

Abstract
We conducted a comprehensive genomic analysis of the temporal response of yeast to anaerobiosis (six generations) and subsequent aerobic recovery (≈2 generations) to reveal metabolic-state (galactose versus glucose)-dependent differences in gene network activity and function. Analysis of variance showed that far fewer genes responded (raw P value of ≤10−8) to the O2 shifts in glucose (1,603 genes) than in galactose (2,388 genes). Gene network analysis reveals that this difference is due largely to the failure of “stress”-activated networks controlled by Msn2/4, Fhl1, MCB, SCB, PAC, and RRPE to transiently respond to the shift to anaerobiosis in glucose as they did in galactose. After ≈1 generation of anaerobiosis, the response was similar in both media, beginning with the deactivation of Hap1 and Hap2/3/4/5 networks involved in mitochondrial functions and the concomitant derepression of Rox1-regulated networks for carbohydrate catabolism and redox regulation and ending (≥2 generations) with the activation of Upc2- and Mot3-regulated networks involved in sterol and cell wall homeostasis. The response to reoxygenation was rapid (<5 min) and similar in both media, dominated by Yap1 networks involved in oxidative stress/redox regulation and the concomitant activation of heme-regulated ones. Our analyses revealed extensive networks of genes subject to combinatorial regulation by both heme-dependent (e.g., Hap1, Hap2/3/4/5, Rox1, Mot3, and Upc2) and heme-independent (e.g., Yap1, Skn7, and Puf3) factors under these conditions. We also uncover novel functions for several cis-regulatory sites and trans-acting factors and define functional regulons involved in the physiological acclimatization to changes in oxygen availability.
Although the majority of yeasts cannot grow in the absence of oxygen, many of the Saccharomyces sensu stricto yeasts, as well as other Saccharomyces members, are facultative anaerobes capable of sustained anaerobic growth (3, 4, 75, 86). From genomic comparisons of obligate aerobic and facultative anaerobic yeasts, it would appear that this capacity coincides with a genome duplication event that occurred ≈100 to 150 million years ago in the Saccharomyces lineage (75, 83), followed by the subsequent evolution of new protein variants and the rewiring of transcriptional networks (44, 93). Interestingly, even under oxygen-replete conditions, these Crabtree-positive yeasts preferentially dissimilate hexoses to the C3 and C2 compounds pyruvate and ethanol. This is due in part to the evolution of a glucose repression circuit, which represses the transcription of respiratory genes in the presence of high concentrations of glucose (reviewed in reference 31). Although thermodynamically less efficient, glucose fermentation provides a much higher power output (ATP · min−1 · glycosyl unit−1) than glucose oxidation, which confers an obvious selective advantage to these fast-growing, ethanol-producing yeasts in certain environments (84). In addition to maximizing fermentation capacity, facultative anaerobic yeasts have had to contend with a number of challenges imposed by anaerobiosis, including an inability to synthesize essential cellular components that require molecular oxygen (e.g., sterols and unsaturated fatty acids) and maintaining cellular redox potential and essential mitochondrial functions in the absence of respiration (reviewed in references 57 and 110). These and other physiological, biochemical, and transcriptional programs have been well studied in the budding yeast Saccharomyces cerevisiae.
As oxygen becomes limiting, cells remodel their metabolism. This includes the retooling of catabolic pathways for reliance on strictly fermentative metabolism, rebalancing cellular energy demand with supply, activating redox regulation pathways, up-regulating anaplerotic pathways for maintaining glutamate production, and, after considerable growth, coping with the depletion of essential cellular components that require oxygen for synthesis and mitigating any deleterious effects from anaerobic end product accumulation (reviewed in references 57 and 91). Classical genetic, physiological, and biochemical analyses, as well as more recent microarray studies (6, 58, 82, 98), have focused primarily on the long-term challenges imposed by anaerobiosis. In addition to identifying biochemical mechanisms for coping with these challenges, these studies revealed that physiological acclimatization is initiated at the level of gene expression, with heme playing a pivotal regulatory role (reviewed in references 57 and 110). According to long-standing models (57, 110), normoxic levels of heme are sufficient to activate transcription factors (e.g., Hap1 and Hap2/3/4/5) that control the expression of aerobically expressed genes. When oxygen availability falls below submicromolar concentrations, heme synthesis declines (60), and cellular levels are diluted with continued cell growth. This results in the deactivation of these factors and the down-regulation of the genes they regulate. Targets of these factors include ROX1, encoding a prevalent repressor of anaerobic genes (58, 99), which in turn controls the expression of UPC2 (58), a prevalent activator of anaerobically expressed genes (106). According to this model, oxygen indirectly controls the expression of these genes through heme, which acts as an on/off switch for the expression of aerobic and anaerobic genes. Although this is a simple model that does not take into account additional known regulatory mechanisms (see, e.g., reference 48), it appears to adequately explain the expression patterns of the majority of oxygen-responsive genes. The model also predicts a substantive delay in the response of heme-regulated gene networks following oxygen depletion, a prediction that has been verified for a number of heme-regulated genes (57). However, few studies have examined the dynamics of the response or conducted global analyses of the transcriptional networks involved.
In a recent transcriptomic temporal study (61), we discovered that the short-term (≤2 generations) response to anaerobiosis consists of two distinct phases when cells are grown on a nonrepressing carbon substrate such as galactose, namely, an acute, transitory phase (≈10 to 60 min) followed by a delayed (>60 min) yet apparently chronic phase. Clustering analyses revealed that the first phase is controlled by Msn2/4-, MCB-, SCB-, PAC-, and RRPE-associated networks responsible for retooling metabolism (respirofermentative to strictly fermentative), balancing energy supply and demand, and regulating the G1/S transition of the cell cycle. Interestingly, similar changes in these gene networks are observed when cells encounter a variety of “environmentally stressful” conditions (15, 34). However, this “stress-like” response is absent when cells are shifted to anaerobiosis on the repressing substrate glucose, in which major changes in dissimilatory pathways are not required, nor are changes in growth rate observed. These results suggest that the “stress” encountered is the abrupt cessation of respiration and the associated energetic changes, not the withdrawal of oxygen per se. Indeed, studies in our laboratory (L.-C. Lai, M. T. Kissinger, and K. E. Kwast, unpublished data) and by others (10) have revealed that simply inhibiting the respiratory chain under aerobiosis produces a transcriptional response similar to that elicited by anaerobiosis when cells are grown in galactose.
In both repressing and nonrepressing carbon sources, more chronic changes in gene networks are observed after a substantive delay (more than one generation) (61). Clustering analyses substantiated that, as expected, these networks are largely controlled by heme-responsive transcription factors. The observed changes include the down-regulation of Hap1 and Hap2/3/4/5 networks associated with mitochondrial functions such as respiration and energy metabolism as well as the up-regulation of Rox1 and Upc2 networks involved in diverse cellular functions required for long-term acclimatization to anaerobiosis, notably, sterol homeostasis and cell wall function (61). However, our previous study of the dynamics of the response provided a limited view of the anaerobic networks, given that its focus was on determining the role of Msn2/4 in the acute, transient phase, and it analyzed just five time points over two generations of growth (61). Moreover, most other genomic studies have focused on chronic changes observed under steady-state anaerobic conditions (6, 58, 82, 98). Thus, the goal here was to conduct a comprehensive temporal analysis of the changes in oxygen-responsive gene network activity, sampling 24 time points over nearly eight generations of growth, when cells were shifted to anaerobiosis and back to aerobiosis under both nonrepressing (galactose) and repressing (glucose) conditions. When combined with transcriptional network analyses, this level of sampling has allowed us to reveal dynamical changes in not only heme-responsive networks but also a diverse array of other networks and has revealed novel insights into the physiological remodeling that is required for acclimatization to changes in oxygen availability.
MATERIALS AND METHODS
Media and growth conditions.
Wild-type S. cerevisiae strain JM43 (MATα leu2-3,112 his4-580 trp1-289 ura3-52 [rho+]) (20) was used in this study. Aerobic and anaerobic batch fermentor cultures were grown as described previously (58, 61) in either a semisynthetic galactose or glucose medium containing Tween 80, ergosterol, and silicon antifoam (SSG-TEA and SSD-TEA, respectively) (12). Liquid precultures were grown at 28°C with shaking (200 rpm) and kept in the early- to mid-exponential growth phase (<100 Klett units; optical density at 600 nm of <1.0) for 3 to 4 days prior to inoculating a New Brunswick BioFlo III fermentor (3.5-liter working volume) (58). The fermentor inoculation volume was adjusted so that the cell density upon final harvesting was ≈60 Klett units. Cultures were allowed to acclimate to fermentor conditions for ≥12 h before harvesting the aerobic control and switching the sparge gas from air to 2.5% CO2 in O2-free N2 (1.2 volumes of gas/volume of medium per min) for six generations of anaerobic growth, followed by 1.6 generations of aerobic recovery (air sparged). The dissolved O2 concentration was maintained and monitored as described previously (58, 61).
To compare the responses in cells grown in glucose medium to those of cells grown galactose medium, in which the growth rate is substantively different (61), samples were harvested after the same relative amount of cell growth as assessed by turbidity measurements (Klett meter) following a change in sparging conditions (61). In total, 14 samples (0, 0.04, 0.08, 0.13, 0.19, 0.25, 0.38, 0.5, 1, 2, 3, 4, 5, and 6 generations) were harvested during the shift to anaerobiosis, and 10 samples (0.03, 0.06, 0.1, 0.13, 0.2, 0.3, 0.4, 0.6, 0.8, and 1.6 generations) were harvested after reoxygenating the medium to normoxic conditions. Three batch fermentor experiments were conducted to complete the full time series, using the samples at zero and six generations to link the subsets, and the full time series was repeated in triplicate for each medium. As mentioned above, cells were kept at low densities to minimize any effects due to changing resource availability during the time courses. Cells were harvested, using a rapid vacuum filtration apparatus (13), onto AcetatePlus membranes (ISC BioExpress, Kaysville, UT) as described previously (61). The filtered cells were washed with either sterile deoxygenated or oxygenated water (as appropriate), flash frozen in liquid N2 within 1 min of initiating the sampling, and stored at −80°C for later RNA isolation.
RNA extraction, cDNA synthesis, and microarray hybridization.
Total RNA was extracted from the filtered cells using hot phenol as described previously (13). Thirty micrograms of total RNA was used for first-strand cDNA synthesis, and microarray target preparation was performed as described elsewhere previously (61). A reference design was used for microarray hybridizations. The references consisted of a pool of equal masses of RNA collected from each time point sampled in galactose or glucose medium. Microarray hybridization, washing, and scanning were conducted as described previously (61). The custom microarrays consisted of the Operon yeast genomic 70-mer oligonucleotide set (version 1.1; QIAGEN, Valencia, CA) spotted in duplicate at a concentration of 20 μM in 150 mM sodium phosphate (pH 8.5), 10 Arabidopsis oligonucleotide spike controls (SpotReport; Stratagene, La Jolla, CA) spotted in quadruplicate, and 10 human and 10 yeast oligonucleotide negative controls spotted in duplicate. The oligonucleotides were printed on Codelink slides (Amersham, Piscataway, NJ) by Microarrays, Inc. (Nashville, TN). Postprint processing was conducted according to the manufacturer's recommendations.
Microarray and statistical analyses.
Data were analyzed as described previously (61). In brief, GenePix Pro software (v4.1) was used for spot identification and fluorescence intensity quantification. After manually flagging and removing spots with aberrant measurements due to array artifacts or poor quality, background fluorescence was subtracted from the median Cy3 and median Cy5 fluorescence intensity values. Any resulting negative intensity values were set to zero, and a constant of 1 fluorescent unit was then added to all intensity values. Outliers were identified and removed using SAS software (SAS Institute Inc., Cary, NC) as described previously (61), and the log2 Cy3 intensity (query cDNA) for all remaining observations on a slide was normalized against the log2 Cy5 intensity (reference cDNA) using locally weighted linear regression (Loess). The linearity of the resulting Cy3 and Cy5 intensities across each slide was compared to that for the Arabidopsis spike controls (fluorescence intensity versus spike mRNA amount [ranging from 0.02 to 2 ng]) (61) and was corrected if necessary (there were zero occurrences here). The log2(Cy3/Cy5) ratio for each spot was calculated, and the mean log2(Cy3/Cy5) ratio across all observations on a slide was normalized to a value of zero. The mean of the normalized log2(Cy3/Cy5) ratio for each gene was then calculated by averaging the duplicate observations on each slide and pooling replicate slides by medium and sampling time.
Statistical analyses were performed as a two-factor analysis of variance (ANOVA) using the SAS MIXED procedure with repeated measures (SAS Institute Inc., Cary, NC). The factors were medium (galactose or glucose) and generation (0 [aerobic control], 0.04, 0.08, 0.13, 0.19, 0.25, 0.38, 0.5, 1, 2, 3, 4, 5, or 6 generations for the anaerobic response and 0 [anaerobic sample after six generations], 0.03, 0.06, 0.1, 0.13, 0.2, 0.3, 0.4, 0.6, 0.8, or 1.6 generations for aerobic recovery). Separate statistical models were run for the response to anaerobiosis and for recovery in each medium. A post hoc step-down Bonferroni P value adjustment was used to minimize the false discovery rate. Postmodel analyses included motif searches using both upstream (−1 to −800 bp) and downstream (+1 to +200 bp) sequences. Several bioinformatic computational programs were used, including regulatory sequence analysis tools (101), MDscan (67), MEME (5), CompareACE (40), and FunSpec (89).
Data clustering and gene network discovery.
The temporal profiles in gene expression were clustered as described previously (61). In brief, the temporal signatures were unbiasedly clustered 10 separate times with a self-organizing map (SOM) algorithm (one-dimensional [1D] ring topology, Pearson correlation) using a range of K values (cluster numbers) from 2 to 50. Two quality assessment metrics were calculated from the results obtained to determine an appropriate K value for recovering the gene network structure: consensus share (CS) and the feature (motif) configuration statistic (FCS) (61). Note that to avoid confusion with the motif conservation statistic presented previously by Kellis et al. (50), henceforth, we refer to what was originally presented as the motif configuration statistic (61) as the FCS to reflect the general applicability of using this metric to assess the configuration of any features, in this case, transcription factor binding motifs. CS is the percentage of genes (not gene pairs) that were consistently grouped together over 10 replicate clusterings using random seeding for initiating the SOM; it provides an indication of the robustness of the clustering results and the extent of structure in the data as a function of K. Genes that were not consistently grouped together over 10 replicate clusterings for a given K value were placed in a separate category and excluded from FCS calculations. FCS is the probability that the observed configuration of a transcription factor motif (TFM) among gene clusters arose by chance alone from the multinomial distribution dictated by cluster sizes (61). In total, we examined the distribution of a compiled list of 2,603 consensus binding sequences (see Table S1 in the supplemental material) (1, 2, 7-9, 11, 16-18, 21, 22, 24, 26, 28, 33, 35, 36, 38, 39, 42, 45, 47, 50-52, 55, 56, 59, 62-66, 68-71, 73, 74, 76-78, 81, 85, 92, 95-97, 100, 102) among the gene clusters generated for each value of K and compared this configuration to that generated by randomly distributing the observed motif counts 106 replicate times among genes within gene clusters by using a Monte Carlo approach (61). An average FCS P value for all TFMs was then calculated for each value of K. By comparing the values of FCS and CS over a range of K values (2 to 50 here), we determined the value of K for which the algorithm consistently groups the temporal profiles (high CS value) in a manner that results in the least probable configuration of TFMs among gene clusters (lowest FCS P value). Additional details of this approach are described elsewhere (61; A. L. Kosorukoff and K. E. Kwast, unpublished data).
In addition to examining the configuration of consensus binding sequences among gene clusters and calculating their hypergeometric enrichment P values, we also used MDscan (67) to identify additional overrepresented sequences in each of the gene clusters. For training, we used a set of ≈30 expression profiles, but no more than 50% of any cluster, that were closest to the mean expression profile in each cluster. CompareACE (40) was used to calculate similarity indices of identified sequences for known transcription factor binding site matrices (38, 40). FunSpec (89) was used to calculate hypergeometric P values for enriched MIPS (Munich Information Center for Protein Sequences; http://mips.gsf.de/genre/proj/yeast) functional categories in each gene cluster or for subgroups of genes identified with MDscan.
Microarray accession numbers.
Microarray data reported in this paper are available in the GEO database under accession numbers GSE2246 for the full galactose data set and GSE2267 for the full glucose data set.
RESULTS
Dynamics of the response to anaerobiosis.
As shown in Fig. 1, several minutes are required to purge O2 from the medium after switching the sparge gas from air to 2.5% CO2 in O2-free N2 (left panel) and to resaturate the medium to normoxic conditions after switching back to air (right panel). Previous research (57) has shown that 1 μM O2 is a critical threshold for the expression of a large number of aerobic and hypoxic genes, with maximal expression of the former above this value and that of the latter below this value. This concentration is reached roughly 10 min after the shift to N2, corresponding to about 0.04 generations in galactose and 0.08 generations in glucose, and within 5 min after reoxygenation, corresponding to about 0.03 generations in galactose and 0.04 generations in glucose.
FIG. 1.

Transient changes in oxygen concentrations during the shift to anaerobiosis (left panel) and the shift back to aerobiosis (right panel). The change in the dissolved O2 concentration (μM) is plotted as a function of time over the first 10 minutes after switching the sparge gas from air to 2.5% CO2 in O2-free N2 (left panel) and then back to air after 24 h of anaerobiosis (right panel). The O2 concentration was calculated from the dissolved oxygen level measured with a 12-mm Ingold polarographic O2 sensor and is based upon the solubility of O2 in the media at 28°C and ambient barometric pressure.
Statistical analyses of the expression changes (ANOVA using the SAS MIXED procedure with repeated measures) revealed a remarkable 2,388 genes whose transcript levels responded significantly (P < 0.01) to shifts in oxygen availability at one or more time points in galactose medium (complete results are provided in Table S2 in the supplemental material and under GEO accession number GSE2246). Note that a post hoc step-down Bonferroni procedure was used to minimize the false discovery rate, which is a stringent cut of the data, with an adjusted P value of 0.01 corresponding to a raw P value of ≈10−8 and a minimum average expression level difference of 1.75-fold. Of these genes, 2,092 responded significantly to the shift to anaerobiosis (1,102 down-regulated, 853 up-regulated, and 45 both up- and down-regulated at different time points), and 1,218 responded significantly to the shift back to aerobiosis (682 up-regulated, 491 down-regulated, and 45 both up- and down-regulated). In glucose, far fewer genes (1,603 in total) responded to the shifts: 1,337 genes responded to the shift to anaerobiosis (560 up-regulated, 733 down-regulated, and 44 both up- and down-regulated), and 991 genes responded to reoxygenation (594 up-regulated, 372 down-regulated, and 25 both up- and down-regulated) (complete results are provided in Table S3 in the supplemental material and in the GEO database under accession number GSE2267).
From these analyses, it is clear that a large fraction of the S. cerevisiae genome is oxygen responsive and that the metabolic state of the cell (i.e., glucose fermentation versus galactose mixed respirofermentation) greatly influences the genes that respond. The results also suggest that more genes respond to the shift to anaerobiosis than to the subsequent shift back to aerobiosis. This is due, in part, to the fact that transcript levels of some genes had not yet returned to their preanaerobic (i.e., steady-state aerobic) levels by the end of the time course (1.6 generations of aerobic recovery), and fewer time points were collected during aerobic recovery (10 compared to 14 for anaerobiosis), which reduces the statistical power for resolving differences. Regardless, given that the goal of this study was to identify oxygen-responsive gene networks, all genes that responded significantly to either shift within a given medium were pooled for gene network analyses.
With respect to overall carbon source-dependent differences, Fig. 2 compares the genes that responded significantly to the oxygen shifts in each medium. Although the majority of genes are common to both sets, a large fraction (nearly half) of those identified in galactose are unique to this medium, whereas a much smaller fraction (one-fifth) are unique to glucose. Of the 1,296 genes in common, a surprising 66% (724/1,296) were found to have a significant (P < 0.01) medium effect as assessed by ANOVA. However, examination of the gross patterns of up- and down-regulation with respect to changes in oxygen availability revealed that the majority of genes (456/724) exhibited similar expression patterns in both media. Thus, many of the medium effects uncovered are due to fine-scale temporal differences in the responses of genes in the two media.
FIG. 2.
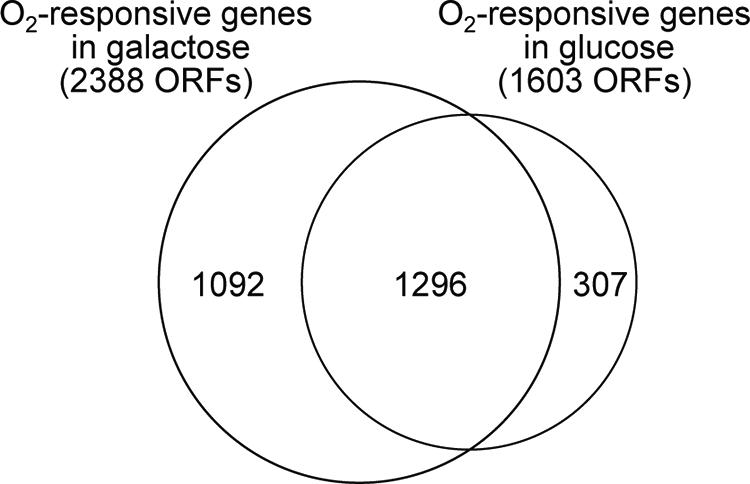
Comparison of oxygen-responsive genes identified in galactose and glucose media. The figure shows the overlap in genes that were found to respond significantly (P < 0.01) to the shifts in oxygen availability in galactose and glucose media. ORFs, open reading frames.
Figure 3 shows the overall dynamics of the response in each medium, with black bars indicating genes that responded to the shifts for the first time at the indicated time point and gray bars indicating those that had responded at an earlier time point but that continued to be differentially expressed with respect to the controls. From Fig. 3, it is clear that the dynamics of the response to reoxygenation (B and D) are similar in the two media but that the response to anaerobiosis is not (A and C). As shown in Fig. 3A, the anaerobic response in galactose is biphasic. The first phase is comprised of a large set of genes (1,068 of 2,092 genes) that exhibit an acute (0.04 to 0.08 generations) yet transient (<0.38 generations) response, with maximum numbers of newly responding genes (Fig. 3A, black bars) appearing at 0.08 generations. After a considerable delay (≥0.25 generations), a second, smaller set of genes (544 genes) was then differentially expressed, most for the duration of anaerobiosis (six generations). Peak numbers of newly responding genes in this phase appeared after two generations of anaerobic growth. In contrast, the anaerobic response in glucose was largely monophasic (Fig. 3C), with increasing numbers of newly responding genes up to three generations. As in galactose, most of these genes continued to be differentially expressed for the duration of anoxia. In comparison, the response to reoxygenation was more rapid and similar in the two media, with maximal numbers of newly responding genes appearing between 0.06 and 0.2 generations (Fig. 3B and D). Most of these genes continued to be differentially expressed for the duration of the time course. Although similar carbon source-dependent differences in the acute response to anaerobiosis were noted in our previous study (61), which examined the samples at 0, 0.04, 0.08, 0.19, and 2 generations, a much clearer picture of both the transient and chronic anaerobic response is afforded by the large number of time points (24 in total) examined in this study and the increased statistical power that they afford in resolving transcript differences by ANOVA. Moreover, unlike the response to anaerobiosis, these results suggest that the metabolic state of the cell has less of an overall effect on the dynamics of the response to reoxygenation. To further dissect the dynamics of these responses, we separately clustered the temporal signatures in the two media using a novel approach (61) to recover the gene networks involved.
FIG. 3.

Dynamics of oxygen-responsive gene induction and repression during acclimatization to anaerobiosis and subsequent recovery. The numbers of genes that responded significantly (P < 0.01) to the shifts in O2 availability in galactose (A and B) and glucose (C and D) media are plotted as a function of time (generations) after the shifts. Genes are divided into those that were significantly up-regulated and those that were significantly down-regulated. Black bars indicate the number of genes that were identified for the first time at that time point to exhibit a significant change in expression from that of the aerobic (A and C) or anaerobic (B and D) controls. Gray bars indicate the number of genes that were differentially expressed in the sample but that had already been identified to have responded significantly to the shift in O2 concentration at an earlier time point. The combined height of the black and gray bars is the total number of genes at each time point that showed a significant difference in expression relative to controls.
Gene network analyses.
To identify the networks, we separately clustered the temporal profiles of O2-reponsive genes identified in each medium (2,388 genes for galactose and 1,603 genes for glucose) using a number of different clustering algorithms, distant metrics, and cluster numbers (K). From the results obtained, we determined the most appropriate approach by evaluating clustering quality using two metrics: CS and FCS (61). CS is the percentage of genes that were identically clustered over replicate runs (n = 10) of an algorithm using a random seed for initiation, whereas FCS is the probability that the observed configuration of transcription factor motifs (2,603 TFMs in total) (see Table S1 in the supplemental material) among gene clusters arose by chance alone from their multinomial distributions dictated by cluster sizes (61). High CS values are indicative of robust clustering results, and low FCS values are indicative of highly improbable motif configurations based on chance alone. By comparing the results for CS and FCS for different approaches, we can determine the most suitable combination of algorithms, distance metrics, and number of clusters for recovering gene network structure.
Oxygen-responsive gene networks identified in galactose medium.
A comparison of the performance of K means, K medoids, and SOM using Manhattan, Euclidean, Sup, and correlation as distance metrics indicated that the SOM algorithm with one-dimensional ring topology and standard correlation produces superior results (data not shown; see reference 61). Figure 4 shows the quality of the SOM clustering results as assessed by CS and FCS for values of K ranging from 2 to 50. From this figure, it is clear that far more structure is recovered from the temporal profiles for K values of <28, as evidenced by the precipitous decrease in CS (Fig. 4, dashed line, right ordinate) and the corresponding increase in FCS (solid line, left ordinate) with increasing K values above 28. A sharp fall in CS and a rise in FCS is predicted as the number of clusters allowed exceeds that supported by the structure contained within the temporal profiles. In addition, the substantive variability observed in CS and FCS for K values between 2 and 28 might also be expected as the algorithm partitions the temporal profiles for K values that are either supported by the underlying network structure (high CS and low FCS) or are unsupported (low CS and high FCS). For a K value of 18, the temporal profiles are robustly partitioned (CS = 97%) in a manner that results in the least probable configuration of TFMs among gene clusters compared to random chance (i.e., the minimum average FCS P value [0.31]). Thus, 18 is the optimum K value for the criteria selected, and we discuss the recovered networks in the sections that follow.
FIG. 4.
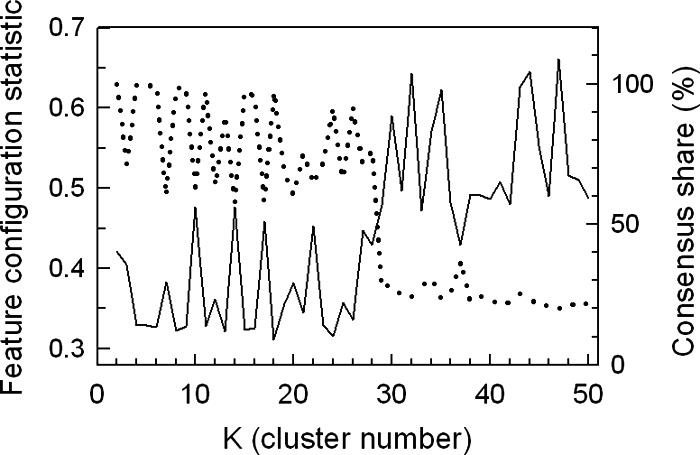
Assessment of clustering quality using the FCS and CS for oxygen-responsive genes identified in galactose medium. The temporal profiles of genes that responded significantly (P < 0.01) to the shifts in oxygen availability in galactose medium (SSG-TEA) were clustered 10 times using an SOM algorithm with 1D ring topology and Pearson correlation as the distance metric. The average FCS P values (solid line, left ordinate) for 2,603 transcription factor consensus binding sequences (TFMs) and CS (dotted line, right ordinate) are plotted as a function of cluster number (K). CS is the percentage of genes that were consistently grouped together over 10 runs of the algorithm. FCS is the probability that the observed configuration of TFMs among gene clusters arose by chance alone from the multinomial distribution dictated by cluster sizes.
As shown in the heat map of the clustered profiles (Fig. 5), the SOM algorithm nicely partitions these signatures into temporally shifted groups beginning with those that were acutely down-regulated in N2 (galactose cluster 1 [Cg1] to Cg6), followed by those with a more delayed response (Cg7 to Cg9). These groups are followed by clusters of genes whose expression was also up-regulated in air (Cg10 to Cg12), those that were acutely up-regulated in N2 (Cg13 to Cg15), and, finally, groups that exhibited an increasing delay in their anaerobic up-regulation (Cg16 to Cg18). Because of the 1D ring topology, the temporal signatures of genes in adjacent clusters are most similar to each other, with Cg18 to Cg1 serving to close the ring. The 64 profiles shown in Cg0 are genes that were not consistently placed in the same cluster over 10 replicate runs of the SOM algorithm, and thus, they were excluded from FCS calculations. In regard to statistical evaluations, the right panel of Fig. 5 shows the same temporal profiles as the left panel but with an overlay that masks gene expression changes that were not found to be significantly different from their respective controls by ANOVA (aerobic sample for anaerobiosis and sixth-generation anaerobic sample for aerobiosis). Finally, in regard to the recovered transcriptional networks, Table 1 is a partial list of TFMs and MIPS functional categories that were significantly [−log10(P) ≥ 2; i.e., P ≤ 0.01] enriched in each of the clusters (complete results and gene-to-cluster membership are provided in Table S2 of the supplemental material). Table 1 also shows the sequence logos from MDscan (67) and hypergeometric P values for MIPS functional category enrichment. Given that our approach first clusters the gene expression profiles based solely on a correlative index of their temporal similarity and then determines what value of K results in the least probable configuration of TFMs among clusters (lowest FCS P value), it is reasonable to presume that individual clusters represent distinct gene networks controlled by specific trans-acting factors. Thus, we discuss each of the clusters separately below and then compare these networks to those identified from clustering the profiles of genes that significantly responded to the oxygen shifts in glucose medium.
FIG. 5.

Heat maps and statistical comparisons of oxygen-responsive genes identified in galactose medium. The temporal profiles of genes that responded significantly (P < 0.01) to the shift to either anaerobiosis or aerobiosis in SSG-TEA medium were clustered using an SOM algorithm with 1D ring topology (K = 18). The left panel shows the temporal signatures, and the right panel shows the same temporal signatures but with a statistical overlay that masks gene expression changes that were not significantly (P > 0.01) different from the controls (aerobic sample for anaerobiosis and sixth-generation anaerobic sample for aerobiosis). Cluster 0 contains genes that were not consistently placed in the same cluster over 10 replicate runs of the SOM algorithm. Green indicates down-regulated expression, and red indicates up-regulated expression. Bars to the right of the heat map indicate genes that also responded significantly (P < 0.01) to the shift in O2 availability in glucose medium.
TABLE 1.
Selected list of enriched consensus sequence motifs (TFMs), MDscan sequence logos, and MIPS functional categories in clusters of genes differentially expressed in response to O2 availability in galactose mediume
 |
TABLE 1.
Continued
 |
TABLE 1.
Continued
 |
P value [−log10(P)] for TFM enrichment relative to clustered genes (2,388)/P value [−log10(P)] for TFM enrichment relative to the genome.
P value [−log10(P)] for MIPS functional category enrichment.
STRE (AGGGG) has been listed as the binding site for Msn2, Msn4, Gis1, and/or Rph1 by different authors (see citations above).
See Table S2 in the supplemental material for an explanation of numbered motifs. F, functional motif; C, coherent motif (17).
SOM clustering with 1D ring topology (K = 18).
Networks transiently down-regulated during anaerobiosis in galactose.
Cg1 contains genes that were acutely (0.04 generations) yet transiently (<0.025 generations) down-regulated in N2 and then chronically up-regulated after a delay (≥0.38 generations) (Fig. 5). Examination of the statistical overlay heat map (right panel of Fig. 5) indicates that few of these genes responded significantly (P < 0.01) to reoxygenation. In terms of function (Table 1), many genes are involved in amino acid (e.g., Arg, Ile, Lys, Thr, Pro, Met, and Gly) and fatty acid metabolism, and motifs (e.g., F6 [AAAGAAA]) (17) or transcription factor binding sites (e.g., ARO80 and BAS1) associated with such genes were significantly enriched. The inhibition of respiration leads to a substantial restructuring of pathways for amino acid biosynthesis and nitrogen metabolism, particularly through Hap2/3/4/5- and Rtg1/3-regulated networks that affect peroxisome function, the glyoxylate cycle, anaplerotic pathways, and portions of the tricarboxylic acid (TCA) cycle leading to glutamate synthesis (reviewed in reference 14). The delayed up-regulation of many of these genes may be associated with such functions, whereas their transient down-regulation is most likely part of an energy-balancing measure elicited during the slowing of growth and the switch to strictly fermentative metabolism on galactose (61). In support of the latter, several TFMs associated with cell cycle regulation, including NRG1, SCB, SWI4, and SWI6 (see Table S2 in the supplemental material for a full list), were significantly enriched, which would provide a link to their acute down-regulation during the slowing of the cell cycle under anaerobiosis. Moreover, few of these transiently responding genes were down-regulated during the transition to anaerobiosis in glucose (discussed below), conditions for which there is no evidence of perturbation of the cell cycle (61).
Cg2 to Cg5 contain the majority of genes that were acutely yet transiently down-regulated in response to anaerobiosis (Fig. 5). Although many genes appear to respond to reoxygenation in a similar manner, few of the observed changes were statistically significant (Fig. 5, right panel). The response of most of these genes was unique to galactose medium, as indicated by the bars on the right side of Fig. 5 (O2 effect in glucose [Glu]). The first genes to respond (Cg2) were those involved in DNA processing, recombination, repair, and other processes associated with the G1/S transition of the cell cycle (Table 1). Remarkably, nearly 40% of these genes have been characterized as G1 specific in previous microarray studies of the cell cycle (96). Predictably, the vast majority of the genes contain TFMs (e.g., FHL1, MCB, SCB, SWI4, SWI6, and MBP1) (see Table S2 in the supplemental material for a full list) for factors associated with G1/S. Members of this cluster include major regulators of G1 (e.g., CDC45, CLB5, CLB6, and CLN1) and genes involved in chromatin remodeling, chromosome replication, DNA replication/repair, checkpoint function, and bud site selection/emergence (see Table S2 in the supplemental material). From a functional viewpoint, the down-regulation of G1-specific genes and a delay in the G1/S transition (i.e., before START) is predictable, given that the acute withdrawal of oxygen results in an abrupt decrease in ATP production during the cessation of respiration, and mass and energy need to be reassessed before committing to another round of the cell cycle (61). The response is also predictably transient, given that the cells quickly (∼1 h) (data not shown) reach a new steady-state growth rate supported solely by galactose fermentation.
The response of genes in Cg3 is similar to that of genes in Cg2 but is shifted to slightly later times (Fig. 5). Given this slight temporal difference, it is remarkable how clearly delineated these two gene clusters are in terms of both function and regulation (Table 1). Most genes in Cg3 contain PAC, ABF1, SCB, and/or RRPE motifs and are involved in early steps of cytoplasmic ribosomal biogenesis, particularly rRNA and tRNA synthesis/processing. Similarly, the response of genes in Cg4 is almost identical to that of genes in Cg3, yet a remarkable 87% of genes contain binding sites for Fhl1, sites that are not enriched in Cg3. Given this enrichment, it is not surprising to find that a large number of these genes (62 in total) encode structural constituents of the cytoplasmic ribosomes. Other genes are involved in translation and/or ribosomal function, including the processing of both 20S and 27S pre-rRNAs and 35S primary transcripts, as well as in initiating translation (e.g., FUN12, GCD11, HCR1, RPG1, SUI1, TIF3, TIF34, and TIF4631). Finally, whereas the acute response of genes in Cg5 is similar to that of genes in Cg4, many genes in Cg5 exhibit delayed chronic down-regulation under anaerobiosis. A large number of genes are also involved in ribosome biogenesis, and about half of the genes contain an FHL1-like motif as identified by MDscan. In addition, members include a small group of genes involved in purine and pyrimidine anabolism (e.g., ADE4, ADE5/7, ADE6, FUR1, HPT1, MTD1, and URA4) that were significantly up-regulated during aerobic recovery.
Overall, despite very little difference in the temporal response of these transiently down-regulated genes in Cg2 to Cg5, it is clear from the differential enrichment of TFMs and MIPS functional categories that our clustering approach divides them into distinct gene networks. As discussed in more detail below, many of these gene networks respond to “environmentally stressful” conditions in a similar manner (15, 34). However, here, it is clear that they respond to the abrupt cessation of galactose-supported respiration and associated energetic changes and not the withdrawal of oxygen per se, given that they fail to respond to the anaerobic shift in glucose medium (see O2 effect in Glu in Fig. 5 and Discussion). Thus, as was proposed previously (61), the transient repression of Fhl1-regulated genes involved in ribosomal function as well as PAC- and RRPE-regulated networks involved in rRNA and tRNA processing/transcription appear to be associated with reducing the energy demand as part of a balancing measure elicited during the cessation of respiration and the switch to strictly fermentative growth. Moreover, the transient energetic crisis apparently results in the down-regulation of MCB and SCB networks and a delay in the progression of the cell cycle at G1 as energy and mass are reassessed before committing to another round.
Networks chronically down-regulated during anaerobiosis in galactose.
Cg6 contains genes that were acutely down-regulated for the duration of anaerobiosis (Fig. 5). Given this response, many genes are predictably associated with mitochondrial function including transport, genome stability, metal ion homeostasis, and adenosine anabolism. Surprisingly, the most prevalent TFMs were those for Skn7, which is involved in the response to oxidative stress, hyperosmolarity, and heat shock (87). Given that this factor's function, along with Yap1, is to mount an oxidative stress response when electron flow is inhibited (23), it seems unlikely that it is the predominant regulator of this cluster or that it couples in a novel way to other factors in perhaps a redox-dependent manner. With the exception of several genes involved in adenosine anabolism (ADE1, ADE12, ADE13, and ADE17) and glycine metabolism (GCV1, GCV2, GCV3, and SHM2), few of these genes were significantly up-regulated during aerobic recovery. MDscan showed an STRE-like motif (correlation = 0.9), which is found in about half of the genes. However, the consensus sequence is unlike any TFM in databases we have searched. Thus, the factor(s) responsible for down-regulating these genes during anaerobiosis is not readily apparent from these analyses.
Genes in Cg7 were transiently up-regulated and then chronically down-regulated for the duration of anaerobiosis, with many genes exhibiting a slow return to preanoxic levels after reoxygenation. Nearly all the genes are involved in mitochondrial function and particularly in protein synthesis and processing, including import, folding, secretion, and targeting. Members of this cluster include 75% of the genes that encode structural constituents of the mitochondrial ribosomes. Despite this tight functional clustering, however, the trans-acting factor(s) responsible for this response is not readily apparent. The expression profiles suggest two distinct regulatory phases. Given the frequency of occurrence and enrichment P values for ADR1 and HSF1 motifs (Table 1), it is possible that these factors play a role in their transient up-regulation, but they are unlikely candidates for down-regulating these genes under anaerobiosis. Rather, delayed yet chronic anaerobic down-regulation is a temporal signature reminiscent of positive regulation by heme even though no associated TFMs are significantly enriched. The absence of identifiable 5′ cis-regulatory sites has been noted previously for similar sets of presumably coregulated genes involved in mitochondrial protein synthesis and related functions (40, 41, 44). Interestingly, the most significantly (P < 10−32) enriched motifs were 3′-untranslated sites for Puf3, an mRNA-binding protein that regulates translation and mRNA decay (46), and 40% of these genes have been shown previously to be regulated by this factor (35). Recent phylogenomic analyses of a number of sequenced yeasts have shed some light on the regulation of these genes, suggesting an apparent loss of ancestral 5′ cis-regulatory sites specifically for mitochondrial ribosome genes in facultative anaerobic yeasts (43). Studies have also shown that proper mitochondrial functioning requires an intricate balance between RNA synthesis and degradation (90). Exactly how this balance is achieved and what factors are responsible for controlling their transcription are currently unclear.
Cg8 contains genes that were transiently up-regulated and then chronically down-regulated, with few genes responding significantly during aerobic recovery. Like Cg6 and Cg7, these genes are primarily involved in mitochondrial function, and they include much of respiratory complex V (ATP1, ATP2, ATP3, ATP5, ATP7, ATP10, ATP14, ATP16, ATP17, and ATP18) and the TCA cycle (ACO1, IDH2, KGD1, LSC1, LSC2, SDH1, SDH2, and SDH4). A remarkable 99% of these genes contain motifs for the homeodomain protein Pho2 (also known as Bas2 and Grf10), and 86% contain motifs for one of its binding partners, Swi5. Together, theses factors are known to activate HO expression, and thus, it is unclear what role they might play here. However, the low probability (P = 5 × 10−7) of finding both of these motifs in 86% of the genes in this cluster alone provides strong circumstantial evidence that these sites are somehow involved in regulating expression, whether through Pho2 and Swi5 or other factors that may bind to such motifs.
Interestingly, Cg9 contains genes for most of the rest of the respiratory chain, including the flavin adenine dinucleotide-dependent glycerol-3-phosphate dehydrogenase (GUT2), NADH dehydrogenase (NDE1), NADH-ubiquinone oxidoreductase (NDI1), and much of complexes III (COR1, QCR2, QCR7, QCR8, QCR9, and QCR10) and IV (COX4, COX5A, COX6, COX7, COX8, COX12, and COX13). Their expression differs from those in Cg8 in showing a much more rapid return to normoxic levels upon reoxygenation, a signature indicative of positive regulation by heme. Indeed, heme-responsive factors appear to be the predominant regulators, with Hap2/3/4/5 binding sites in 76% of these genes and Hap1 binding sites in 35% of these genes. Many are also known to be glucose repressed. Thus, it is not surprising to find significant enrichment for TFMs involved in this process (e.g., RGT1 and MIG1), even though they are predicted to be inactive under these experimental conditions.
Overall, it is clear that our network discovery approach results in tight functional clusters of genes involved in mitochondrial functions that were chronically down-regulated under anaerobiosis. For several clusters, promoter analyses using directed (i.e., specific TFM searches) or matrix-assisted searches either failed to reveal the factors most likely to be responsible for the observed expression patterns or suggest novel regulatory roles for specific cis-regulatory sites and/or trans-acting factors. These analyses also further implicate the importance of posttranscriptional processing (e.g., by Puf3) in regulating transcript levels of specific sets of genes, for example, those involved in mitochondrial protein synthesis (Cg7). From an examination of the temporal responses, they also reveal distinct differences in timing and regulation during the reestablishment of specific mitochondrial pathways after reoxygenation; for example, the rapid up-regulation of heme-responsive components (i.e., Hap2/3/4/5 and Hap1 networks in Cg9) of the respiratory chain (complexes III and IV) responsible for generating the proton-motive force followed by the more delayed up-regulation of components of the TCA cycle and FoF1 ATP synthase (Cg8) by different regulatory networks.
Networks rapidly up-regulated during aerobic recovery in galactose.
Cg10 to Cg12 contain the majority of genes that were rapidly up-regulated upon reoxygenation (Fig. 5). In response to anaerobiosis, they exhibit divergent temporal signatures, with genes in Cg10 and Cg11 chronically down-regulated and many genes in Cg12 transiently up-regulated. Many of these genes are involved in processes that either directly utilize oxygen or protect cells from by-products of oxygen metabolism. For example, a large number of genes are involved in sterol, unsaturated fatty acid, and heme biosynthesis as well as respiratory and peroxisome function. Cg10 contains genes involved in diverse cellular processes, notably, thiamine biosynthesis (SNO2, SNO3, THI5, THI11, THI12, and THI13), lipid metabolism (FAS2, IZH2, IZH4, LSB6, MDH3, OAF1, OLE1, OSH7, SFK1, and TAZ1), and the oxidative stress response (e.g., AHP1, DDR2, GRX2, SOD1, SOD2, and TSA1). MSN2/4 and HAP1 motifs were the most prevalent and significantly enriched (Table 1). What additional factor(s) may be involved in controlling the expression of a large percentage (44%) of these genes that lack either motif is unclear given the low occurrence of other motifs known to regulate such functional categories of genes (e.g., SKN7 and YAP1).
Cg11 contains genes that responded rapidly to reoxygenation, including a number of transcription factors important for mediating this response (e.g., CIN5, MSN2, MGA1, SPT23, and YAP7) as well as heme-responsive transcription factors (e.g., ROX1 and MOT3). Members include much of the early pathway for sterol synthesis (e.g., ERG8, ERG12, ERG13, ERG20, HMG1, and MVD1), genes for oxidative stress and/or redox regulation (e.g., CTT1, GPX2, SRX1, and TRX2), and other genes for metal ion homeostasis and/or respiratory function (e.g., COX15, COX19, CYC7, HEM2, ICT1, ISU2, IZH1, and YDR506C). Many of these genes have been shown to be Hap1 and/or Yap1 regulated, and motifs for these factors, although not particularly prevalent, were significantly enriched (Table 1).
Cg12 contains genes that were rapidly yet transiently up-regulated after reoxygenation. Interestingly, members include the entire pathway for the de novo synthesis of homocysteine (SUL1, SUL2, MET3, MET14, MET16, ECM17, MET10, and MET17) and S-adenosyl-methionine (AdoMet) (MET6, SAM1, and SAM2) as well as other genes involved in sulfur metabolism (e.g., STR3, MMP1, MUP1, MUP3, and MXR1). Homocysteine is required not only as a precursor for the synthesis of glutathione but also for ergosterol through AdoMet, which condenses with zymosterol to form fecosterol. During anoxia, ergosterol cannot be synthesized due to a lack of oxygen, and squalene (an intermediate) is accumulated to high levels. Upon reoxygenation, ergosterol is rapidly synthesized (53) due, in part, to the chronic up-regulation of the latter portion of the biosynthetic pathway under anaerobiosis (see Cg17 and Cg18, discussed below). Thus, the de novo synthesis of homocysteine from extracellular sulfate may be an absolute requirement for rapidly increasing both glutathione and AdoMet for ergosterol synthesis during reoxygenation. In support of this, genes for glutathione synthesis (CYS3, CYS4, and GSH1) and reduction (TRR1) are members of this cluster, as are others involved in the oxidative stress response (e.g., CCP1, CTA1, DDR48, MXR1, OXR1, and YAP1). Yap1 is likely the predominant regulator of this cluster, and 70% of the genes contain consensus binding sites (Table 1). Given the preponderance of genes for the synthesis of methionine and other amino acids, there is also predictable enrichment for motifs (e.g., GCN4, MET31, and CBF1) that regulate these genes even though they are predicted to be “inactive” here. Finally, it is interesting that some of these genes were also transiently up-regulated in response to anaerobiosis. Some reports suggest, paradoxically, that a shift to anaerobiosis results in transient oxidative stress in yeast (27). Whereas it is possible that low levels of reactive oxygen species may play a role in signaling, genes that exhibit this temporal signature appear to be associated specifically with redox regulation. For example, in addition to the aforementioned genes leading to glutathione biosynthesis, other genes (e.g., GSH1 and ZWF1) involved in redox regulation were transiently induced during both the anaerobic and aerobic shifts, whereas genes directly involved in mitigating reactive oxygen species (e.g., AHP1, CCP1, CTA1, CTT1, GPX2, PRX1, SOD1, SOD2, and TSA1) responded only to reoxygenation.
Networks transiently up-regulated during anaerobiosis in galactose.
Cg13 and Cg14 contain genes that were transiently up-regulated during the acute phase of the anaerobic response. Many genes in Cg14 also exhibit delayed, chronic up-regulation (Fig. 5). Nearly all genes contain Msn2/4 binding sites (Table 1) and many have been shown to be induced by these factors in response to anaerobiosis in knockout studies conducted using this yeast strain (61). In addition to MSN2/4 motifs, 71% of the genes in Cg14 also contain ROX1 motifs, which is consistent with the chronic up-regulation of many of these genes following their acute, transient induction by Msn2/4. As indicated in Fig. 5, many genes in Cg13 and Cg14 are not differentially expressed in response to the O2 shifts in glucose medium, suggesting that they respond to the abrupt cessation of respiration rather oxygen deprivation per se (10, 61). In support of this, many of these genes are apparently involved in increasing cellular energy currency during the metabolic switch. Members include sensors of nutritional status (e.g., PSK1), genes for regulating glycogen (GDB1, GIP2, GLC3, GLC8, GSY2, PCL6, RIM11, and YPI1) and trehalose reserves (NTH1, NTH2, TPS1, TPS2, TPS3, and TSL1), and genes for hexose transport (GAL2, HXT3, HXT4, HXT6, HXT7, HXT11, HXT13, HXT15, HXT16, HXT17, and MAL11), dissimilation (GLK1, HXK1, and PYK2), and regulation (GAL3, GAL10, MAL13, MDH2, RGT2, and SNF3). Other genes are involved in secondary catabolism (GRE3, SUC2, and YJR096W) and the negative regulation of gluconeogenesis (FYV10, GID7, RMD5, VID28, VID30, and UBC8). Finally, nearly half of the genes for autophagy (e.g., ATG2, ATG3, ATG4, ATG8, ATG9, ATG20, and SNX4) as well as a large number of genes involved in protein folding, sorting, and targeting; proteasomal degradation; and vacuolar function are also members of these clusters, providing further evidence of a global response to an energetic crisis. In addition to the above-mentioned genes involved in the regulation of energy reserves, other genes subject to dual regulation by Msn2/4 and Rox1 (Cg14) are associated with carbohydrate transport and metabolism (e.g., FSP2, GAL2, GAL10, GDB1, GIP2, GLK1, GPH1, HXT4, HXT6, HXT7, HXT11, HXT15, HXT16, LAT1, MAL13, MDH2, NGG1, PGM2, RGT2, RTG2, SNF3, TPS1, TPS3, YGR287C, and YIL172C [with a MIPS enrichment P value of ≤2.5 × 10−10]), consistent with previous genomic analyses of the individual knockout strains (58, 61).
Networks chronically up-regulated during anaerobiosis in galactose.
Genes in Cg15 and Cg16 were more chronically up-regulated under anaerobiosis and rapidly returned to preanoxic levels after reoxygenation (Fig. 5). This expression pattern suggests negative regulation by heme (57), and indeed, over 70% of these genes have ROX1-like sequences in their promoters (Table 1). Moreover, the function of many of these genes fits well with the role of Rox1 determined in previous studies (58), specifically in regulating carbohydrate utilization and redox balance. Members include genes encoding nearly all of glycolysis (HXK2 [HXK1 and GLK1 in Cg14], PGI1, PFK1, PFK2, PFK26, FBA1, TPI1, GPD2, TDH1, TDH2, PGK1, GPM1, GPM2, GPM3, ENO1, ENO2, CDC19 [PYK2 in Cg14], and DLD3), and for regenerating NAD+ from the reduction of acetaldehyde (ADH1, ADH2, ADH3, and ADH5), dihydroxyacetone phosphate (GPD2), and fumarate (OSM1 and YEL047C [also BRO1 in Cg0, whose temporal profile is most highly correlated with this cluster]). In addition, a number of retrograde (RTG)-responsive genes and other genes involved in anaplerotic functions for glutamate synthesis and nitrogen homeostasis (e.g., APE3, CIT2, COQ6, DAL5, DLD3, GDH3, GPM1, PUT1, SDH3, and UGA1) are members of these clusters.
Finally, genes in Cg17 and Cg18 also exhibit chronic anaerobic up-regulation but only after a substantive delay (≥2 generations). After reoxygenation, many are either further induced or remain elevated for some time after the shift. The former expression pattern has been dubbed “delayed anaerobic” (18), and many of these genes predictably contain Upc2 consensus binding sites in their promoters. Previous studies with rox1 null strains have shown that the expression of UPC2 is negatively regulated by Rox1 (58), and thus, a substantive delay in the induction of Upc2-regulated genes is predicted based upon models of heme dilution under anaerobiosis. Indeed, UPC2 is a member of Cg18. In addition to UPC2 motifs, YAP1, HAP2/3/4/5, and ROX1 motifs are significantly enriched in Cg17, suggesting that many of these genes are subject to combinatorial regulation, i.e., anaerobic up-regulation as a result of Upc2 activation and/or Rox1 derepression and aerobic induction as a result of Yap1 and/or Hap2/3/4/5 activation. Some of the genes that were most strongly induced upon reoxygenation are involved in ergosterol synthesis, and nearly all of these genes contain HAP2/3/4/5 and/or YAP1 sites in addition to UPC2 motifs. The majority of genes in Cg18 contain both MOT3 and UPC2 motifs. Mot3 is involved in controlling a number of anaerobically expressed genes, including many genes involved in cell wall maintenance and sterol biosynthesis (54, 94).
In terms of overall function, most genes in Cg17 and Cg18 are involved in sterol homeostasis (e.g., ARE1, ATF2, AUS1, CYB5, ERG1, ERG2, ERG3, ERG5, ERG6, ERG7, ERG11, ERG24, ERG25, ERG26, ERG28, HES1, IDI1, NCP1, PDR11, SUT2, TGL1, UPC2, and YEH1) and cell wall maintenance (e.g., AGA1, DAN1, DAN2, DAN3, DAN4, DFG16, GNT1, GSC2, KRE9, KTR1, KTR2, KTR4, PAU1, PAU2, PAU3, PAU4, PAU5, PAU6, PAU7, PLB1, PMT3, PMT5, PST1, RCR1, SAG1, SIM1, SUN4, TIR1, TIR2, TIR3, TIR4, and other members of the seripauperin gene family). Other genes are involved in mitochondrial processes (e.g., BI2, CCS1, FMP34, GLT1, GTT1, HEM13, MSS1, and SCM4), particularly transport (AAC3, ATM1, ATO3, ODC2, ORT1, POR2, and PTK1). The functional role of many of these networks under anaerobiosis has been reviewed previously (58). In brief, modifications in cell wall porosity are required for the import of ergosterol and unsaturated fatty acids under anaerobiosis as well as the export of potentially toxic end products of anaerobic metabolism. This involves the remodeling of a large complex of Upc2-, Mot3-, and Rox1-regulated gene networks.
Oxygen-responsive gene networks identified in glucose medium.
Using the same network recovery approach as that used with the galactose data set, we clustered the temporal profiles of genes that responded significantly (P < 0.01) to the oxygen shifts in glucose medium (1,603 genes in total). From Fig. 6, it is clear that the optimum K value for exploring the transcriptional networks is 13, as it results in the lowest FCS P value (0.40) with high CS (99.6%). Given that far fewer genes responded in glucose than in galactose, and entire networks of transiently responding genes controlled by Msn2/4, Fhl1, SCB, MCB, and PAC are absent, as indicated in Fig. 5, fewer network-defined clusters were expected for the glucose set (see Table S3 in the supplemental material for a full list and for gene-to-cluster membership). As shown in the heat maps of Fig. 7, the SOM algorithm nicely partitions the profiles into temporally shifted groups, beginning with those that were primarily up-regulated during aerobic recovery (dextrose cluster 1 [Cd1] to Cd3), followed by genes that were primarily down-regulated under anaerobiosis (Cd4 to Cd8). These genes are followed by ones that were primarily up-regulated under anaerobiosis with increasing delays (Cd9 to Cd13). Given that the majority of these genes have a similar response in galactose and were discussed in this context, we limit our discussion here to additional regulatory insight gained through the clustering of these temporal profiles and differences in the responses of specific networks in the two media.
FIG. 6.
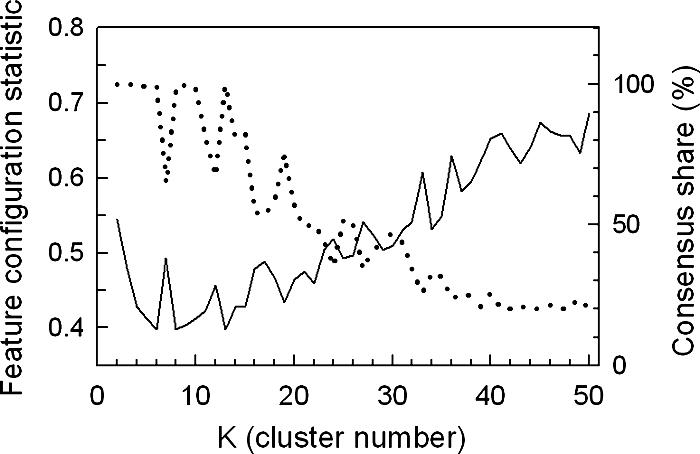
Assessment of clustering quality using the FCS and CS for oxygen-responsive genes identified in glucose medium. The temporal profiles of genes that responded significantly (P < 0.01) to the shifts in oxygen availability in glucose medium (SSD-TEA) were clustered 10 times using an SOM algorithm with 1D ring topology and Pearson correlation as the distance metric. The average FCS P values (solid line, left ordinate) for 2,603 transcription factor consensus binding sequences (TFMs) and CS (dotted line, right ordinate) are plotted as a function of the cluster number (K).
FIG. 7.

Heat maps and statistical comparisons of oxygen-responsive genes identified in glucose medium. The temporal profiles of genes that responded significantly (P < 0.01) to the shift to either anaerobiosis or aerobiosis in SSD-TEA medium were clustered using an SOM algorithm with 1D ring topology (K = 13). The left panel shows the temporal signatures, and the right panel shows the same temporal signatures but with a statistical overlay that masks gene expression changes that were not significantly (P > 0.01) different from the controls (aerobic sample for anaerobiosis and sixth-generation anaerobic sample for aerobiosis). Cluster 0 contains genes that exhibited unstable cluster membership. Green indicates down-regulated expression, and red indicates up-regulated expression. Bars to the right of the heat map indicate genes that also responded significantly (P < 0.01) to the shift in O2 availability in galactose medium.
Networks transiently up-regulated during aerobic recovery in glucose.
Most of the genes that were transiently up-regulated in response to reoxygenation are found in Cd1 to Cd3 (Fig. 7). Despite the similarities in their expression profiles, it is clear from the differential enrichment of TFMs and functional categories (Table 2) that these gene clusters are regulated by distinct transcriptional networks. Most genes in Cd1 are found in galactose cluster Cg12, and both clusters are enriched for YAP1, GCN4, and YAP7 motifs. Predictably, many of these genes are associated with oxidative stress and/or redox regulation (e.g., AAD4, AAD6, AAD16, CCP1, CTA1, CTT1, GTT2, SNQ2, and TRR1) and, as discussed above, with rapidly increasing the de novo synthesis of homocysteine for both glutathione and ergosterol synthesis upon reoxygenation (Table 2). Interestingly, members include a number of genes in the latter portion of the ergosterol biosynthetic pathway (e.g., ERG2, ERG5, ERG7, ERG9, ERG26, and ERG27), which were chronically up-regulated in galactose (Cg18) but not in glucose. These results suggest that they are differentially regulated in the two media, perhaps reflecting the different functional states of mitochondria in these media (88). In support of this, it has been reported that the de novo synthesis of sterols may be required for the induction of respiratory genes during the transition from anaerobic to aerobic conditions (91). Thus, the chronic anaerobic up-regulation of genes for converting squalene to ergosterol makes functional sense under non-catabolite-repressed conditions (i.e., in galactose), where the reestablishment of respiration is critical for energy production.
TABLE 2.
Selected list of enriched consensus sequence motifs (TFMs), MDscan sequence logos, and MIPS functional categories in clusters of genes differentially expressed in response to O2 availability in glucose mediume
 |
TABLE 2.
Continued
 |
P value [−log10(P)] for TFM enrichment relative to clustered genes (1,603)/P value [−log10(P)] for TFM enrichment relative to the genome. NS, not significant.
P value [−log10(P)] for MIPS functional category enrichment.
STRE (AGGGG) has been listed as the binding site for Msn2, Msn4, Gis1, and/or Rph1 by different authors (see citations above).
See Table S3 in the supplemental material for an explanation of the numbered motifs: F, functional motif; C, coherent motif (17); O, see reference 50.
SOM clustering with 1D ring topology (K = 13).
Genes in Cd2 were chronically down-regulated under anaerobiosis and rapidly induced upon reoxygenation. They are widely dispersed among the galactose clusters, again indicating some differences in their responses in the two media. Members include genes for sterol homeostasis (e.g., ARE2, ERG8, ERG12, ERG13, ERG20, HMG1, MVD1, NSG2, PDR16, and OYE3), redox regulation and/or oxidative stress (e.g., FMO1, GPX2, GRE2, GRX2, MXR1, PRX1, SOD2, SRX1, TRX2, YAP1, and ZWF1), detoxification, and lipid, fatty acid, and isoprenoid metabolism, as indicated in Table 2. Despite substantive overlap in functional groups between Cd1 and Cd2, it is clear from promoter analyses this is not true of their regulation. The expression profiles suggest positive regulation by heme, but the factor(s) responsible is not apparent from promoter analyses. The most significantly enriched motifs were, surprisingly, those for SPT15 (TBP) and a putative regulatory site associated with the cell cycle, namely, C49 (TAGAAA) (17). AT-rich sites are particularly abundant in this cluster. However, it is unclear whether these sites are functional and, if so, what factor may bind to them.
Cd3 contains a large fraction of mitochondrially targeted genes for respiration and metal ion homeostasis that were chronically down-regulated under anaerobiosis and rapidly induced upon reoxygenation in both media (see Cg9 above). Both their temporal signatures and function suggest positive regulation by heme, and HAP1 motifs, although not the most prevalent, were the most significantly enriched (Table 2). STE12 and RTG1 motifs were marginally enriched and are found in a large number of these genes but are unlikely to be predominant regulators of this cluster based upon known mechanisms of regulation.
Networks chronically down-regulated during anaerobiosis in glucose.
Cd4 to Cd8 contain most of the remainder of the genes that were chronically down-regulated during anaerobiosis. Predictably, many of these genes are involved in mitochondrial functions including respiration and metal ion homeostasis (Cd4), mitochondrial ribosome biogenesis and protein synthesis (Cd5), nucleotide metabolism and energetics (Cd6), and the TCA cycle (Cd7). In contrast, members of Cd8 are involved primarily in the dissimilation of C5 and C6 compounds and reserve energy metabolism. The temporal responses of genes in Cd4 and Cd5 are similar in the two media, with many found in Cg9 and Cg7, respectively. As was the case for similar clusters of mitochondrially associated genes from the galactose response, the factors responsible for controlling their expression are not readily apparent from promoter analyses. Although motifs for both Hap1 (Cd4) and Hap2/3/4/5 (Cd5) are enriched, few genes contain such motifs. In Cd5, a remarkable 97% of the genes contain Gcr1 binding sites, yet it is an unlikely candidate for down-regulating these mitochondrially associated genes during anoxia. Rather, as was seen in Cg7, there is remarkable enrichment for 3′ PUF3 and 3′ Motif6 sites, suggesting that these genes may be regulated predominantly at the posttranscriptional level.
Some of the genes in Cd6 were acutely yet transiently up-regulated during anoxia and then chronically down-regulated. Notably, these genes include a coherent group for purine biosynthesis and import (e.g., ADE1, ADE2, ADE4, ADE5/7, ADE6, ADE12, ADE13, ADE17, ADK2, FCY2, FCY22, HPT1, IMD3, MTD1, and TPN1). Given this finding, it is not surprising to find enrichment for BAS1 sites, which might account for the transient up-regulation of this subgroup. However, the factor(s) responsible for the chronic anaerobic down-regulation of most of these genes is not readily apparent. Nearly 40% of these genes are unique to glucose medium, although they are involved in functional processes similar to those of the rest of the genes in this cluster that also responded to the O2 shifts in galactose.
A large fraction of genes in Cd7 and Cd8 appear to be differentially regulated in the two media (Fig. 7), given that many of these genes exhibit opposite patterns of up- and down-regulation during anaerobiosis. For example, nearly half of the genes in Cd7 and Cd8, which were transiently or chronically down-regulated in glucose, are found in Cg13 and Cg14, clusters that exhibit transient up-regulation in galactose. Members include genes for carbohydrate import and dissimilation and for mitochondrial functions as well as a number of RTG-responsive genes involved in anaplerotic functions for glutamate synthesis, nitrogen homeostasis, and amino acid synthesis. Many of these genes have been shown to be Msn2/4 regulated (15, 34, 61), and both clusters are significantly enriched for consensus binding sites. Whereas their transient up-regulation in response to anoxia in galactose has been shown to be due to Msn2/4 activation (61), what factor(s) accounts for their down-regulation in glucose is unclear. In addition to MSN2/4 motifs, 99% of genes in Cd7 contain ADR1 sites, and 72% have RTG1 sites, which fits well with the functional regulons found in these clusters but, based on known regulatory mechanisms (108), cannot explain their down-regulation here.
Networks up-regulated during anaerobiosis in glucose.
Cd9 to Cd11 contain the majority of genes that were up-regulated during anaerobiosis. Genes in Cd9 and Cd10 are primarily found in galactose clusters Cg14 to Cg16, and they exhibit similar behaviors in the two media. Cd9 is enriched for both MSN2/4 (STRE) and ROX1 motifs, which fits with their temporal response observed in galactose (Cg14). However, in glucose, these genes exhibit only delayed, chronic up-regulation, a signature indicative of derepression by Rox1 and the absence of Msn2/4 activation (61). In Cd10, a remarkable 94% of the genes have ROX1 sites, and both Cd9 and Cd10 are enriched for functional categories associated with this regulator, particularly carbohydrate import and utilization. Finally, genes in Cd11 exhibit a substantive delay (≤3 generations) in their anaerobic up-regulation, a signature indicative of regulation by Upc2, whose binding sites were the most significantly enriched. Most of these genes are members of galactose cluster Cg11, and they include much of the seripauperin gene family as well as other genes involved in cell wall function and sterol and lipid metabolism as discussed above.
The response of genes in Cd12 is markedly dissimilar in the two media. One-third of the genes are unique to the glucose shift, and the remaining members are widely distributed across nearly all of the galactose clusters. These genes are chronically induced after a substantive delay (≈3 generations), a temporal signature suggesting negative regulation by heme. However, few of these genes contain binding sites for heme-regulated transcription factors (e.g., only 11% contain ROX1 sites) (P = 0.005). Rather, the most prevalent and significantly enriched TFMs are associated with cell cycle control and ribosomal biogenesis (e.g., MCM1, MCB, SCB, ABF1, PAC, and RRPE) (see Table S3 in the supplemental material). Accordingly, many of these genes are found in Cg3 and Cg4, clusters that were significantly enriched for such motifs yet transiently down-regulated in response to anoxia in galactose. Many of these genes are involved in amino acid biosynthesis and metabolism (AAT1, ALT2, ARG8, ARG80, ARO2, ARO8, ASP1, BAT2, CHA1, CPA1, DIP5, DYS1, GCN20, GLT1, HMT1, HPA3, ILV1, ILV3, LEU1, LEU3, LEU4, LYS2, LYS4, PRO2, PRS3, SSH4, THR4, TRP1, TRP3, TRP5, TYR1, and WRS1). These genes were either transiently down-regulated or failed to respond in the galactose shift. Other members of this cluster are involved in rRNA transcription/synthesis and processes associated with the G1/S transition (Table 2). Given the preponderance of genes involved in the synthesis of amino acids, proteins, and nucleotides (e.g., AAH1, DCD1, SDT1, PRS3, and URA1), these results suggest the depletion of nitrogen-containing and possibly phosphate-containing (e.g., PHO11 and PHO12) cellular components after three generations of glucose-supported anaerobic growth, requiring the up-regulation of these pathways before committing to another round of the cell cycle. In support of this, members include a number of genes induced by nitrogen or amino acid starvation (e.g., DFG16, GCN20, YVH1, ZPR1, and YJL200C) and other genes for controlling the G1/S transition (e.g., CKS1, CLN2, CTR9, SDA1, TAF10, and YTM1). What may account for their differential regulation and response in the two media is currently unclear.
Finally, the responses of genes in Cd13 are similar in the two media (see Cg17 and Cg18 above), consisting of delayed anaerobic induction and further induction upon reoxygenation. Many of these genes are involved in amino acid biosynthesis (ARG1, ARG4, ARG5/6, CAN1, HIS5, HOM3, LYS1, MET2, MET10, MET13, ODC2, and TRP2), with predictable enrichment for TFMs (e.g., ARO80 and GCN4) that regulate such genes. In addition, a large number of these genes are involved in sterol homeostasis (e.g., CYB5, ERG1, ERG3, ERG4, ERG6, ERG11, ERG24, ERG25, ERG28, and NCP1). In terms of overall regulation, YAP1 motifs are the most prevalent, which is consistent with their induction during aerobic recovery. Moreover, 40% of these genes contain Upc2 consensus binding sites, consistent with their delayed anaerobic up-regulation, although the hypergeometric enrichment P value for these sites is only 0.037. Regardless, a number of these genes have been shown to be Upc2 regulated in genomic studies (106), and many of these genes are involved in functions consistent with regulation by this factor (specifically, sterol homeostasis). Thus, many genes in this cluster are apparently subject to dual regulation over the time course, namely, Upc2-dependent anaerobic induction followed by Yap1 activation upon reoxygenation. Comparisons of additional gene networks that show differences in catabolite-repressed (glucose) and nonrepressed (galactose) cells are discussed below.
DISCUSSION
Gene network identification.
Given that the quality of our report is dependent on the analytical methods used for the evaluation of data, we begin our discussion by describing the rationale and some of the advantages of our clustering approach for gene network identification. Although unsupervised learning or clustering has emerged as a ubiquitous tool for analyzing genome-wide expression profiles (see, e.g., references 30, 32, and 96), the quality of the clustering results is often difficult to evaluate, and each clustering algorithm has tunable parameters with no obvious way to choose appropriate values (reviewed in reference 37). Moreover, most algorithms require the number of clusters to be predetermined, yet this value is rarely known and, thus, is often arrived at by subjective criteria. Our clustering approach uses statistical evaluation to systematically address these challenges in order to choose appropriate parameter settings, including the algorithm, distance or similarity metric, and number of clusters for recovering gene network structure.
In brief, we use the CS metric to narrow the large list of potential parameter settings to those combinations that consistently recover the most fine-grained structure (highest K values with, say, a CS of >0.95) from unbiased clustering of the gene expression profiles alone. Among the promising candidates, we then examined the configuration of TFMs among gene clusters to determine which settings result in robust yet highly improbable motif configurations based on chance alone (low FCS P values). Our TFM list is comprehensive (2,603 in total) and purposely includes both putative and experimentally defined sites as well as all known sequence variants in order to maximize the probability that it contains all regulatory sites to which transcription factors that are active under the experimental conditions bind. cis-regulatory sites that are “active” will have a highly biased configuration among network-defined gene clusters (low FCS P values), whereas those that are inactive should have a more random configuration (high FCS P values). We calculate an average FCS P value across all motifs in our list, which serves two functions: first, it minimizes the impact of any false positives that by chance have low FCS P values for some clustering configurations, and second, it identifies a clustering configuration that is a compromise solution for all motifs that are potentially “active” under the experimental conditions examined. By examining both metrics in concert, we are able to choose clustering parameter settings (including K) that consistently (high CS value) result in the least probable configuration by chance of all TFMs among gene clusters (lowest average FCS P value) and, thus, a configuration that is most likely to capture the true gene network structure.
In evaluating the clustering results, it should be obvious that our approach cannot definitively identify the transcription factors responsible for the observed expression patterns but should in most cases identify the most plausible candidate(s). It is predicted that the most prevalent and significantly enriched TFMs in each cluster are those most likely to be responsible. However, interpretation must rely on a prudent examination of additional knowledge of mechanisms of activation/deactivation, conditions in which the activity of the factor is known to change, functional annotations, and previous experimental results that have defined such sites or target genes. Obviously, if the binding sites for active regulators are not contained in our TFM list, we cannot assess their distribution and will likely underestimate the number of active networks. We also conducted matrix-assisted searches using MDscan to identify additional sites, but here the results primarily confirmed enrichment for cis-regulatory sites that were contained in our list. In several gene clusters defined here, we also saw predictable enrichment for regulatory sites that are known to control specific subgroups of genes within a cluster even though they are “inactive” under these conditions, making it more difficult, in cases where they are numerous, to identify the most plausible regulator(s) of the entire cluster. For several clusters, promoter analyses either failed to reveal the factors responsible or suggest novel regulatory roles for specific cis-regulatory sites and/or trans-acting factors, suppositions that can be tested with further study. Our analyses also implicate the importance of posttranscriptional processing in regulating transcript levels of some groups of genes, for example, those that are Puf3 regulated and involved mitochondrial protein synthesis, and, thus, emphasize the benefit of conducting 3′ as well as 5′ searches.
Overall, one of the most striking features to come from these analyses is the large number of genes subject to combinatorial regulation under the experimental conditions examined. This showcases a distinct advantage of our gene network approach over other methods that, for example, regress the temporal profiles of genes onto candidate cis-regulatory sites (19). An excellent example is Cg14. In response to anoxia, these genes were transiently induced by Msn2/4 and then chronically derepressed by a loss of Rox1 activity; upon reoxygenation, they rapidly returned to their preanoxic levels as a result of renewed Rox1 repression. Cg14 is flanked by clusters that are regulated by Msn2/4 (Cg13) or Rox1 (Cg15) alone. For the majority of these genes, we have independent verification that they are indeed regulated by these factors, given that we previously conducted transcriptomic analysis of rox1 and msn2/4 null strains in the same genetic background and under similar experimental conditions (58, 61). Most of the genes in Cg14 were also clustered together in the glucose set (Cd9), and both MSN2/4 and ROX1 motifs were again significantly enriched. However, in glucose, their temporal signatures are indicative of Rox1 regulation alone, a result consistent with our previous analyses showing that Msn2/4 are not active under these conditions (61). Although an examination of the temporal signatures in conjunction with the enriched motifs and knowledge of their modes of regulation would lead to a similar conclusion, there is an obvious advantage in comparing the response under multiple experimental conditions, as was done here.
When nearly identical expression profiles are produced by the activity of different transcription factors, the clustering algorithm may not be able to differentiate between them, and clusters could contain a mixture of genes controlled by multiple factors. However, with our approach, even slight differences in the temporal responses of different networks should yield distinct clustering divisions, given that their separation will result in a lower FCS P value. An excellent example is the transiently down-regulated networks in Cg2 to Cg5, which differ little in terms of their temporal signatures and yet are clearly delineated networks based on the differential enrichment of TFMs and MIPS functional categories in each. These results further highlight advantages of our gene network discovery approach over other clustering methods that use subjective criteria for evaluation. In ongoing studies in our laboratory, we seek further refinements, for example by filtering out false positives for “active” cis-regulatory sites in genes whose profiles do not fit the mean of all genes that contain such sites. In addition, we will take into account profiles for genes that contain different combinations of active cis-regulatory sites and use Bayesian priors for sites that are known to be “active” under the experimental conditions examined. With this brief introduction to our gene network discovery approach, we now turn to the changes in gene network activity that are observed when cells acclimatize to changes in the availability of environmental oxygen.
Genomic remodeling in response to changes in O2 availability.
A fairly complete picture of the genomic remodeling required for yeast to acclimatize to changes in oxygen availability emerges from this study. Moreover, by comparing the responses under both catabolite-repressing (glucose) and non-catabolite-repressing (galactose) conditions, we reveal commonalities as well as substantive differences in gene network activity that result from differences in the metabolic state of the cells. Under nonrepressing conditions, in which energy requirements are met using mixed respirofermentative metabolism, MCB and SCB networks (Cg1 to Cg3) are the first to be negatively affected after about 0.04 generations (10 min) of anaerobiosis. This results in the down-regulation of genes involved in DNA replication/repair and other processes associated with the G1/S transition of the cell cycle. Concomitantly, PAC/RRPE (Cg3 and Cg4)- and Fhl1 (Cg4 and Cg5)-associated networks involved in cytoplasmic rRNA processing and protein synthesis are negatively affected, while Msn2/4-regulated networks (Cg13 and Cg14) involved in the import and utilization of primary and alternative carbon sources and reserve energy metabolism are activated. In addition, genes involved in mitochondrial functions including ribosomal biogenesis (Cg7) and early components of the respiratory chain (Cg8 and Cg9) are also transiently induced, although for these networks it is unclear what factors are directly responsible.
Overall, this remodeling activity suggests the simultaneous exploration/utilization of available carbon sources while sparing energetic demand by arresting the de novo synthesis of the cytoplasmic translational machinery. The response is predictably transitory, given that the cells quickly (≈1 h) reach a new steady-state growth rate supported solely by galactose fermentation. Although the response is similar to that observed during nutrient limitation or a switch to lower-quality carbon sources (79, 80), it affects a specific subset of genes associated with the balancing of energetic supply and demand and the G1/S transition of the cell cycle. Of the 819 genes identified in the environmental stress response (34), 501 genes (61%) were differentially expressed with similar kinetics and expression patterns during this metabolic transition. Those genes involved in mitochondrial functions that were transiently induced in Cg7 to Cg9 are not part of the environmental stress response and appear to respond directly to the inhibition of respiration. Although the initial event that triggers these changes is the abrupt decline in oxygen availability, the signal that is responsible for eliciting changes in gene network activity appears to be metabolic in origin and linked to the cessation of respiration, given that a similar response is evoked under normoxic conditions with treatment with either antimycin A (Lai et al., unpublished) or myxothiazol (10). Moreover, a similar response is not invoked in catabolite-repressed cells in which the respiratory capacity is repressed, and no change in growth rate is observed during the transition from aerobic to anaerobic conditions. Ongoing studies in our laboratory are aimed at identifying the sensor(s) for the change in energetic status and the signaling pathways responsible for changes in gene network activity.
After ≈0.25 generations of anaerobiosis in both media, we began to see evidence of changes in heme-dependent transcription factor activity. This includes the chronic down-regulation of Hap1- and Hap2/3/4/5-regulated networks (Cg9 to Cg11 and Cd3 to Cd4) involved in mitochondrial functions and the concomitant derepression of Rox1-regulated functions (Cg14 to Cg16 and Cd9 to Cd10) involved in carbohydrate utilization and redox balance. After ≈0.38 generations, mRNA levels of a large group of genes (Cg7 and Cd5) involved in mitochondrial protein synthesis and energetics decline for the duration of anaerobiosis. These genes are part of a functional regulon that have apparently lost their ancestral 5′ cis-regulatory sites (43) and appear to be posttranscriptionally regulated by Puf3. Finally, after ≈2 generations of anaerobiosis, the last networks to respond are Upc2- and Mot3-regulated networks involved in sterol, cell wall, and unsaturated fatty acid homeostasis. Overall, changes in the activities of these heme-dependent networks are predictably slow to occur in response to anaerobiosis, given that heme is not thought to be degraded in this species, and, thus, pools of heme in the nucleus are slowly diluted with anaerobic growth (reviewed in reference 57). Changes in the activities of these gene networks are essential for surviving anaerobiosis, and we discuss their functional roles in detail in the supplemental material (see “functional regulons”).
Unlike the anaerobic shift, in which temporal changes in the activities of a number of different networks were fairly well separated, upon reoxygenation, both heme-dependent networks and those that are activated by changes in oxygen free-radical concentrations respond simultaneously. Remarkably, within 0.03 to 0.06 generations after the shift to normoxia, Yap1 (Cg11 to Cg12 and Cd1 and Cd13) and Msn2/4 (Cg10) networks involved in controlling oxidative defenses are activated concomitantly with Hap1 networks (Cg10 to Cg11 and Cd3) involved in oxygen-utilizing pathways. Note that several of these gene clusters are enriched for combinations of these regulatory factors, suggesting that there may be little difference in the temporal signatures dictated by these different factors. For example, genes in Cg11 are rapidly up-regulated in response to reoxygenation, and the majority of the genes have either Hap1 or Yap1 sites but not both. At the same time, we see evidence for the heme-dependent repression of genes that were chronically up-regulated under anaerobiosis, especially those that are regulated by Rox1 (Cg14 to Cg16 and Cd9 to Cd10). The rapid response of both heme-induced and heme-repressed networks is predictable, given that the heme biosynthetic pathway appears to remain intact under anaerobiosis, and, thus, heme is rapidly synthesized upon reoxygenation (60). Interestingly, transcript levels of many Upc2-regulated genes remain elevated for some time after the aerobic shift or are further induced by Yap1 and/or Hap2/3/4/5 (Cg17 and Cd13). Overall, although many of the changes in the activities of these gene networks were predicted based upon models of regulation, this is the first time that their temporal response has been captured following reoxygenation. A comparison of this response to that elicited by treatment with oxidants such as diamide or H2O2 (15, 34) yields poor overlap save for genes that are directly involved in reducing oxygen by-products and regulating redox potential. These results suggest there is effective mitigation of any “stress” induced by reoxygenation, that is before any damage has occurred to cellular components and repair pathways must be activated.
In regard to the overall effects of carbon sources on the O2-dependent responses, a comparison of the clustering results in the two media reveals consistent enrichment for binding sites of transcription factors that are known to regulate the majority of oxygen-responsive genes. These include factors that control genes involved in respiration (Hap1 and Hap2/3/4/5), carbohydrate usage and anaerobic redox balance (Rox1), sterol and cell wall maintenance (Upc2 and Mot3), and the oxidative stress response (Yap1, Skn7, Yap7, and Msn2/4). As noted above, most of the gene networks that were differentially expressed in the two media are unique to galactose and involved in the switch from mixed respirofermentative to strictly fermentative metabolism. These include Msn2/4-regulated ones involved in bolstering energy production during the switch and those for down-regulating the protein synthetic capacity and controlling the G1/S transition (e.g., MCB, SCB, Fhl1, PAC, and RRPE). A number of other differences were noted, which can be attributed to the different functional states of these cells. These include the chronic anaerobic up-regulation of much of the ergosterol biosynthetic pathway under non-catabolite-repressed conditions, highlighting the importance of neosynthesized ergosterol in reestablishing respiration during the transition from anaerobic to aerobic conditions (91). Others include the up-regulation of genes involved in amino acid biosynthesis and the G1/S transition of the cell cycle after three generations of anaerobic growth in glucose alone by as-yet-uncharacterized mechanisms. For additional comparisons of these oxygen-responsive networks and the functional responses, refer to the supplemental material.
Finally, the present study identifies target gene networks for many different effectors as well as some of their interactions. However, the sensory and signaling pathways that converge and control these effectors are only partially known. Accumulating evidence suggests that they exert multiplex control on the effectors. Such multiplex signal regulation is apparently more prevalent than once thought and is no longer the prerogative of the cell cycle alone (49). For example, Msn2/4 is regulated by TOR, PKA, and Snf1 (25, 72), whereas Rim15 is regulated by TOR, PKA, Sch9, and Pho85 (105). We see evidence of such signals in coordinating the response of the gene clusters discussed above. However, it is clear that future studies must integrate these data with proteomic (see, e.g., reference 29) and metabolomic analyses of the response to effectively uncover the sensing and signaling networks involved.
Supplementary Material
Acknowledgments
We are deeply grateful to Jun Liu and Xin Lu for providing a preliminary look at the transcription factor activity profiles for the galactose data set using MotifRegressor.
This work was supported by National Institutes of Health grant RO1-GM59826 to K.E.K.
Footnotes
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1.Acton, T. B., H. Zhong, and A. K. Vershon. 1997. DNA-binding specificity of Mcm1: operator mutations that alter DNA-bending and transcriptional activities by a MADS box protein. Mol. Cell. Biol. 17:1881-1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson, J. S. J., and R. Parker. 2000. Computational identification of cis-acting elements affecting post-transcriptional control of gene expression in Saccharomyces cerevisiae. Nucleic Acids Res. 28:1604-1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andreasen, A., and T. Stier. 1953. Anaerobic nutrition of Saccharomyces cerevisiae. I. Ergosterol requirement for growth in a defined medium. J. Cell. Comp. Physiol. 41:23-36. [DOI] [PubMed] [Google Scholar]
- 4.Andreasen, A., and T. Stier. 1954. Anaerobic nutrition of Saccharomyces cerevisiae. II. Unsaturated fatty acid requirement for growth in defined medium. J. Cell. Comp. Physiol. 43:271-281. [DOI] [PubMed] [Google Scholar]
- 5.Bailey, T. L., and C. Elkan. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2:28-36. [PubMed] [Google Scholar]
- 6.Becerra, M., L. J. Lombardia-Ferreira, N. C. Hauser, J. D. Hoheisel, B. Tizon, and M. E. Cerdan. 2002. The yeast transcriptome in aerobic and hypoxic conditions: effects of hap1, rox1, rox3 and srb10 deletions. Mol. Microbiol. 43:545-555. [DOI] [PubMed] [Google Scholar]
- 7.Bernstein, B. E., J. K. Tong, and S. L. Schreiber. 2000. Genomewide studies of histone deacetylase function in yeast. Proc. Natl. Acad. Sci. USA 97:13708-13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blinder, D., P. W. Coschigano, and B. Magasanik. 1996. Interaction of the GATA factor Gln3p with the nitrogen regulator Ure2p in Saccharomyces cerevisiae. J. Bacteriol. 178:4734-4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boorstein, W. R., and E. A. Craig. 1990. Regulation of a yeast HSP70 gene by a cAMP responsive transcriptional control element. EMBO J. 9:2543-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bourges, I., S. Horan, and B. Meunier. 2005. Effect of inhibition of the BC1 complex on gene expression profile in yeast. J. Biol. Chem. 280:29743-29749. [DOI] [PubMed] [Google Scholar]
- 11.Buchman, A. R., W. J. Kimmerly, J. Rine, and R. D. Kornberg. 1988. Two DNA-binding factors recognize specific sequences at silencers, upstream activating sequences, autonomously replicating sequences, and telomeres in Saccharomyces cerevisiae. Mol. Cell. Biol. 8:210-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burke, P. V., K. E. Kwast, F. Everts, and R. O. Poyton. 1998. A fermentor system for regulating oxygen at low concentrations in cultures of Saccharomyces cerevisiae. Appl. Environ. Microbiol. 64:1040-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burke, P. V., L. C. Lai, and K. E. Kwast. 2004. A rapid filtration apparatus for harvesting cells under controlled conditions for use in genome-wide temporal profiling studies. Anal. Biochem. 328:29-34. [DOI] [PubMed] [Google Scholar]
- 14.Butow, R. A., and N. G. Avadhani. 2004. Mitochondrial signaling: the retrograde response. Mol. Cell 14:1-15. [DOI] [PubMed] [Google Scholar]
- 15.Causton, H. C., B. Ren, S. S. Koh, C. T. Harbison, E. Kanin, E. G. Jennings, T. I. Lee, H. L. True, E. S. Lander, and R. A. Young. 2001. Remodeling of yeast genome expression in response to environmental changes. Mol. Biol. Cell 12:323-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi, J. Y., J. Stukey, S. Y. Hwang, and C. E. Martin. 1996. Regulatory elements that control transcription activation and unsaturated fatty acid-mediated repression of the Saccharomyces cerevisiae OLE1 gene. J. Biol. Chem. 271:3581-3589. [DOI] [PubMed] [Google Scholar]
- 17.Cliften, P., P. Sudarsanam, A. Desikan, L. Fulton, B. Fulton, J. Majors, R. Waterston, B. A. Cohen, and M. Johnston. 2003. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science 301:71-76. [DOI] [PubMed] [Google Scholar]
- 18.Cohen, B. D., O. Sertil, N. E. Abramova, K. J. Davies, and C. V. Lowry. 2001. Induction and repression of DAN1 and the family of anaerobic mannoprotein genes in Saccharomyces cerevisiae occurs through a complex array of regulatory sites. Nucleic Acids Res. 29:799-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conlon, E. M., X. S. Liu, J. D. Lieb, and J. S. Liu. 2003. Integrating regulatory motif discovery and genome-wide expression analysis. Proc. Natl. Acad. Sci. USA 100:3339-3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cumsky, M. G., C. Ko, C. E. Trueblood, and R. O. Poyton. 1985. Two nonidentical forms of subunit V are functional in yeast cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 82:2235-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cyert, M. S. 2003. Calcineurin signaling in Saccharomyces cerevisiae: how yeast go crazy in response to stress. Biochem. Biophys. Res. Commun. 311:1143-1150. [DOI] [PubMed] [Google Scholar]
- 22.Deckert, J., A. M. Torres, S. M. Hwang, A. J. Kastaniotis, and R. S. Zitomer. 1998. The anatomy of a hypoxic operator in Saccharomyces cerevisiae. Genetics 150:1429-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demasi, A. P., G. A. Pereira, and L. E. Netto. 2006. Yeast oxidative stress response. FEBS J. 273:805-816. [DOI] [PubMed] [Google Scholar]
- 24.DeRisi, J. L., V. R. Iyer, and P. O. Brown. 1997. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278:680-686. [DOI] [PubMed] [Google Scholar]
- 25.De Wever, V., W. Reiter, A. Ballarini, G. Ammerer, and C. Brocard. 2005. A dual role for PP1 in shaping the Msn2-dependent transcriptional response to glucose starvation. EMBO J. 24:4115-4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Winde, J. H., and L. A. Grivell. 1992. Global regulation of mitochondrial biogenesis in Saccharomyces cerevisiae: ABF1 and CPF1 play opposite roles in regulating expression of the QCR8 gene, which encodes subunit VIII of the mitochondrial ubiquinol-cytochrome c oxidoreductase. Mol. Cell. Biol. 12:2872-2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dirmeier, R., K. O'Brien, M. Engle, A. Dodd, E. Spears, and R. O. Poyton. 2004. Measurement of oxidative stress in cells exposed to hypoxia and other changes in oxygen concentration. Methods Enzymol. 381:589-603. [DOI] [PubMed] [Google Scholar]
- 28.Dohrmann, P. R., G. Butler, K. Tamai, S. Dorland, J. R. Greene, D. J. Thiele, and D. J. Stillman. 1992. Parallel pathways of gene regulation: homologous regulators SWI5 and ACE2 differentially control transcription of HO and chitinase. Genes Dev. 6:93-104. [DOI] [PubMed] [Google Scholar]
- 29.Du, Y., B. A. Parks, S. Sohn, K. E. Kwast, and N. L. Kelleher. 2006. Top-down approaches for measuring expression ratios of intact yeast proteins using Fourier transform mass spectrometry. Anal. Chem. 78:686-694. [DOI] [PubMed] [Google Scholar]
- 30.Eisen, M. B., P. T. Spellman, P. O. Brown, and D. Botstein. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95:14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gancedo, J. M. 1998. Yeast carbon catabolite repression. Microbiol. Mol. Biol. Rev. 62:334-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gasch, A. P., and M. B. Eisen. 2002. Exploring the conditional coregulation of yeast gene expression through fuzzy k-means clustering. Genome Biol. 3:RESEARCH0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gasch, A. P., A. M. Moses, D. Y. Chiang, H. B. Fraser, M. Berardini, and M. B. Eisen. 2004. Conservation and evolution of cis-regulatory systems in ascomycete fungi. PLoS Biol. 2:e398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gasch, A. P., P. T. Spellman, C. M. Kao, O. Carmel-Harel, M. B. Eisen, G. Storz, D. Botstein, and P. O. Brown. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11:4241-4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerber, A. P., D. Herschlag, and P. O. Brown. 2004. Extensive association of functionally and cytotopically related mRNAs with Puf family RNA-binding proteins in yeast. PLoS Biol. 2:E79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grishin, A. V., M. Rothenberg, M. A. Downs, and K. J. Blumer. 1998. Mot3, a Zn finger transcription factor that modulates gene expression and attenuates mating pheromone signaling in Saccharomyces cerevisiae. Genetics 149:879-892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Handl, J., J. Knowles, and D. B. Kell. 2005. Computational cluster validation in post-genomic data analysis. Bioinformatics 21:3201-3212. [DOI] [PubMed] [Google Scholar]
- 38.Harbison, C. T., D. B. Gordon, T. I. Lee, N. J. Rinaldi, K. D. Macisaac, T. W. Danford, N. M. Hannett, J. B. Tagne, D. B. Reynolds, J. Yoo, E. G. Jennings, J. Zeitlinger, D. K. Pokholok, M. Kellis, P. A. Rolfe, K. T. Takusagawa, E. S. Lander, D. K. Gifford, E. Fraenkel, and R. A. Young. 2004. Transcriptional regulatory code of a eukaryotic genome. Nature 431:99-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horak, C. E., N. M. Luscombe, J. Qian, P. Bertone, S. Piccirrillo, M. Gerstein, and M. Snyder. 2002. Complex transcriptional circuitry at the G1/S transition in Saccharomyces cerevisiae. Genes Dev. 16:3017-3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hughes, J. D., P. W. Estep, S. Tavazoie, and G. M. Church. 2000. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 296:1205-1214. [DOI] [PubMed] [Google Scholar]
- 41.Hughes, T. R., M. J. Marton, A. R. Jones, C. J. Roberts, R. Stoughton, C. D. Armour, H. A. Bennett, E. Coffey, H. Dai, Y. D. He, M. J. Kidd, A. M. King, M. R. Meyer, D. Slade, P. Y. Lum, S. B. Stepaniants, D. D. Shoemaker, D. Gachotte, K. Chakraburtty, J. Simon, M. Bard, and S. H. Friend. 2000. Functional discovery via a compendium of expression profiles. Cell 102:109-126. [DOI] [PubMed] [Google Scholar]
- 42.Idrissi, F. Z., and B. Pina. 1999. Functional divergence between the half-sites of the DNA-binding sequence for the yeast transcriptional regulator Rap1p. Biochem. J. 341:477-482. [PMC free article] [PubMed] [Google Scholar]
- 43.Ihmels, J., S. Bergmann, M. Gerami-Nejad, I. Yanai, M. McClellan, J. Berman, and N. Barkai. 2005. Rewiring of the yeast transcriptional network through the evolution of motif usage. Science 309:938-940. [DOI] [PubMed] [Google Scholar]
- 44.Ihmels, J., G. Friedlander, S. Bergmann, O. Sarig, Y. Ziv, and N. Barkai. 2002. Revealing modular organization in the yeast transcriptional network. Nat. Genet. 31:370-377. [DOI] [PubMed] [Google Scholar]
- 45.Iyer, V. R., C. E. Horak, C. S. Scafe, D. Botstein, M. Snyder, and P. O. Brown. 2001. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature 409:533-538. [DOI] [PubMed] [Google Scholar]
- 46.Jackson, J. S., Jr., S. S. Houshmandi, L. F. Lopez, and W. M. Olivas. 2004. Recruitment of the Puf3 protein to its mRNA target for regulation of mRNA decay in yeast. RNA 10:1625-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jang, Y. K., L. Wang, and G. B. Sancar. 1999. RPH1 and GIS1 are damage-responsive repressors of PHR1. Mol. Cell. Biol. 19:7630-7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang, Y., M. J. Vasconcelles, S. Wretzel, A. Light, C. E. Martin, and M. A. Goldberg. 2001. MGA2 is involved in the low-oxygen response element-dependent hypoxic induction of genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 21:6161-6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jorgensen, P., J. L. Nishikawa, B. J. Breitkreutz, and M. Tyers. 2002. Systematic identification of pathways that couple cell growth and division in yeast. Science 297:395-400. [DOI] [PubMed] [Google Scholar]
- 50.Kellis, M., N. Patterson, M. Endrizzi, B. Birren, and E. S. Lander. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423:241-254. [DOI] [PubMed] [Google Scholar]
- 51.Kent, N. A., S. M. Eibert, and J. Mellor. 2004. Cbf1p is required for chromatin remodeling at promoter-proximal CACGTG motifs in yeast. J. Biol. Chem. 279:27116-27123. [DOI] [PubMed] [Google Scholar]
- 52.King, D. A., L. Zhang, L. Guarente, and R. Marmorstein. 1999. Structure of a HAP1-DNA complex reveals dramatically asymmetric DNA binding by a homodimeric protein. Nat. Struct. Biol. 6:64-71. [DOI] [PubMed] [Google Scholar]
- 53.Klein, H. P., N. R. Eaton, and J. C. Murphy. 1954. Net synthesis of sterols in resting cells of Saccharomyces cerevisiae. Biochim. Biophys. Acta 13:591. [DOI] [PubMed] [Google Scholar]
- 54.Klinkenberg, L. G., T. A. Mennella, K. Luetkenhaus, and R. S. Zitomer. 2005. Combinatorial repression of the hypoxic genes of Saccharomyces cerevisiae by DNA binding proteins Rox1 and Mot3. Eukaryot. Cell 4:649-660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knapp, D., L. Bhoite, D. J. Stillman, and K. Nasmyth. 1996. The transcription factor Swi5 regulates expression of the cyclin kinase inhibitor p40SIC1. Mol. Cell. Biol. 16:5701-5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Koerkamp, M. G., M. Rep, H. J. Bussemaker, G. P. Hardy, A. Mul, K. Piekarska, C. A. Szigyarto, J. M. De Mattos, and H. F. Tabak. 2002. Dissection of transient oxidative stress response in Saccharomyces cerevisiae by using DNA microarrays. Mol. Biol. Cell 13:2783-2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kwast, K. E., P. V. Burke, and R. O. Poyton. 1998. Oxygen sensing and the transcriptional regulation of oxygen-responsive genes in yeast. J. Exp. Biol. 201:1177-1195. [DOI] [PubMed] [Google Scholar]
- 58.Kwast, K. E., L. C. Lai, N. Menda, D. T. James III, S. Aref, and P. V. Burke. 2002. Genomic analyses of anaerobically induced genes in Saccharomyces cerevisiae: functional role of Rox1 and other factors in mediating the anoxic response. J. Bacteriol. 184:250-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laabs, T. L., D. D. Markwardt, M. G. Slattery, L. L. Newcomb, D. J. Stillman, and W. Heideman. 2003. ACE2 is required for daughter cell-specific G1 delay in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 100:10275-10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Labbe-Bois, R., and P. Labbe. 1990. Tetrapyrrole and heme biosynthesis in the yeast Saccharomyces cerevisiae, p. 235-285. In H. A. Dailey (ed.), Biosynthesis of heme and chlorophylls. McGraw-Hill, New York, N.Y.
- 61.Lai, L. C., A. L. Kosorukoff, P. V. Burke, and K. E. Kwast. 2005. Dynamical remodeling of the transcriptome during short-term anaerobiosis in Saccharomyces cerevisiae: differential response and role of Msn2 and/or Msn4 and other factors in galactose and glucose media. Mol. Cell. Biol. 25:4075-4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lamb, T. M., W. Xu, A. Diamond, and A. P. Mitchell. 2001. Alkaline response genes of Saccharomyces cerevisiae and their relationship to the RIM101 pathway. J. Biol. Chem. 276:1850-1856. [DOI] [PubMed] [Google Scholar]
- 63.Le Crom, S., F. Devaux, P. Marc, X. Zhang, W. S. Moye-Rowley, and C. Jacq. 2002. New insights into the pleiotropic drug resistance network from genome-wide characterization of the YRR1 transcription factor regulation system. Mol. Cell. Biol. 22:2642-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee, T. I., N. J. Rinaldi, F. Robert, D. T. Odom, Z. Bar-Joseph, G. K. Gerber, N. M. Hannett, C. T. Harbison, C. M. Thompson, I. Simon, J. Zeitlinger, E. G. Jennings, H. L. Murray, D. B. Gordon, B. Ren, J. J. Wyrick, J. B. Tagne, T. L. Volkert, E. Fraenkel, D. K. Gifford, and R. A. Young. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298:799-804. [DOI] [PubMed] [Google Scholar]
- 65.Lieb, J. D., X. Liu, D. Botstein, and P. O. Brown. 2001. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat. Genet. 28:327-334. [DOI] [PubMed] [Google Scholar]
- 66.Liesen, T., C. P. Hollenberg, and J. J. Heinisch. 1996. ERA, a novel cis-acting element required for autoregulation and ethanol repression of PDC1 transcription in Saccharomyces cerevisiae. Mol. Microbiol. 21:621-632. [DOI] [PubMed] [Google Scholar]
- 67.Liu, X. S., D. L. Brutlag, and J. S. Liu. 2002. An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat. Biotechnol. 20:835-839. [DOI] [PubMed] [Google Scholar]
- 68.Lundin, M., J. O. Nehlin, and H. Ronne. 1994. Importance of a flanking AT-rich region in target site recognition by the GC box-binding zinc finger protein MIG1. Mol. Cell. Biol. 14:1979-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lyons, T. J., N. Y. Villa, L. M. Regalla, B. R. Kupchak, A. Vagstad, and D. J. Eide. 2004. Metalloregulation of yeast membrane steroid receptor homologs. Proc. Natl. Acad. Sci. USA 101:5506-5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Machado, A. K., B. A. Morgan, and G. F. Merrill. 1997. Thioredoxin reductase-dependent inhibition of MCB cell cycle box activity in Saccharomyces cerevisiae. J. Biol. Chem. 272:17045-17054. [DOI] [PubMed] [Google Scholar]
- 71.Mai, B., S. Miles, and L. L. Breeden. 2002. Characterization of the ECB binding complex responsible for the M/G1-specific transcription of CLN3 and SWI4. Mol. Cell. Biol. 22:430-441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mayordomo, I., F. Estruch, and P. Sanz. 2002. Convergence of the target of rapamycin and the Snf1 protein kinase pathways in the regulation of the subcellular localization of Msn2, a transcriptional activator of STRE (stress response element)-regulated genes. J. Biol. Chem. 277:35650-35656. [DOI] [PubMed] [Google Scholar]
- 73.McDaniels, C. P. J., L. T. Jensen, C. Srinivasan, D. R. Winge, and T. D. Tullius. 1999. The yeast transcription factor Mac1 binds to DNA in a modular fashion. J. Biol. Chem. 274:26962-26967. [DOI] [PubMed] [Google Scholar]
- 74.McIntosh, P. B., I. A. Taylor, S. J. Smerdon, T. A. Frenkiel, and A. N. Lane. 1999. 1H, 15N and 13C assignments of the DNA binding domain of transcription factor Mbp1 from S. cerevisiae in both its free and the DNA bound forms, and 1H assignments of the free DNA. J. Biomol. NMR 13:397-398. [DOI] [PubMed] [Google Scholar]
- 75.Moller, K., L. Olsson, and J. Piskur. 2001. Ability for anaerobic growth is not sufficient for development of the petite phenotype in Saccharomyces kluyveri. J. Bacteriol. 183:2485-2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.O'Brien, R. M., N. Halmi, P. E. Stromstedt, R. L. Printz, and D. K. Granner. 1995. Expression cloning of a zinc-finger cyclic AMP-response-element-binding protein. Biochem. J. 312:17-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oshiro, J., G. S. Han, W. M. Iwanyshyn, K. Conover, and G. M. Carman. 2003. Regulation of the yeast DPP1-encoded diacylglycerol pyrophosphate phosphatase by transcription factor Gis1p. J. Biol. Chem. 278:31495-31503. [DOI] [PubMed] [Google Scholar]
- 78.Ozsarac, N., M. J. Straffon, H. E. Dalton, and I. W. Dawes. 1997. Regulation of gene expression during meiosis in Saccharomyces cerevisiae: SPR3 is controlled by both ABFI and a new sporulation control element. Mol. Cell. Biol. 17:1152-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parrou, J. L., B. Enjalbert, L. Plourde, A. Bauche, B. Gonzalez, and J. Francois. 1999. Dynamic responses of reserve carbohydrate metabolism under carbon and nitrogen limitations in Saccharomyces cerevisiae. Yeast 15:191-203. [DOI] [PubMed] [Google Scholar]
- 80.Parrou, J. L., M. A. Teste, and J. Francois. 1997. Effects of various types of stress on the metabolism of reserve carbohydrates in Saccharomyces cerevisiae: genetic evidence for a stress-induced recycling of glycogen and trehalose. Microbiology 143:1891-1900. [DOI] [PubMed] [Google Scholar]
- 81.Patil, C. K., H. Li, and P. Walter. 2004. Gcn4p and novel upstream activating sequences regulate targets of the unfolded protein response. PLoS Biol. 2:E246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Piper, M. D., P. Daran-Lapujade, C. Bro, B. Regenberg, S. Knudsen, J. Nielsen, and J. T. Pronk. 2002. Reproducibility of oligonucleotide microarray transcriptome analyses. An interlaboratory comparison using chemostat cultures of Saccharomyces cerevisiae. J. Biol. Chem. 277:37001-37008. [DOI] [PubMed] [Google Scholar]
- 83.Piskur, J. 2001. Origin of the duplicated regions in the yeast genomes. Trends Genet. 17:302-303. [DOI] [PubMed] [Google Scholar]
- 84.Piskur, J., and R. B. Langkjaer. 2004. Yeast genome sequencing: the power of comparative genomics. Mol. Microbiol. 53:381-389. [DOI] [PubMed] [Google Scholar]
- 85.Pritsker, M., Y. C. Liu, M. A. Beer, and S. Tavazoie. 2004. Whole-genome discovery of transcription factor binding sites by network-level conservation. Genome Res. 14:99-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pronk, J. T., S. H. Yde, and J. P. van Dijken. 1996. Pyruvate metabolism in Saccharomyces cerevisiae. Yeast 12:1607-1633. [DOI] [PubMed] [Google Scholar]
- 87.Raitt, D. C., A. L. Johnson, A. M. Erkine, K. Makino, B. Morgan, D. S. Gross, and L. H. Johnston. 2000. The Skn7 response regulator of Saccharomyces cerevisiae interacts with Hsf1 in vivo and is required for the induction of heat shock genes by oxidative stress. Mol. Biol. Cell 11:2335-2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reiner, S., D. Micolod, G. Zellnig, and R. Schneiter. 2006. A genomewide screen reveals a role of mitochondria in anaerobic uptake of sterols in yeast. Mol. Biol. Cell 17:90-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Robinson, M. D., J. Grigull, N. Mohammad, and T. R. Hughes. 2002. FunSpec: a web-based cluster interpreter for yeast. BMC Bioinform. 3:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rogowska, A. T., O. Puchta, A. M. Czarnecka, A. Kaniak, P. P. Stepien, and P. Golik. 2005. Balance between transcription and RNA degradation is vital for Saccharomyces cerevisiae mitochondria: reduced transcription rescues the phenotype of deficient RNA degradation. Mol. Biol. Cell 17:1184-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rosenfeld, E., and B. Beauvoit. 2003. Role of the non-respiratory pathways in the utilization of molecular oxygen by Saccharomyces cerevisiae. Yeast 20:1115-1144. [DOI] [PubMed] [Google Scholar]
- 92.Roth, S., J. Kumme, and H. J. Schuller. 2004. Transcriptional activators Cat8 and Sip4 discriminate between sequence variants of the carbon source-responsive promoter element in the yeast Saccharomyces cerevisiae. Curr. Genet. 45:121-128. [DOI] [PubMed] [Google Scholar]
- 93.Seoighe, C., and K. H. Wolfe. 1999. Yeast genome evolution in the post-genome era. Curr. Opin. Microbiol. 2:548-554. [DOI] [PubMed] [Google Scholar]
- 94.Sertil, O., R. Kapoor, B. D. Cohen, N. Abramova, and C. V. Lowry. 2003. Synergistic repression of anaerobic genes by Mot3 and Rox1 in Saccharomyces cerevisiae. Nucleic Acids Res. 31:5831-5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shakoury-Elizeh, M., J. Tiedeman, J. Rashford, T. Ferea, J. Demeter, E. Garcia, R. Rolfes, P. O. Brown, D. Botstein, and C. C. Philpott. 2004. Transcriptional remodeling in response to iron deprivation in Saccharomyces cerevisiae. Mol. Biol. Cell 15:1233-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Spellman, P. T., G. Sherlock, M. Q. Zhang, V. R. Iyer, K. Anders, M. B. Eisen, P. O. Brown, D. Botstein, and B. Futcher. 1998. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9:3273-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Svetlov, V. V., and T. G. Cooper. 1995. Compilation and characteristics of dedicated transcription factors in Saccharomyces cerevisiae. Yeast 11:1439-1484. [DOI] [PubMed] [Google Scholar]
- 98.ter Linde, J. J., H. Liang, R. W. Davis, H. Y. Steensma, J. P. van Dijken, and J. T. Pronk. 1999. Genome-wide transcriptional analysis of aerobic and anaerobic chemostat cultures of Saccharomyces cerevisiae. J. Bacteriol. 181:7409-7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.ter Linde, J. J., and H. Y. Steensma. 2002. A microarray-assisted screen for potential Hap1 and Rox1 target genes in Saccharomyces cerevisiae. Yeast 19:825-840. [DOI] [PubMed] [Google Scholar]
- 100.van Dyk, D., G. Hansson, I. S. Pretorius, and F. F. Bauer. 2003. Cellular differentiation in response to nutrient availability: the repressor of meiosis, Rme1p, positively regulates invasive growth in Saccharomyces cerevisiae. Genetics 165:1045-1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.van Helden, J., B. Andre, and J. Collado-Vides. 2000. A web site for the computational analysis of yeast regulatory sequences. Yeast 16:177-187. [DOI] [PubMed] [Google Scholar]
- 102.Vasconcelles, M. J., Y. Jiang, K. McDaid, L. Gilooly, S. Wretzel, D. L. Porter, C. E. Martin, and M. A. Goldberg. 2001. Identification and characterization of a low oxygen response element involved in the hypoxic induction of a family of Saccharomyces cerevisiae genes. Implications for the conservation of oxygen sensing in eukaryotes. J. Biol. Chem. 276:14374-14384. [DOI] [PubMed] [Google Scholar]
- 103.Vik, A., and J. Rine. 2001. Upc2p and Ecm22p, dual regulators of sterol biosynthesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 21:6395-6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vuidepot, A. L., F. Bontems, M. Gervais, B. Guiard, E. Shechter, and J. Y. Lallemand. 1997. NMR analysis of CYP1(HAP1) DNA binding domain-CYC1 upstream activation sequence interactions: recognition of a CGG trinucleotide and of an additional thymine 5 bp downstream by the zinc cluster and the N-terminal extremity of the protein. Nucleic Acids Res. 25:3042-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wanke, V., I. Pedruzzi, E. Cameroni, F. Dubouloz, and C. De Virgilio. 2005. Regulation of G0 entry by the Pho80-Pho85 cyclin-CDK complex. EMBO J. 24:4271-4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wilcox, L. J., D. A. Balderes, B. Wharton, A. H. Tinkelenberg, G. Rao, and S. L. Sturley. 2002. Transcriptional profiling identifies two members of the ATP-binding cassette transporter superfamily required for sterol uptake in yeast. J. Biol. Chem. 277:32466-32472. [DOI] [PubMed] [Google Scholar]
- 107.Xing, B., and M. J. van der Laan. 2005. A statistical method for constructing transcriptional regulatory networks using gene expression and sequence data. J. Comput. Biol. 12:229-246. [DOI] [PubMed] [Google Scholar]
- 108.Young, E. T., K. M. Dombek, C. Tachibana, and T. Ideker. 2003. Multiple pathways are co-regulated by the protein kinase Snf1 and the transcription factors Adr1 and Cat8. J. Biol. Chem. 278:26146-26158. [DOI] [PubMed] [Google Scholar]
- 109.Zhu, J., and M. Q. Zhang. 1999. SCPD: a promoter database of the yeast Saccharomyces cerevisiae. Bioinformatics 15:607-611. [DOI] [PubMed] [Google Scholar]
- 110.Zitomer, R. S., and C. V. Lowry. 1992. Regulation of gene expression by oxygen in Saccharomyces cerevisiae. Microbiol. Rev. 56:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.