterozygote animals expressed ≈50% of the activity observed in wild-type mice. In brain, insulysin mRNA levels (Fig. 3A) and immunoreactive protein (Fig. 3B) in heterozygote IDE+/− mice also were approximately half of those detected in wild-type littermate controls, and the γ-endorphin-generating activity was 56 ± 9% of wild type (n = 6 determined from three independent sets of mice consisting of two IDE−/+ and one IDE+/+ sex-matched littermates). No insulysin protein (Fig. 3C) or mRNA (not shown) is detected in brain from IDE−/− mice.
Figure 2.

Liver insulysin activity in IDE gene-trap mice. Shown are HPLC chromatograms of assays of insulysin activity in liver extracts from an IDE+/+ mouse (trace B), an IDE+/− mouse (trace C), and an IDE−/− mouse (trace D), as well as a control incubation with the substrate β-endorphin (β-Ep) alone (trace A). The chromatograms are offset so that all peaks can be seen. Peak a, β-Ep-(17–31); peak b, β-Ep-(18–31); peak c, β-Ep-(1–17) (γ-endorphin), peak d, β-Ep-(1–18); peak e, an unidentified secondary cleavage product.
Figure 3.

Brain insulysin mRNA and immunoreactive protein levels are decreased in heterozygote IDE−/+ mice. (A) Relative insulysin mRNA levels in two sets of mice consisting of one wild-type (WT) and two heterozygote (IDE−/+) sex-matched littermates were determined by real-time PCR using the comparative cycle threshold method, assigning a value of 1 to the WT sample in each set. Insulysin mRNA levels were normalized to 18S rRNA levels in each sample. (B) Two different quantities of brain homogenate protein (10 and 5 μg) from two IDE+/+ and two IDE−/+ mice were resolved by SDS/PAGE and analyzed by Western blotting with the insulysin peptide-specific antibody, hIDE7–24. The blot is representative of three independent comparisons from IDE−/+ and IDE+/+ mice. (C) Equal quantities of brain homogenate protein (30 μg) from an IDE−/− mouse and wild-type, IDE+/+ littermate were resolved by SDS/PAGE and analyzed by Western blotting with the antibody elicited with recombinant rat insulysin.
After characterization of insulysin mRNA, protein, and activity levels in the insulysin gene-trap mice, we examined the effect of absent or diminished insulysin on Aβ accumulation in the brain. Both Aβ40 (Fig. 4A) and Aβ42 (Fig. 4B) were increased, 1.6- and 1.4-fold, respectively, in IDE−/− mice compared with wild-type littermate controls, with means and SDs for the combined age groups of 1.81 ± 0.17 (n = 7) compared with 1.17 ± 0.16 (n = 5) pmol/g, respectively, for Aβ40 and 0.57 ± 0.03 (n = 7) compared with 0.42 ± 0.04 (n = 5) pmol/g, respectively, for Aβ42. Intermediate increases were observed in heterozygous IDE−/+ mice; Aβ40 was 1.57 ± 0.17 pmol/g and Aβ42 was 0.51 ± 0.04 pmol/g with n = 7 for both. Older (20-wk) adult mice had somewhat higher Aβ40 and Aβ42 levels than young (8- to 10-wk) adult mice in all groups (IDE−/−, IDE−/+, and IDE+/+), but similar increases occurred with insulysin deficiency. In both age groups, the mean accumulation of both the Aβ40 and Aβ42 peptides (listed in the legend to Fig. 4) was significantly different among the three groups (IDE−/−, IDE−/+, and IDE+/+) and inversely correlated with copy number of the gene-trap allele.
Figure 4.

Aβ peptide accumulation is increased in insulysin-deficient mice. Aβ40 (A) and Aβ42 (B) were quantified by ELISA in brain diethylamine extracts. Two-way ANOVA revealed a significant effect of both IDE genotype and age on the steady-state levels of Aβ40 (P < 0.0001 for both effects) and Aβ42 (P < 0.001 and P = 0.002, respectively). The post hoc Student–Neuman–Keuls test indicated that, for both peptides, the mean values for the three different genotypes were statistically different from each other. In young, adult (8- to 10-wk) IDE+/+ (n = 3), IDE+/− (n = 3), and IDE−/− (n = 4) mice, the mean Aβ40 levels were 1.05, 1.40, and 1.70 pmol/g, respectively, and the mean Aβ42 levels were 0.39, 0.48, and 0.56 pmol/g. In the older (20-wk) IDE+/+ (n = 2), IDE+/− (n = 4), and IDE−/− (n = 3) mice, the mean Aβ40 levels were 1.36, 1.69, and 1.97 pmol/g, respectively, whereas the mean Aβ42 levels were 0.46, 0.53, and 0.59 pmol/g.
To examine the influence of insulysin activity on APP and other APP metabolites, we examined the steady-state levels of the APP holoprotein and CTFs. No change in the levels of either the APP holoprotein (Fig. 5A) or CTFβ (not shown) was detected in the brains of the insulysin-deficient mice relative to wild-type littermates. There was, however, a robust, ≈6-fold increase in the level of CTFγ, the γ-secretase-generated C-terminal fragment of APP, in the insulysin-deficient mice (Fig. 5B).
Figure 5.

APP levels are unchanged in insulysin-deficient mice, whereas CTFγ fragment levels are elevated. APP (A) and CTFγ (B) were detected by Western blot analysis using the O443 antibody against the C-terminal 20 residues of APP, and their relative levels were determined by densitometry of the scanned images. The values presented for both peptides are the mean ± SEM from eight IDE+/+ and six IDE−/− mice. Representative Western blots are shown in C.
Discussion
The increase in brain Aβ peptide levels observed in insulysin-deficient mice occurred without any change in the concentration of APP or CTFβ, the immediate precursor of the Aβ peptides. These findings provide strong evidence that insulysin degrades Aβ peptides in vivo and contributes to the maintenance of steady-state levels of Aβ in brain. The increased accumulation of Aβ peptides in heterozygous IDE−/+ mice in which insulysin activity is diminished, but not absent, is of particular note because it suggests that the enzyme activity, rather than substrate availability, is rate-limiting in insulysin degradation of Aβ peptides. Thus, polymorphisms that result in even modest decreases in insulysin activity are predicted to result in increased Aβ accumulation. Furthermore, the ≈50% decrease in insulysin mRNA, protein, and activity in the heterozygote IDE+/− gene-trap mice relative to wild-type littermates indicates that there is no compensatory mechanism to increase insulysin expression. The magnitude of Aβ elevation that results from modulation of insulysin activity in this model is likely to pose a significant increase in the risk of AD. The increase in the Aβ peptide levels in the homozygous IDE−/− mice is similar in magnitude to that seen in presenilin mutation models of AD (19, 20), which increase Aβ production and dramatically accelerate the onset of AD-like pathology in APP transgenic mice (21, 22). Because of the long onset in most cases of sporadic AD, the effect of even modest increases in Aβ levels accumulated over time also is thought to be highly significant in the development of AD. Insulysin has been shown in vitro to degrade a limited number of peptides in addition to Aβ (9, 10). Thus, inhibition of insulysin-catalyzed Aβ degradation could occur by substrate competition during chronic elevation of insulin, β-endorphin, or other insulysin substrates, and these conditions also may pose a risk for AD. Conversely, any increase in insulysin activity may result in the decreased accumulation of Aβ peptides.
With this report, insulysin joins neprilysin (23) and, most recently, endothelin-converting enzyme (24) as proteases with a documented in vivo role in regulating steady-state Aβ peptide levels in brain. It is likely that distinct Aβ pools are catabolized by the different enzymes. Both intracellular and extracellular Aβ have been implicated in the etiology of AD. Neprilysin is a membrane-bound ectoenzyme and is suggested to be particularly important in the catabolism of extracellular Aβ (25, 26). Insulysin is predominately a cytosolic protein, but a portion is targeted to peroxisomes and even smaller amounts are found on the plasma membrane or are secreted (7, 27–29). Thus, in principal, both intracellular and extracellular Aβ could be targets of insulysin-catalyzed Aβ degradation. In CHO cells expressing wild-type human APP, insulysin was found to degrade both nonionic detergent-soluble and detergent-insoluble intracellular Aβ (30). Neprilysin was most active in the degradation of extracellular Aβ and had no activity toward the detergent-soluble intracellular Aβ pool.
The additional finding in our studies that CTFγ levels are increased almost 6-fold in the IDE−/− mice is consistent with the recent report that insulysin can degrade CTFγ in vitro (31) and extends the in vitro studies to the in vivo situation in the brain. The much greater increase in steady-state levels of CTFγ in the IDE−/− mice relative to the 1.4- and 1.6-fold increases in the Aβ peptides indicates that insulysin may be more important quantitatively in CTFγ degradation. This finding is consistent with the suggestion that insulysin is one of several enzymes responsible for Aβ degradation. Although the function of CTFγ (also referred to as AICD, APP-intracellular domain) is not yet known, it has been suggested to be a transcriptional regulator (31, 32). Furthermore, smaller, caspase-generated fragments of CTFγ have been shown to be toxic to neurons in vivo (33–35). Thus, an increase in CTFγ levels resulting from insulysin deficiency also may represent a risk for neurologic disease. The effects of the accumulation of CTFγ on brain function and its role in the pathogenesis of AD are important questions for future studies. The insulysin-deficient mouse seems to represent an excellent model system for such studies.
In summary, our studies on the insulysin-deficient mouse provide strong experimental evidence that two metabolites of APP processing, Aβ and CTFγ, are physiologically important substrates for insulysin and are elevated when insulysin activity is diminished. Although the consequences of elevated CTFγ presently are not understood, based on the large body of evidence linking increased Aβ peptide levels and AD risk, these studies suggest a mechanistic link between changes in insulysin activity (genetic or acquired) and an increased risk of late-onset AD.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (National Institute on Drug Abuse DA 08801 to B.C.M., L.B.H., and D.L.T.) and the Alzheimer's Association (to E.A.E., K.S., and L.B.H.), a Mayo Alzheimer's Disease Research Center pilot grant (to E.A.E.), and the Mayo Foundation for Medical Education and Research (to C.B.E.).
Abbreviations
- AD
Alzheimer's disease
- Aβ
amyloid-β
- APP
amyloid precursor protein
- CTF
C-terminal fragment
- DIG
digoxigenin
- IDE
insulin-degrading enzyme
References
- 1.Hardy J, Selkoe D J. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 2.Sambamurti K, Greig N H, Lahiri D K. Neuromol Med. 2002;1:1–31. doi: 10.1385/NMM:1:1:1. [DOI] [PubMed] [Google Scholar]
- 3.Kamal A, Almenar-Queralt A, LeBlanc J F, Roberts E A, Goldstein L S. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 4.Nunan J, Small D H. FEBS Lett. 2000;483:6–10. doi: 10.1016/s0014-5793(00)02076-7. [DOI] [PubMed] [Google Scholar]
- 5.Kurochkin I V, Goto S. FEBS Lett. 1994;345:33–37. doi: 10.1016/0014-5793(94)00387-4. [DOI] [PubMed] [Google Scholar]
- 6.Qiu W Q, Ye Z, Kholodenko D, Seubert P, Selkoe D J. J Biol Chem. 1997;272:6641–6646. doi: 10.1074/jbc.272.10.6641. [DOI] [PubMed] [Google Scholar]
- 7.Qiu W Q, Walsh D M, Ye Z, Vekrellis K, Zhang J, Podlisny M B, Rosner M R, Safavi A, Hersh L B, Selkoe D J. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 8.Mirsky I A. Recent Prog Horm Res. 1957;13:429–435. [PubMed] [Google Scholar]
- 9.Becker A B, Roth R A. Methods Enzymol. 1995;248:693–703. doi: 10.1016/0076-6879(95)48046-3. [DOI] [PubMed] [Google Scholar]
- 10.Safavi A, Miller B C, Cottam L, Hersh L B. Biochemistry. 1996;35:14318–14325. doi: 10.1021/bi960582q. [DOI] [PubMed] [Google Scholar]
- 11.Zambrowicz B P, Friedrich G A, Buxton E C, Lilleberg S L, Person C, Sands A T. Nature. 1998;392:608–611. doi: 10.1038/33423. [DOI] [PubMed] [Google Scholar]
- 12.Chomczynski P, Saachi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 13.Nguyen K, Miller B C. J Immunol. 2002;168:4440–4445. doi: 10.4049/jimmunol.168.9.4440. [DOI] [PubMed] [Google Scholar]
- 14.Duff K, Eckman C, Zehr C, Yu X, Prada C M, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, et al. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 15.Haugabook S J, Le T, Yager D, Zenk B, Healy B M, Eckman E A, Prada C, Younkin L, Murphy P, Pinnix I, et al. FASEB J. 2001;15:16–18. doi: 10.1096/fj.00-0528fje. [DOI] [PubMed] [Google Scholar]
- 16.Pinnix I, Musunuru U, Tun H, Sridharan A, Golde T, Eckman C, Ziani-Cherif C, Onstead L, Sambamurti K. J Biol Chem. 2001;276:481–487. doi: 10.1074/jbc.M005968200. [DOI] [PubMed] [Google Scholar]
- 17.Affholter J A, Fried V A, Roth R A. Science. 1988;242:1415–1418. doi: 10.1126/science.3059494. [DOI] [PubMed] [Google Scholar]
- 18.Baumeister H, Muller D, Rehbein M, Richter D. FEBS Lett. 1993;317:250–254. doi: 10.1016/0014-5793(93)81286-9. [DOI] [PubMed] [Google Scholar]
- 19.Nakano Y, Kondoh G, Kudo T, Imaizumi K, Kato M, Miyazaki J I, Tohyama M, Takeda J, Takeda M. Eur J Neurosci. 1999;11:2577–2581. doi: 10.1046/j.1460-9568.1999.00698.x. [DOI] [PubMed] [Google Scholar]
- 20.Holcomb L, Gordon M N, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, et al. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 21.Borchelt D R, Thinakaran G, Eckman C B, Lee M K, Davenport F, Ratovitsky T, Prada C M, Kim G, Seekins S, Yager D, et al. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 22.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, et al. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 23.Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard N P, Gerard C, Hama E, Lee H J, Saido T C. Science. 2001;292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 24.Eckman E A, Watson M, Marlow L, Sambamurti K, Eckman C B. J Biol Chem. 2003;278:2081–2084. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- 25.Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee H J, Hama E, Sekine-Aizawa Y, et al. Nat Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 26.Fukami S, Watanabe K, Iwata N, Haraoka J, Lu B, Gerard N P, Gerard C, Fraser P, Westaway D, St. George-Hyslop P, et al. Neurosci Res. 2002;43:39–56. doi: 10.1016/s0168-0102(02)00015-9. [DOI] [PubMed] [Google Scholar]
- 27.Seta K A, Roth R A. Biochem Biophys Res Commun. 1997;231:167–171. doi: 10.1006/bbrc.1997.6066. [DOI] [PubMed] [Google Scholar]
- 28.Vekrellis K, Ye Z, Qiu W Q, Walsh D, Hartley D, Chesneau V, Rosner M R, Selkoe D J. J Neurosci. 2000;20:1657–1665. doi: 10.1523/JNEUROSCI.20-05-01657.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller B C, Thiele D, Hersh L B, Cottam G L. Immunopharmacology. 1996;31:151–161. doi: 10.1016/0162-3109(95)00046-1. [DOI] [PubMed] [Google Scholar]
- 30.Sudoh S, Frosch M, Wolf B. Biochemistry. 2002;41:1091–1099. doi: 10.1021/bi011193l. [DOI] [PubMed] [Google Scholar]
- 31.Edbauer D, Willem M, Lammich S, Steiner H, Haass C. J Biol Chem. 2002;277:13389–13393. doi: 10.1074/jbc.M111571200. [DOI] [PubMed] [Google Scholar]
- 32.Cao X, Sudhof T C. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 33.Passer B, Pellegrini L, Russo C, Siegel R M, Lenardo M J, Schettini G, Bachmann M, Tabaton M, D'Adamio L. J Alzheimers Dis. 2000;2:289–301. doi: 10.3233/jad-2000-23-408. [DOI] [PubMed] [Google Scholar]
- 34.Lu D C, Rabizadeh S, Chandra S, Shayya R F, Ellerby L M, Ye X, Salvesen G S, Koo E H, Bredesen D E. Nat Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 35.Bertrand E, Brouillet E, Caille I, Bouillot C, Cole G, Prochiantz A, Allinquant B. Mol Cell Neurosci. 2001;18:503–511. doi: 10.1006/mcne.2001.1030. [DOI] [PubMed] [Google Scholar]
- 36.Selkoe D J. Neuron. 2001;32:177–180. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- 37.Carson J A, Turner A J. J Neurochem. 2002;81:1–8. doi: 10.1046/j.1471-4159.2002.00855.x. [DOI] [PubMed] [Google Scholar]
- 38.Mukherjee A, Hersh L B. J Alzheimers Dis. 2002;4:341–348. doi: 10.3233/jad-2002-4501. [DOI] [PubMed] [Google Scholar]