

Abstract
The strictly anaerobic pathogenic bacterium Clostridium difficile occurs in the human gut and is able to thrive from fermentation of leucine. Thereby the amino acid is both oxidized to isovalerate plus CO2 and reduced to isocaproate. In the reductive branch of this pathway, the dehydration of (R)-2-hydroxyisocaproyl-coenzyme A (CoA) to (E)-2-isocaprenoyl-CoA is probably catalyzed via radical intermediates. The dehydratase requires activation by an ATP-dependent one-electron transfer (J. Kim, D. Darley, and W. Buckel, FEBS J. 272:550-561, 2005). Prior to the dehydration, a dehydrogenase and a CoA transferase are supposed to be involved in the formation of (R)-2-hydroxyisocaproyl-CoA. Deduced amino acid sequences of ldhA and hadA from the genome of C. difficile showed high identities to d-lactate dehydrogenase and family III CoA transferase, respectively. Both putative genes encoding the dehydrogenase and CoA transferase were cloned and overexpressed in Escherichia coli; the recombinant Strep tag II fusion proteins were purified to homogeneity and characterized. The substrate specificity of the monomeric LdhA (36.5 kDa) indicated that 2-oxoisocaproate (Km = 68 μM, k cat = 31 s−1) and NADH were the native substrates. For the reverse reaction, the enzyme accepted (R)- but not (S)-2-hydroxyisocaproate and therefore was named (R)-2-hydroxyisocaproate dehydrogenase. HadA showed CoA transferase activity with (R)-2-hydroxyisocaproyl-CoA as a donor and isocaproate or (E)-2-isocaprenoate as an acceptor. By site-directed mutagenesis, the conserved D171 was identified as an essential catalytic residue probably involved in the formation of a mixed anhydride with the acyl group of the thioester substrate. However, neither hydroxylamine nor sodium borohydride, both of which are inactivators of the CoA transferase, modified this residue. The dehydrogenase and the CoA transferase fit well into the proposed pathway of leucine reduction to isocaproate.
Clostridia and a few other anaerobes can exploit amino acids as an energy source in the absence of a terminal electron acceptor, such as oxygen, nitrate, or sulfate. In the famous Stickland reaction, one amino acid serves as an oxidant and a different one as reductant (36). Other fermentations use the same amino acid as both electron donor and acceptor (1). Thus, Clostridium difficile oxidizes l-leucine to isovalerate (3-methylbutyrate) plus CO2 (oxidative branch, see equation 1) and reduces the amino acid to isocaproate (4-methylpentanoate) (reductive branch, see equation 2) (6, 14). This bacterium is a gram-positive, strictly anaerobic spore-forming human pathogen, which is a major cause of antibiotic-associated diarrhea and the causative agent of pseudomembranous colitis (4, 34).
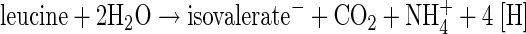 |
(1) |
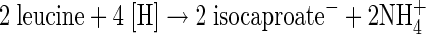 |
(2) |
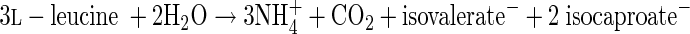 |
(3) |
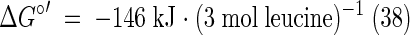 |
(4) |
The oxidative branch of leucine fermentation has not been characterized in detail. The conversion of l-leucine to 2-oxoisocaproate most likely occurs by amino transfer to 2-oxoglutarate and oxidative deamination of the resulting glutamate (Fig. 1A). An alternative way to 2-oxoisocaproate could be a direct oxidative deamination of leucine. Further oxidation of 2-oxoisocaproate, probably by ferredoxin, leads to CO2 plus isovaleryl coenzyme A (CoA), from which isovalerate and ATP are formed via substrate level phosphorylation.
FIG. 1.

Enzymes (A) and their genes (B) involved in the reductive branch of l-leucine fermentation by C. difficile. (1) Leucine aminotransferase and glutamate dehydrogenase; (2) ldhA, (R)-2-hydroxyisocaproate dehydrogenase; (3) hadA, 2-hydroxyisocaproate CoA transferase; (4) hadI, activator of dehydratase; (5) hadBC, 2-hydroxyisocaproyl-CoA dehydratase; (6) acdB, acyl-CoA dehydrogenase, and etfBA, electron transfer proteins. 2-OG, 2-oxogluatrate; Glu, glutamate. Enzymes 4 and 5 (20) and 2 and 3 (this work) have been purified and characterized.
The proposed pathway of the reductive branch of leucine fermentation is shown in Fig. 1A. The first step, the oxidation of leucine to 2-oxoisocaproate, is the same as in the oxidative branch. Reduction of 2-oxoisocaproate by NADH leads to (R)-2-hydroxyisocaproate, which is converted to the thiol ester by CoA transfer to yield (R)-2-hydroxyisocaproyl-CoA, the substrate of the subsequent dehydration to 2-isocaprenoyl-CoA. The latter reaction is chemically difficult because the β-hydrogen to be eliminated as a proton is not acidic (pK, ca. 40). The acidification of this hydrogen requires radical chemistry mediated by two enzymes, a dehydratase (HadBC) and an ATP- and ferredoxin-dependent activator, also called archerase (HadI). The purification and characterization of both enzymes have been previously reported (20, 30a). Four other open reading frames (hadA, acdB, and etfBA) form a transcriptional unit with hadIBC (Fig. 1B) (12). The amino acid sequence alignment indicated that HadA belongs to family III of the CoA transferases, which could catalyze the formation of (R)-2-hydroxyisocaproyl-CoA (16). The three additional genes, acdB and etfBA, might encode the enzymes mediating the reduction of 2-isocaprenoyl-CoA to 2-isocaproyl-CoA by NADH, since the deduced amino acid sequences are homologous to short-chain acyl-CoA dehydrogenases and to the two subunits of electron transferring flavoproteins, respectively. Upstream of hadA, the open reading frame ldhA, running in the opposite direction, was detected, whose translation identified it as a member of the NAD+-dependent d-lactate dehydrogenase family (2).
In summary, the reductive branch contains one oxidative and two reductive steps. Thus, 2 mol of leucine has to be reduced to 2 mol of isocaproate (equation 2) to balance both oxidative steps in the conversion of 1 mol of leucine to isovalerate plus CO2 (equation 1). Since in the reductive branch no ATP is formed via substrate level phosphorylation, only 1 mol of ATP is conserved from 3 mol of leucine. The free energy of the fermentation of 3 leucines allows, however, the conservation of 2 ATP (≤−70 kJ mol−1 ATP) (38). Therefore, the organism could use additional ion gradient phosphorylation, which might be generated via the oxidation of reduced ferredoxin by NAD+ (3).
In this work, we describe the cloning of ldhA and hadA and their expression in Escherichia coli as N- and C-terminal Strep tag II fusion proteins, respectively. The purification and characterization of the produced enzymes identified LdhA as NAD+-dependent (R)-2-hydroxyisocaproate dehydrogenase and HadA as (R)-2-hydroxyisocaproate CoA transferase, which catalyzes the conversion of 2-oxoisocaproate to (R)-2-hydroxyisocaproyl-CoA, the substrate of the dehydration in the reductive pathway of l-leucine in C. difficile.
MATERIALS AND METHODS
Materials.
Clostridium difficile (DSM 1296T) was purchased from the Deutsche Sammlung für Mikroorganismen und Zellkulturen (DMSZ, Braunschweig, Germany). Expression vectors pASK-IBA3 and pASK-IBA7 and a Strep-Tactin Sepharose column were from IBA GmbH (Göttingen, Germany). The enzymes for DNA manipulation were obtained from New England Biolabs (Frankfurt am Main, Germany), ABgene (Hamburg, Germany), and Amersham Biosciences (Freiburg, Germany). PCR and sequencing primers labeled at their 5′ ends with the infrared dye IRD-41 were purchased from MWG (Ebersberg, Germany). Protein molecular mass markers and DNA size markers were from Amersham Biosciences.
Chemicals and synthesis of CoA esters.
Pyruvate, isocaproate (4-methylpentanoate), 2-oxoisocaproate (4-methyl-2-oxopentanoate), 2-oxobutyrate, 2-oxoisovalerate (3-methyl-2-oxobutyrate), 2-oxopentanoate, 2-oxohexanoate, and phenylpyruvate were obtained from Sigma Aldrich (München, Germany). d- and l-leucine were deaminated with nitrous acid to (R)- and (S)-2-hydroxyisocaproate, respectively (10). (E)-2-Isocaprenoate (4-methyl-2-pentenoic acid) was synthesized from isobutyraldehyde with sodium diethyl malonate (15). (R)-2-Hydroxyisocaproyl-CoA, (S)-2-hydroxyisocaproyl-CoA, (E)-2-isocaprenoyl-CoA, and isocaproyl-CoA were prepared by following the modified anhydrous 1,1′-carbonyldiimidazole synthesis method (19). The molecular masses of the CoA esters were confirmed by matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectrometry as described in references 20 and 32.
Gene cloning.
Routine manipulation of plasmid DNA, PCR, and construction of recombinant plasmids were performed as described in reference 29. C. difficile was cultivated as described previously (31), and the chromosomal DNA was isolated using standard techniques. Proofreading polymerase (Extensor Hi-Fidelity PCR enzyme mix from ABgene, Hamburg, Germany) was used for the PCR amplification of the open reading frames (ORFs) ldhA and hadA using the following primers containing the BsaI restriction site [GGTCTC(N)1, underlined]: FldhA, 5′-ATGGTAGGTCTCAGCGCAAAATACTAGTATTTGGAGCACGCG-3′; RldhA, 5′-ATGGTAGGTCTCATATCAATTTACTCTATTAGTAGCAGTTCCTG-3′; FhadA, 5′-ATGGTAGGTCTCAAATGCTTTTAGAAGGAGTTAAAGTAGTAGA-3′; RhadA, 5′-ATGGTAGGTCTCAGCGCTATATCTTACAACTTTACTATCTTTAAAG-3′. The amplified fragments, 1.0 kb (ldhA) and 1.2 kb (hadA) in length, were cloned into the BsaI restriction site of the expression vectors pASK-IBA7 and pASK-IBA3, resulting in N- and C-terminal Strep tag II (Trp-Ser-His-Pro-Gln-Phe-Glu-Lys) fusion proteins, respectively. Three clones from three separate PCRs were sequenced to exclude reading errors of the polymerase. The plasmid constructs were named p7ldhA (pASK-IBA7::ldhA) and p3hadA (pASK-IBA3::hadA).
Preparation of HadA variants (D171N and D171A).
The mismatch (underlined) oligonucleotide primers were designed as follows: FD171N, 5′-CAGCAGCAGGATTTGGTAACCACTATGCAGGTCTAG-3′; RD171N, 5′-CTAGACCTGCATAGTGGTTACCAAATCCTGCTGCTG-3′; FD171A, 5′-CAGCAGGATTTGGTGCGCACTATGCAGGTCTAG-3′; RD171A, 5′-CTAGACCTGCATAGTGCGCACCAAATCCTGCTG-3′. Each mutant plasmid was constructed by following the protocol of the QuikChange II site-directed mutagenesis kit from Amersham Biosciences (Freiburg, Germany). The point mutation was confirmed by sequencing the gene; secondary mutations were not detected. The mutant plasmids were handled exactly as described below for the wild-type plasmid p3hadA.
Gene expression and protein purification.
For the expression of genes, p7ldhA or p3hadA was transformed into E. coli BL21-CodonPlus(DE3) harboring rare codon tRNA genes (arg, ileY, and leuW) for expression of AT-rich genes. An overnight preculture of 50 ml at 37°C (p7ldhA) or 30°C (p3hadA) was used to inoculate 1 liter Standard I medium (Merck, Darmstadt, Germany) containing antibiotics (ampicillin, 100 μg · ml−1; chloramphenicol, 50 μg · ml−1) and grown at the same temperature under aerobic conditions. When the culture reached the middle exponential phase, D 578 = 0.5 to 0.7, gene expression was induced with anhydrotetracycline (200 μg · liter−1). Cells were harvested 3 h after induction, washed, and suspended in 3 volumes of equilibration buffer (100 mM Tris-HCl, pH 8.0, 1 mM EDTA, and 300 mM NaCl). Cells were broken by sonication, and cell debris was removed by ultracentrifugation at 100,000 × g for 1 h. The supernatant was loaded by gravity flow onto a 5-ml Strep-Tactin Sepharose column, which was washed with 5 column volumes of equilibration buffer. The enzyme was eluted with equilibration buffer containing 3 mM d-dethiobiotin. The enzyme could be stored at −80°C for at least 2 months without any significant loss of activity.
Enzyme activity assay.
(R)-2-Hydroxyisocaproate dehydrogenase activity was measured in cuvettes of 1.0-ml total volume containing 50 mM Tris-HCl, pH 8.0, 0.2 mM NADH, and 1.0 mM 2-oxoisocaproate at ambient temperature. After addition of the enzyme, the absorbance decrease of NADH was monitored at 340 nm (ɛ = 6.3 mM−1 cm−1) (41). The back reaction was measured under the same conditions with (R)-2-hydroxyisocaproate and NAD+ as substrates. To increase the sensitivity at low NAD+ or (R)-2-hydroxyisocaproate concentrations, the formazan method was used. Thereby, the cuvette contained, in addition, 1.0 mM iodonitrotetrazolium chloride [2-(4-iodophenyl)-3-(4-nitrophenyl)-5-phenyltetrazolium chloride], 60 nM meldolablau (8-dimethylamino-2,3-benzophenoxazine), and 0.1% Triton X-100. The formation of the red formazan was followed at 492 nm (ɛ = 19.4 mM−1 cm−1) (27).
2-Hydroxyisocaproate CoA transferase was measured using the difference of the millimolar absorbance between (R)-2-hydroxyisocaproyl-CoA (ɛ260 = 16 mM−1 cm−1) and (E)-2-isocaprenoyl-CoA (ɛ260 = 22 mM−1 cm−1) (9) in a total volume of 1.0 ml 50 mM potassium phosphate, pH 7.0, at room temperature. The absorbance increase due to the formation of (E)-2-isocaprenoyl-CoA from (E)-2-isocaprenoate and (R)-2-hydroxyisocaproyl-CoA was monitored at 260 nm (Δɛ260 = 6 mM−1 cm−1). Unless otherwise indicated, the concentration of the CoA thioesters was 200 μM and that of the acids was 1 mM.
Inactivation of CoA transferase.
The purified HadA (320 μg) was incubated with 20 mM NaBH4 or 200 mM hydroxylamine at pH 7.0 in the presence of 200 μM (R)-2-hydroxyisocaproyl-CoA. After 30 min of incubation at ambient temperature, CoA transferase activity was measured from small samples as described above (see references 7 and 13).
Generation and separation of peptides.
The inactivated HadA (2 nmol) was prepared as described above, and the buffer was exchanged by gel filtration on Sephadex G25 equilibrated with 50 mM ammonium acetate, pH 7.0, 10% (vol/vol) acetonitrile. The protein was digested with 2% (wt/wt) trypsin for 4 h at 37°C and lyophilized. The peptides were redissolved in 100 μl 4 M guanidinium hydrochloride, 0.1% (vol/vol) trifluoroacetic acid and partially separated on a Superdex peptide column (HR 10/30; Amersham) equilibrated with 10% (vol/vol) acetonitrile, 0.1% (vol/vol) trifluoroacetic acid. The effluent was collected in 500-μl fractions and analyzed by MALDI-TOF mass spectrometry as previously described (30).
Molecular masses of native enzymes.
The apparent molecular masses of the enzymes were determined by gel filtration on a Superose 6 column in 150 mM NaCl and 50 mM Tris-HCl, pH 8.0, at a flow rate of 0.5 ml/min. Amylase, aldolase, bovine serum albumin, catalase, and cytochrome c were used for calibration. The molecular mass standards were obtained from Roche Molecular Biochemicals (Mannheim, Germany).
Other biochemical methods.
Protein concentration was determined with the Bio-Rad protein assay. Bovine serum albumin was used as a standard (5). Sodium dodecyl sulfate (SDS)-polyacrylamide gels were stained with Coomassie brilliant blue.
RESULTS
Cloning and expression of the genes.
The previously identified ORFs, ldhA and hadA, were amplified by PCR using proofreading polymerase with pairs of primers described in Materials and Methods. The amplified genes were ligated into the expression vectors pASK-IBA7 and pASK-IBA3, resulting in the plasmids p7ldhA and p3hadA, respectively. To obtain an authentic nucleotide sequence, three clones from three individual PCRs were sequenced. The gene ldhA was composed of 993 bp (GenBank accession number AY772817). Although three nucleotide bases (T552, C891, and C945) did not match with those of the C. difficile strain 630 (C, T, and T) from the Sanger Centre, the deduced amino acids were identical. The nucleotide sequence of hadA was 1,194 bp long (GenBank accession number AY772818), with two silent mismatches (C813T and A456G). For expression of the genes, the plasmid constructs p7ldhA and p3hadA were transformed into E. coli BL21-CodonPlus(DE3), and the proteins were produced as described in Materials and Methods. The recombinant proteins were purified by one-step affinity chromatography on Strep-Tactin Sepharose columns, on which the Strep tag II peptide fused to the N terminus of LdhA and to the C terminus of HadA could bind. From our experience, none of the NAD+-dependent hydroxy acid dehydrogenases and CoA transferases from anaerobic bacteria have been sensitive to oxygen (7, 13, 30, 33, 40). Therefore, recombinant LdhA and HadA were purified and characterized under oxic conditions.
(R)-2-Hydroxyisocaproate dehydrogenase.
The purified LdhA Strep tag II fusion protein showed a band of 37 kDa on SDS-polyacrylamide gel electrophoresis (PAGE) (Fig. 2), which agreed well with the calculated mass of the deduced amino acid sequence (36.5 kDa plus 1 kDa of Strep tag II peptide). The recombinant protein behaved as a monomer (36 ± 1 kDa) on the gel filtration column Superose 6. The enzyme activity was measured under aerobic conditions by monitoring the absorbance decrease of NADH at 340 nm after addition of a 2-oxo acid; NADPH did not serve as a cosubstrate. NADH reduced 2-oxoisocaproate (4-methyl-2-oxopentanoate), 2-oxopentanoate, 2-oxohexanoate, and phenylpyruvate to the corresponding 2-hydroxy acids by NADH, while 2-oxoisovalerate (3-methyl-2-oxobutyrate) and 2-oxobutyrate were not accepted as substrates (Table 1). Apparent Km and V max values were determined for each substrate, and comparing the catalytic parameters, the native substrate of the dehydrogenase is most likely 2-oxoisocaproate (Km = 68 μM; k cat = 31 s−1; k cat/Km = 4.6 × 105 M−1 s−1). For the reverse reaction, (R)- and (S)-2-hydroxyisocaproate were tested in the presence of 1.0 mM NAD+; NADP+ gave no measurable activity. The dehydrogenase activity was obtained only with the R-enantiomer with a Km of 2.8 mM, a k cat of 51 s−1, and a k cat/Km ratio of 1.8 × 104 M−1 s−1. Therefore, the enzyme was named (R)-2-hydroxyisocaproate dehydrogenase. The Km values for NAD+ (56 μM) and NADH (31 μM) with (R)-2-hydroxyisocaproate and 2-oxoisocaproate as second substrates, respectively, were also determined. They are about an order of magnitude lower than the concentrations of the coenzymes used for the determination for the Km values of the carboxylates. Hence, saturating concentrations of the coenzymes were applied.
FIG. 2.

Purification of recombinant (R)-2-hydroxyisocaproate dehydrogenase fused at its N terminus with the Strep tag II peptide. SDS-PAGE (15%) was stained with Coomassie brilliant blue. M, molecular mass marker; UI, uninduced cell extract; I, cell extract induced with anhydrotetracycline (200 μg/liter); FT, flowthrough of the Strep-Tactin Sepharose affinity column; lanes 1 to 3, fractions (5 ml) obtained by elution with 3 mM d-dethiobiotin.
TABLE 1.
Substrate specificity of recombinant (R)-2-hydroxyisocaproate dehydrogenase fused at its N-terminus with the Strep tag II peptide a
| Substrate | Km (mM) | kcat (s−1) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| (R)-2-Hydroxyisocaproate | 2.80 ± 0.03 | 51 ± 2 | 1.8 × 104 |
| 2-Oxoisocaproate | 0.068 ± 0.004 | 31 ± 1 | 4.6 × 105 |
| 2-Oxopentanoate | 0.084 ± 0.005 | 22 ± 1 | 2.6 × 105 |
| 2-Oxohexanoate | 5.0 ± 0.5 | 33 ± 2 | 6.6 × 103 |
| Phenylpyruvate | 10 ± 1 | 63 ± 3 | 6.3 × 103 |
| NAD+ | 0.056 ± 0.001 | 25 ± 1 | 4.5 × 105 |
| NADH | 0.031 ± 0.001 | 49 ± 2 | 1.6 × 106 |
All assays were performed in 100 mM Tris-HCl, pH 8.0, with 0.2 mM NADH or 1 mM NAD+. The K m for NAD+ was measured with 10 mM (R)-2-hydroxyisocaproate, and that for NADH was measured with 1 mM 2-oxoisocaproate. No reactions could be observed with 10 mM (S)-2-hydroxyisocaproate and 1 mM NAD+, 10 mM pyruvate or 10 mM 2-oxoisovalerate and 0.2 mM NADH, and 1.0 mM 2-oxoisocaproate and 0.2 mM NADPH.
2-Hydroxyisocaproate CoA transferase.
In the first trial, no CoA transferase activity was detected using the protein prepared with a Strep tag II fused to the N terminus. We assumed that the structure of the active site of the enzyme was disturbed by the Strep tag II peptide. By C-terminal tag fusion, however, an active enzyme was obtained. The fusion protein had molecular masses of 43 kDa, as determined by SDS-PAGE (Fig. 3), and 86 ± 2 kDa, as determined by size-exclusion chromatography, indicating a homodimeric quaternary structure. The CoA transferase activity was measured under oxic conditions using the difference of the molar absorbance (Δɛ260 = 6 mM−1 cm−1) between α,β-unsaturated and saturated CoA thioesters. Due to the high background absorbance, detailed kinetic studies could not be performed. A specific activity of 1.4 U/mg was obtained for the CoA transfer from 200 μM (R)-2-hydroxyisocaproyl-CoA to 1.0 mM (E)-2-isocaprenoate, and a specific activity of 1.3 U/mg was obtained with 200 μM isocaproyl-CoA as the CoA donor. Although this specific activity is low for a good enzyme, the V max values of the family III CoA transferases specific for phenyllactate (13) and oxalate (18), which are kinetically much better characterized, are not much higher (≤6 U/mg). Therefore, we assume that the low specific activity is an intrinsic property of family III CoA transferases and not due to the fusion of the C terminus with the Strep tag II peptide. The formation of (R)-2-hydroxyisocaproyl-CoA (mass = 883.33 Da) from isocaproyl-CoA (mass = 867.30 Da) and (R)-2-hydroxyisocaproate could be detected by MALDI-TOF mass spectrometry (Fig. 4). The spectrophotometric determination of the latter CoA transfer was not possible because of the almost complete absence of an absorbance difference between acyl-CoA (λmax = 233 nm; ɛ = 4.5 mM−1 cm−1) (9) and 2-hydroxyacyl-CoA (λmax = 235 nm; ɛ = 4.7 mM−1 cm−1) (21). Other CoA thioesters, such as acetyl-CoA or butyryl-CoA, were not accepted as substrates. Unexpectedly, CoA transfers from chemically synthesized (E)-2-isocaprenoyl-CoA to (R)-2-hydroxyisocaproate or to isocaproate could not be observed. The synthesized (E)-2-isocaprenoyl-CoA, however, revealed the correct mass by MALDI-TOF mass spectrometry (mass = 865.30 Da). Furthermore, under anoxic conditions, a cell extract from C. difficile catalyzed the NADH-dependent reduction of this compound with a specific activity of 3 U/mg; after exposing the extract to air for 15 h at 0°C, the activity was lost. Therefore, it can be assumed that this compound is (E)-2-isocaprenoyl-CoA, which is reduced to isocaproyl-CoA according to Fig. 1A. In the last step of this pathway, isocaproyl-CoA serves as a CoA donor for (R)-2-hydroxyisocaproate. In another experiment, however, 0.1 mM (R)-2-hydroxyisocaproyl-CoA was incubated with 1.0 mM (E)-2-isocaprenoate. The addition of CoA transferase caused an increase in absorbance at 260 nm until equilibrium was reached. At this point, 1.0 mM (R)-2-hydroxyisocaproate was added, and a decrease in absorbance was observed until the new equilibrium was established. Hence, this experiment indicates that the reaction is reversible and that the unsaturated CoA derivative generated and consumed again in this assay is not identical to the chemically synthesized (E)-2-isocaprenoyl-CoA.
FIG. 3.

Purification of 2-hydroxyisocaproate CoA transferase fused at its C terminus with the Strep tag II peptide. SDS-PAGE (15%) was stained with Coomassie brilliant blue. M, molecular mass marker; UI, uninduced cell extract; I, cell extract induced with anhydrotetracycline (200 μg/liter); FT, flowthrough of the Strep-Tactin Sepharose affinity column; lanes 1 and 2, fractions (5 ml) obtained by elution with 3 mM d-dethiobiotin.
FIG. 4.

MALDI-TOF mass spectrometry for CoAS− transfer from isocaproyl-CoA to (R)-2-hydroxyisocaproate catalyzed by the HadA Strep-tag II fusion. Isocaproyl-CoA (0.2 μmol) was incubated with 0.2 U HadA and (R)-2-hydroxyisocaproate (1 mmol) in 50 mM potassium phosphate buffer, pH 7.0, at ambient temperature in a total volume of 1 ml. After 30 min of incubation, the reaction product was purified through a Sep-pak C18 cartridge (Waters, Eschborn, Germany), and the molecular masses (isocaproyl-CoA, 867.30; (R)-2-hydroxyisocaproyl-CoA, 883.33) were determined by MALDI-TOF mass spectrometry as described in references 12 and 19.
Inactivation of HadA.
According to the amino acid sequence alignment, 2-hydroxyisocaproate CoA transferase belongs to family III (16), whose reaction mechanism was proposed to proceed via formation of a covalent anhydride intermediate between a conserved aspartate residue and the acyl group of the CoA thioester substrate (18, 35). If this anhydride is cleaved by a nucleophile other than water, which attacks at the carbonyl of the aspartate, the enzyme should become inactive. There are reports of an unambiguous partial inactivation of family III CoA transferases by NaBH4 or hydroxylamine (13, 22), whose protocol we applied to the recombinant 2-hydroxyisocaproate CoA transferase. Therefore, the enzyme was incubated with 20 mM NaBH4 or 200 mM hydroxylamine at pH 7.0 in the presence of 200 μM (R)-2-hydroxyisocaproyl-CoA. After removal of the small molecules by gel filtration, the transferase samples showed 88% or 94% inactivation by NaBH4 or hydroxylamine, respectively, while no inactivation was observed under the same conditions in the absence of (R)-2-hydroxyisocaproyl-CoA. To check whether these two reagents had modified the conserved aspartate residue D171 (184, according to the numbering in Fig. 5), we determined the mass of the tryptic peptide G159-K187 (G172-K201 in Fig. 5) by MALDI-TOF mass spectrometry (m/z = 2,813; calculated average m/z = 2,812) (Fig. 5). In both cases, however, we found the same mass as that of the untreated enzyme. No peak was observed at an m/z of −14 (treatment with NaBH4) or at an m/z of +15 (treatment with hydroxylamine). Incubation of the CoA transferase with 2-hydroxyisocaproyl-CoA did not result in the formation of an enzyme-CoA thioester, as observed with family I CoA transferases (7). Afterwards, the excess of the CoA derivative was removed by a passage through Sephadex G-25, and the obtained protein was analyzed by MALDI-TOF mass spectrometry and UV spectroscopy. No increase in the mass or the absorbance at 260 nm could be detected.
FIG. 5.

Amino acid sequence alignment for family III CoA transferases. HadA, 2-hydroxyisocaproate CoA transferase from C. difficile; FldA, phenyllactate CoA transferase from C. sporogenes; Frc, formate CoA transferases from O. formigenes; BbsF, one subunit of benzylsuccinate CoA transferase from T. aromatica; CaiB, carnitine CoA transferase from E. coli. Identical amino acid residues are shown with a gray background, and the Asp residue forming anhydride intermediates is indicated by an asterisk.
Since the conserved D171 appeared not to be modified by inactivation with borohydride or hydroxylamine, we assessed its participation in catalysis by site-specific mutagenesis. Two variant enzymes, D171N and D171A, were constructed. Like the active enzyme, both variants contained the Strep tag II fused to the C terminus and were readily purified by affinity chromatography. As a control, the wild-type enzyme was prepared in an identical manner. Whereas, in the standard assay, 2 μg of the wild-type enzyme exhibited a ΔA of 0.23 min−1, 20 μg of each mutant gave no activity (<0.001 min−1). Hence, both variants are more than 2,000-fold less active. Family I glutaconate CoA transferase contains a glutamate residue (βE54) at the active site, which forms a CoA thiol ester during catalysis (23). The βE54Q variant exhibited 1% activity, which could be increased to almost that of the wild type (82%) after incubation with both substrates at 37°C for 40 h. Whereas the activity of the variant reached a maximum at 40 h, that of the wild type passed through a maximum (140%) at 12 h and returned to its original value (100%) at 40 h (25). When this experiment was performed with the 2-hydroxyisocaproate CoA transferase in the presence of 0.5 mM 2-hydroxyisocaproate and 10 mM isocaprenoate at pH 8.0 and 25°C, the wild type also became activated after 1 h (120%) and afterwards returned to its original value (100%). Both variants, however, remained completely inactive during the whole incubation time of 40 h.
DISCUSSION
The amino acid sequence of (R)-2-hydroxyisocaproate dehydrogenase LdhA shows high identities to other d-2- or (R)-2-hydroxyacid dehydrogenases such as the d-lactate dehydrogenases from Lactobacillus pentosus (45%) and Lactobacillus bulgaricus (40%), d-2-hydroxyisocaproate dehydrogenase (d-HicDH) from Lactobacillus casei (38%), and (R)-2-hydroxyglutarate dehydrogenase (HGDH) from Acidaminococcus fermentans (36%). These enzymes belong to the d-lactate dehydrogenase (D-LDH) family, which has a substrate stereospecificity different from that of the counterpart l-lactate dehydrogenase. These two families of enzymes are only distantly related to each other. The crystal structures of d-LDH from L. pentosus (37) and L. bulgarius (28), d-HicDH from L. casei (11), and HGDH from A. fermentans (26) identified key conserved catalytic amino acid residues, especially E265 and H297, which act as general acids to protonate at the substrate carbonyl oxygen, while R236 fixes the substrate carboxylate anion by a salt bridge. In d-HicDH, L51 corresponding to Y52 in d-LDH and R52 in HGDH are suggested as key residues for the differences in substrate specificity. Replacement of Y52 by leucine in d-LDH from L. pentosus caused a change of the substrate specificity from d-lactate to d-2-hydroxyisocaproate (39). This key residue is not conserved in LdhA but occupied by a glutamine, which might explain in part the much broader substrate specificity of d-HicDH. No activity could be observed with 3-methyl-2-oxobutyrate (2-oxoisovalerate), which might be due to the limited space available for the methyl group at carbon 3 formed by F300 and I309 (Y298 and M307, respectively, in d-HicDH from L. casei). The aromatic amino acids F300 in LdhA and Y298 in d-HicDH could, however, be responsible for the high activities observed for these enzymes with phenylpyruvate as substrate, which may bind via π-π interactions.
Although d-2-hydroxyisocaproate dehydrogenases from lactic acid bacteria have been extensively studied and applied to the synthesis of commercially valuable chiral compounds (17), their role in metabolism is unknown. This report shows that (R)-2-hydroxyisocaproate should be the native product of LdhA catalyzing the NADH-dependent reduction of 2-oxoisocaproate, a step in the reductive branch of l-leucine fermentation by C. difficile (Fig. 1A). The fermentation of amino acids via their (R)-2-hydroxy acids appears to be a rich source for d-2-hydroxyacid dehydrogenases, as are d-lactate dehydrogenase in alanine fermentation by Clostridium propionicum (30), (R)-2-hydroxyglutarate dehydrogenase in glutamate fermentation by A. fermentans and Fusobacterium nucleatum (26), (R)-3-phenyllactate dehydrogenase in phenylalanine fermentation by Clostridium sporogenes (13), and (R)-2-hydroxyisocaproate dehydrogenase (LdhA) in leucine fermentation by C. difficile, as shown in this work.
The conversion of (R)-2-hydroxyisocaproate to an electron-withdrawing CoA thioester is essential for the consecutive unusual dehydration, which has been proposed to be initiated by a one-electron reduction of the thioester carbonyl to a ketyl radical anion (8). As shown for the 2-hydroxy acid dehydrogenases, CoA transferases are also present in every pathway of amino acid fermentation via (R)-2-hydroxy acids. Furthermore, their genes are located upstream of those coding for the corresponding 2-hydroxyacyl-CoA dehydratase. This has been established for propionate CoA transferase from C. propionicum (33; T. Selmer, unpublished data), glutaconate CoA transferase from A. fermentans (23), Clostridium symbiosum (D. Brügel, I. Schall, and W. Buckel, unpublished data), and Fusobacterium nucleatum (3), phenyllactate CoA transferase from C. sporogenes (13) and Clostridium botulinum (12), and 2-hydroxyisocaproate CoA transferase, as is shown in Fig. 1. Remarkably, however, propionate and glutaconate CoA transferases are members of family I CoA transferases, whereas phenyllactate and 2-hydroxyisocaproate CoA transferases apparently belong to family III.
Although the highly specific CoA transferase described here readily catalyzed the formation of (R)-2-hydroxyisocaproate and 2-isocaprenoyl-CoA from (R)-2-hydroxyisocaproyl-CoA and (E)-2-isocaprenoate, chemically synthesized (E)-2-isocaprenoyl-CoA did not serve as a CoA donor in the back reaction. In contrast, the enzymatically in situ-produced 2-isocaprenoyl-CoA was accepted as a substrate of the CoA transferase and mediated the CoA transfer from to (R)-2-hydroxyisocaproate. Since synthetic (E)-2-isocaprenoyl-CoA as well as the CoA ester prepared with the CoA transferase also were not hydrated by the dehydratase (20), we conclude that the thereby unavoidable treatments with acid caused an unknown change in the molecule.
The amino acid sequence of 2-hydroxyisocaproate CoA transferase showed identities with family III enzymes: 45% to phenyllactate CoA transferase (FldA) from Clostridium sporogenes, 24% to one subunit of benzylsuccinate CoA transferase (BbsF) from Thauera aromatica, 25% to oxalate CoA transferase (Frc) from Oxalobacter formigenes, and 25% to carnitine CoA transferase (CaiB) from E. coli. Crystal structures of Frc (18) and CaiB (35) revealed the conserved D169 of Frc (D171 in HadA, D184 in Fig. 5) as a key residue for catalysis. Furthermore, a mixed anhydride between that D169 and formate has been directly seen in the structure of Frc and the activities of the D169A, S and E mutants were decreased more than 1,000-fold (19). Trapping or reducing the anhydride between D184 and 2-hydroxyisocaproate at the carbonyl group of HadA could readily explain the almost complete inactivation of the enzyme with hydroxylamine or borohydride in the presence of substrate. Interestingly, the mass of the tryptic peptide containing D171 remained unchanged after such treatment. Possibly, the mixed anhydride between substrate and enzyme was cleaved by hydroxylamine or borohydride at the carbonyl of the substrate. This would lead to unmodified enzyme and either hydroxylamine to the hydroxamic acid of 2-hydroxyisocaproate or borohydride to 4-methylpentane-1,2-diol, both of which could act as competitive inhibitors. But gel filtration should have removed all small molecules and thus might exclude such inhibitions. Hence, these observations call into question either the participation of D171 in catalysis or the proposed mechanism. Our results with the >2,000-fold less-active D171N and D171A variants (cf. the variants of formate CoA transferase, see above) demonstrate, however, that D171 does participate in catalysis but apparently not in the same manner as in family I CoA transferases (24, 25). Whereas, upon incubation of the βE57Q variant of glutaconate CoA transferase with both substrates, the CoA ester and the carboxylate, the enzyme was converted to the wild type by exchange of the amide group by hydroxyl, this was not observed with the D171N variant of 2-hydroxisocaproate CoA transferase. On the other hand, a function of D171 different from the formation of a mixed anhydride also appears unlikely. For a CoA transfer, neither a catalytic base nor an acid is required (32). Currently, we have no reasonable answer to this intriguing question.
Acknowledgments
The work was funded by grants from the Deutsche Forschungsgemeinschaft and the Fonds der Chemischen Industrie.
REFERENCES
- 1. Barker, H. A. 1961. Fermentation of nitrogenous compounds, p. 151-207. In I. C. Gunsalus (ed.), The bacteria, vol. 2. Academic Press, Inc., New York, N.Y. [Google Scholar]
- 2. Bernard, N., K. Johnsen, T. Ferain, D. Garmyn, P. Hols, J. J. Holbrook, and J. Delcour. 1994. NAD+-dependent D-2-hydroxyisocaproate dehydrogenase of Lactobacillus delbrueckii subsp. bulgaricus. Gene cloning and enzyme characterization. Eur. J. Biochem. 224: 439-446. [DOI] [PubMed] [Google Scholar]
- 3. Boiangiu, C. D., E. Jayamani, D. Brügel, G. Herrmann, L. Forzi, R. Hedderich, I. Vgenopoulou, J. Kim, A. J. Pierik, J. Steuber, and W. Buckel. 2005. Sodium ion pumps and hydrogen production in glutamate fermenting anaerobic bacteria. J. Mol. Microbiol. Biotechnol. 10: 105-119. [DOI] [PubMed] [Google Scholar]
- 4. Borriello, S. P., and M. H. Wilcox. 1998. Clostridium difficile infections of the gut: the unanswered questions. J. Antimicrob. Chemother. 41(Suppl. C): 67-69. [DOI] [PubMed] [Google Scholar]
- 5. Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248-254. [DOI] [PubMed] [Google Scholar]
- 6. Britz, M. L., and R. G. Wilkinson. 1982. Leucine dissimilation to isovaleric and isocaproic acids by cell suspensions of amino acid fermenting anaerobes: the Stickland reaction revisited. Can. J. Microbiol. 28: 291-300. [DOI] [PubMed] [Google Scholar]
- 7. Buckel, W., U. Dorn, and R. Semmler. 1981. Glutaconate CoA-transferase from Acidaminococcus fermentans. Eur. J. Biochem. 118: 315-321. [DOI] [PubMed] [Google Scholar]
- 8. Buckel, W., M. Hetzel, and J. Kim. 2004. ATP-driven electron transfer in enzymatic radical reactions. Curr. Opin. Chem. Biol. 8: 462-467. [DOI] [PubMed] [Google Scholar]
- 9. Decker, K. 1959. Die aktivierte Essigsäure. Ferdinand Enke Verlag, Stuttgart, Germany.
- 10. Degerbeck, F., B. Fransson, L. Grehn, and U. Ragnarsson. 1995. Synthesis of 15N-labelled Leu-enkephalins. Acta Chem. Scand. 49: 149-151. [DOI] [PubMed] [Google Scholar]
- 11. Dengler, U., K. Niefind, M. Kiess, and D. Schomburg. 1997. Crystal structure of a ternary complex of D-2-hydroxyisocaproate dehydrogenase from Lactobacillus casei, NAD+ and 2-oxoisocaproate at 1.9 Å resolution. J. Mol. Biol. 267: 640-660. [DOI] [PubMed] [Google Scholar]
- 12. Dickert, S., A. J. Pierik, and W. Buckel. 2002. Molecular characterization of phenyllactate dehydratase and its initiator from Clostridium sporogenes. Mol. Microbiol. 44: 49-60. [DOI] [PubMed] [Google Scholar]
- 13. Dickert, S., A. J. Pierik, D. Linder, and W. Buckel. 2000. The involvement of coenzyme A esters in the dehydration of (R)-phenyllactate to (E)-cinnamate by Clostridium sporogenes. Eur. J. Biochem. 267: 3874-3884. [DOI] [PubMed] [Google Scholar]
- 14. Elsden, S. R., and M. G. Hilton. 1978. Volatile acid production from threonine, valine, leucine and isoleucine by clostridia. Arch. Microbiol. 117: 165-172. [DOI] [PubMed] [Google Scholar]
- 15. Gibson, T. W., and W. F. Erman. 1972. The photochemistry of substituted 1,5-hexadien-3-ones. J. Org. Chem. 37: 1148-1154. [Google Scholar]
- 16. Heider, J. 2001. A new family of CoA-transferases. FEBS Lett. 509: 345-349. [DOI] [PubMed] [Google Scholar]
- 17. Hummel, W., and M. R. Kula. 1989. Dehydrogenases for the synthesis of chiral compounds. Eur. J. Biochem. 184: 1-13. [DOI] [PubMed] [Google Scholar]
- 18. Jonsson, S., S. Ricagno, Y. Lindqvist, and N. G. Richards. 2004. Kinetic and mechanistic characterization of the formyl-CoA transferase from Oxalobacter formigenes. J. Biol. Chem. 279: 36003-36012. [DOI] [PubMed] [Google Scholar]
- 19. Kawaguchi, A., T. Yoshimura, and S. Okuda. 1981. A new method for the preparation of acyl-CoA thioesters. J. Biochem. (Tokyo) 89: 337-339. [DOI] [PubMed] [Google Scholar]
- 20. Kim, J., D. Darley, and W. Buckel. 2005. 2-Hydroxyisocaproyl-CoA dehydratase and its activator from Clostridium difficile. FEBS J. 272: 550-561. [DOI] [PubMed] [Google Scholar]
- 21. Klees, A. G., and W. Buckel. 1991. Synthesis and properties of (R)-2-hydroxyglutaryl-1-CoA. (R)-2-hydroxyglutaryl-5-CoA, an erroneous product of glutaconate CoA-transferase. Biol. Chem. Hoppe-Seyler 372: 319-324. [DOI] [PubMed] [Google Scholar]
- 22. Leutwein, C., and J. Heider. 2001. Succinyl-CoA:(R)-benzylsuccinate CoA-transferase: an enzyme of the anaerobic toluene catabolic pathway in denitrifying bacteria. J. Bacteriol. 183: 4288-4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mack, M., K. Bendrat, O. Zelder, E. Eckel, D. Linder, and W. Buckel. 1994. Location of the two genes encoding glutaconate coenzyme A-transferase at the beginning of the hydroxyglutarate operon in Acidaminococcus fermentans. Eur. J. Biochem. 226: 41-51. [DOI] [PubMed] [Google Scholar]
- 24. Mack, M., and W. Buckel. 1997. Conversion of glutaconate CoA-transferase from Acidaminococcus fermentans into an acyl-CoA hydrolase by site-directed mutagenesis. FEBS Lett. 405: 209-212. [DOI] [PubMed] [Google Scholar]
- 25. Mack, M., and W. Buckel. 1995. Identification of glutamate beta 54 as the covalent-catalytic residue in the active site of glutaconate CoA-transferase from Acidaminococcus fermentans. FEBS Lett. 357: 145-148. [DOI] [PubMed] [Google Scholar]
- 26. Martins, B. M., S. Macedo-Ribeiro, J. Bresser, W. Buckel, and A. Messerschmidt. 2005. Structural basis for stereo-specific catalysis in NAD+-dependent (R)-2-hydroxyglutarate dehydrogenase from Acidaminococcus fermentans. FEBS J. 272: 269-281. [DOI] [PubMed] [Google Scholar]
- 27. Möllering, H., A. W. Wahlefeld, and G. Michal. 1974. Visualisierung NADH-abhängiger Reaktionen, p. 145-153. In H. U. Bergmeyer (ed.), Methoden der enzymatischen Analyse, 3rd ed., vol. 1. Verlag Chemie, Weinheim/Bergstraβe, Germany. [Google Scholar]
- 28. Razeto, A., S. Kochhar, H. Hottinger, M. Dauter, K. S. Wilson, and V. S. Lamzin. 2002. Domain closure, substrate specificity, and catalysis of D-lactate dehydrogenase from Lactobacillus bulgaricus. J. Mol. Biol. 318: 109-119. [DOI] [PubMed] [Google Scholar]
- 29. Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 30. Schweiger, G., and W. Buckel. 1984. On the dehydration of (R)-lactate in the fermentation of alanine to propionate by Clostridium propionicum. FEBS Lett. 171: 79-84. [DOI] [PubMed] [Google Scholar]
- 30a. Sebaihia, M., B. W. Wren, P. Mullany, N. F. Fairweather, N. Minton, R. Stabler, N. R. Thomson, A. P. Roberts, A. M. Cerdeno-Tarraga, H. Wang, M. T. Holden, A. Wright, C. Churcher, M. A. Quail, S. Baker, N. Bason, K. Brooks, T. Chillingworth, A. Cronin, P. Davis, L. Dowd, A. Fraser, T. Feltwell, Z. Hance, S. Holroyd, K. Jagels, S. Moule, K. Mungall, C. Price, E. Rabbinowitsch, S. Sharp, M. Simmonds, K. Stevens, L. Unwin, S. Whithead, B. Dupuy, G. Dougan, B. Barrell, and J. Parkhill. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 38: 779-786. [DOI] [PubMed] [Google Scholar]
- 31. Selmer, T., and P. I. Andrei. 2001. p-Hydroxyphenylacetate decarboxylase from Clostridium difficile. A novel glycyl radical enzyme catalysing the formation of p-cresol. Eur. J. Biochem. 268: 1363-1372. [DOI] [PubMed] [Google Scholar]
- 32. Selmer, T., and W. Buckel. 1999. Oxygen exchange between acetate and the catalytic glutamate residue in glutaconate CoA-transferase from Acidaminococcus fermentans. Implications for the mechanism of CoA-ester hydrolysis. J. Biol. Chem. 274: 20772-20778. [DOI] [PubMed] [Google Scholar]
- 33. Selmer, T., A. Willanzheimer, and M. Hetzel. 2002. Propionate CoA-transferase from Clostridium propionicum. Cloning of the gene and identification of glutamate 324 at the active site. Eur. J. Biochem. 269: 372-380. [DOI] [PubMed] [Google Scholar]
- 34. Spencer, R. C. 1998. Clinical impact and associated costs of Clostridium difficile-associated disease. J. Antimicrob. Chemother. 41(Suppl. C): 5-12. [DOI] [PubMed] [Google Scholar]
- 35. Stenmark, P., D. Gurmu, and P. Nordlund. 2004. Crystal structure of CaiB, a type-III CoA transferase in carnitine metabolism. Biochemistry 43: 13996-14003. [DOI] [PubMed] [Google Scholar]
- 36. Stickland, L. H. 1935. XXXV. Studies in the metabolism of the strict anaerobes (genus Clostridium). II. The reduction of proline. Biochem. J. 29: 288-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stoll, V. S., M. S. Kimber, and E. F. Pai. 1996. Insights into substrate binding by D-2-ketoacid dehydrogenases from the structure of Lactobacillus pentosus D-lactate dehydrogenase. Structure 4: 437-447. [DOI] [PubMed] [Google Scholar]
- 38. Thauer, R. K., K. Jungermann, and K. Decker. 1977. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 41: 100-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tokuda, C., Y. Ishikura, M. Shigematsu, H. Mutoh, S. Tsuzuki, Y. Nakahira, Y. Tamura, T. Shinoda, K. Arai, O. Takahashi, and H. Taguchi. 2003. Conversion of Lactobacillus pentosus d-lactate dehydrogenase to a d-hydroxyisocaproate dehydrogenase through a single amino acid replacement. J. Bacteriol. 185: 5023-5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wohlfarth, G., and W. Buckel. 1985. A sodium ion gradient as energy source for Peptostreptococcus asaccharolyticus. Arch. Microbiol. 142: 128-135. [DOI] [PubMed] [Google Scholar]
- 41. Ziegenhorn, J., M. Senn, and T. Bücher. 1976. Molar absorptivities of beta-NADH and beta-NADPH. Clin. Chem. 22: 151-160. [PubMed] [Google Scholar]