

Abstract
Previous studies have described in vitro serial passage of a Δγ134.5 herpes simplex virus type 1 (HSV-1) strain in SK-N-SH neuroblastoma cells and selection of mutants that have acquired the ability to infect and replicate in this previously nonpermissive cell line. Here we describe the selection of a mutant HSV-1 strain by in vivo serial passage, which prolongs survival in two separate experimental murine brain tumor models. Two conditionally replication-competent Δγ134.5 viruses, M002, which expresses murine interleukin-12, and its parent virus, R3659, were serially passaged within human malignant glioma D54-MG cell lines in vitro or flank tumor xenografts in vivo. The major findings are (i) viruses passaged in vivo demonstrate decreased neurovirulence, whereas those passaged in vitro demonstrate a partial recovery of the neurovirulence associated with HSV-1; and (ii) vvD54-M002, the virus selected after in vivo serial passage of M002 in D54-MG tumors, improves survival in two independent murine brain tumor models compared to the parent (unpassaged) M002. Additionally, in vitro-passaged, but not in vivo-passaged, M002 displayed changes in the protein synthesis profile in previously nonpermissive cell lines, as well as early US11 transcription. Thus, a mutant HSV-1 strain expressing a foreign gene can be selected for enhanced antitumor efficacy via in vivo serial passage within flank D54-MG tumor xenografts. The enhanced antitumor efficacy of vvD54-M002 is not due to restoration of protein synthesis or early US11 expression. This finding emphasizes the contribution of the in vivo tumor environment for selecting novel oncolytic HSV specifically adapted for tumor cell destruction in vivo.
Oncolytic viruses have provided much promise as a novel treatment modality for brain tumors such as glioblastoma multiforme, the most frequently occurring primary brain tumor and one that is extremely recalcitrant to traditional therapies. Manipulation of the herpes simplex virus type 1 (HSV-1) genome has provided several agents demonstrating the ability to specifically target and destroy tumor cells and spread their progeny to new tumor cell targets (24). The thymidine kinase (tk) dlsptk deletion mutant afforded an attenuated neurovirulence and promising efficacy profile, but was not advanced to clinical trials because of toxicity displayed at high titers and resistance to antiherpetic agents from its tk-negative status (16). A disabling lacZ insertion in the UL39 locus, encoding the ribonucleotide reductase large subunit, limited the hrR3 mutant HSV-1 to dividing cells, such as malignant glioma tumor cells, which could provide cellular ribonucleotide reductase in trans (9, 10, 17). Mapping of the neurovirulence phenotype of HSV-1 to the diploid γ134.5 gene provided a valuable tool in the production of antitumor HSV-1 vectors; HSV-1 mutants with γ134.5 deleted were avirulent in the normal brain milieu but able to proliferate after infection of actively cycling cells (7). HSV-1 strains with γ134.5 deleted include G207 (double mutant possessing deletions of both γ134.5 genes and a lacZ insertion disrupting the UL39 locus) (18) and HSV1716 (γ134.5-negative only) (26). These vectors kill tumor cells directly but are incapable of significant replication in postmitotic cells such as neurons. Clinical studies have proven the safety of these vectors for direct administration in the brain (15, 22).
Whereas these viruses were engineered by design to possess particular mutations conferring beneficial phenotypes for use as antitumor agents, recent studies have described acquisition of mutations that overcome certain limitations of the γ134.5-negative status of attenuated HSV-1 via in vitro serial passage. Mohr and Gluzman reported selection of the SUP-1 mutant after passage of Δγ134.5 SPBg5e (derived from the Patton HSV-1 strain) in the nonpermissive SK-N-SH human neuroblastoma cell line in vitro (19). SUP-1 acquired a deletion in the US10-US12 region, resulting in earlier expression of the US11 gene. This shift in US11 expression kinetics precluded phosphorylation of eukaryotic initiation factor-2α (eIF-2α) to permit continued viral protein translation in infected SK-N-SH cells. SUP-1 thus exhibited a protein synthesis phenotype similar to that of γ134.5-positive HSV-1 (5, 6, 11), but retained the neurovirulence profile of its Δγ134.5 parent (20). Similarly, Δγ134.5 R3616, derived from the parent HSV-1 (F) strain, was sequentially passaged in SK-N-SH cells and distinct variants were isolated that were also capable, like SUP-1, of blocking the protein kinase R (PKR)-induced shutoff of protein synthesis (4). These mutations mapped outside of the US10-12 domain, and at least one isolate had acquired a partially restored neurovirulence profile, though its 50% lethal dose (LD50) was still over 3-log-fold higher than that of the wild type. These studies demonstrate that though Δγ134.5 viruses display a safe neurovirulence profile and valuable antitumor properties, their effectiveness as replication-competent oncolytic viruses is limited in certain human tumor cells, and in vitro serial passage can provide a means to acquire mutations facilitating their ability to kill tumor cells.
We extended this approach to select HSV mutants specifically capable of enhanced antitumor activity, particularly against malignant gliomas. The recombinant Δγ134.5 HSV strain M002 carries two copies of the murine interleukin-12 (IL-12) gene in the γ134.5 loci and has demonstrated significant antitumor activity in both murine neuroblastoma and glioma brain tumor models (12, 21). M002 and R3659, the parent virus to M002, were each sequentially passaged in the human malignant glioma cell line D54-MG, which is more resistant to HSV infection than other malignant glioma cell lines (2). We compared serial passage, performed both in vitro in tissue culture and in vivo in flank tumor xenografts, of M002 to that of R3659 since expression of the immunomodulatory cytokine IL-12 could potentially alter the nature and function of the mutations acquired. We hypothesized passage in an in vivo setting would provide selective pressures far different from those encountered in vitro. We found passage of R3659 and M002 in vitro in tissue culture monolayers selected mutants exhibiting slight to moderate neurotoxicity, as compared to the parent viruses, but the viruses were still significantly less neurotoxic than the wild-type HSV-1 (F). Additionally, US11 expression from in vitro-passaged M002 shifted from late in infection to much earlier in the replicative cycle. The in vitro-passaged M002 exhibited a restored protein synthesis phenotype in SK-N-SH and D54-MG cells. These changes did not, however, translate to an improvement in survival of brain tumor-bearing animals in the models tested relative to the parent nonpassaged viruses, which display late US11 expression and shutdown of protein synthesis following SK-N-SH infection. In contrast, the in vivo-passaged M002, which demonstrated decreased neurovirulence compared to the M002 parent after selection in D54 flank tumors, significantly improved survival of tumor-bearing mice in two separate murine experimental brain tumor models: human D54-MG intracranial xenografts in SCID mice and murine Neuro-2a neuroblastoma syngeneic tumors in A/J mice. Thus, these studies emphasize the importance of the in vivo tumor environment for selecting novel oncolytic HSV strains that mediate improved survival in vivo.
MATERIALS AND METHODS
Cell lines and viruses.
Vero, Neuro-2a murine neuroblastoma (derived from A/J mice), U87 human malignant glioma, and SK-N-SH human neuroblastoma cells were obtained from the American Type Culture Collection (Manassas, VA). Human D54-MG cells were obtained from D. Bigner (Duke University, Durham, NC), and 4C8 murine glioma cells were obtained from C. Dyer (E. K. Shriver, New York, NY). Cells were propagated in antibiotic-free modified Eagle's medium (Vero) or Dulbecco's modified Eagle's medium (DMEM)-F12 in a 50:50 mix (as were all other cell lines) supplemented with 7% fetal bovine serum (HyClone, Logan, UT) and 2.6 mM l-glutamine (Life Technologies, Carlsbad, CA). R5104 (6) and the parent recombinant viruses R3659 (14) and M002 (21) have been described elsewhere. HSV-1 (F), the prototypical wild-type virus used for these studies (8), and R3659 were generous gifts from Bernard Roizman (University of Chicago, Chicago, IL).
In vitro and in vivo serial passage of Δγ134.5 HSV.
In vitro-passaged viruses were selected by infecting D54-MG tissue culture monolayers (80 to 90% confluence) with 100 PFU of the parent viruses. Three to 4 days postinfection, when cells reached approximately 100% cytopathic effect, cells and supernatant were harvested and the titer of virus recovered was determined. One hundred PFU of the recovered virus was used to infect a new D54-MG culture. Nine such rounds of serial passage were performed on both R3659 and M002.
For in vivo serial passage, flank tumor xenografts were established in SCID mice by injecting 5 × 106 D54-MG cells via 200 μl 5% methylcellulose. Three tumors at least 100 mm3 were injected with 100 PFU of either parent virus for the first passage. Four days postinoculation, tumors were harvested and pooled and then homogenized into a mixture of sterile milk and DMEM-F12 medium (50:50) and the viral titer was determined on Vero cells. For subsequent passages, 100 μl of tumor homogenate from the previous passage was used for inoculation into new flank tumors for harvest as before. Viruses were monitored during passage for differences in both replication and IL-12 production (for M002-based viruses) in vitro. We noted initial changes in phenotypes fairly early (passages 4 to 6). Due to the probable complexity of selective pressures from the environmental factors associated with a flank tumor, we carried out two extra passages (11 total). Passage was continued for both mutants to ensure stability of selected mutations and that we were working with a sufficiently pure population of viruses.
After passage, high-titer HSV virus stocks were obtained and purified on OptiPrep gradients (AXIS-SHIELD PoC AS, Oslo, Norway). Briefly, Vero cells were infected at low multiplicity of infection (MOI; 0.01). When ∼100% CPE was reached, both culture supernatants and cells were collected and harvested separately. Culture supernatants were subjected to three centrifugation steps: step 1, 1,811 × g (15 min); step 2, transfer of supernatants to Nalgene tubes and centrifugation at 3,568 × g (5 min, JA14 rotor); and step 3, transfer of the final supernatants to Beckman Ultraclear tubes and centrifugation at 53,000 × g (1 h, SW28 rotor). Viral pellets were resuspended in 1× phosphate-buffered saline (PBS) containing 10% glycerol. Cells were scraped, lysed in 0.4 M NaCl, and subjected to multiple centrifugations as described above prior to PBS-10% glycerol suspension. Virus pellets were purified on OptiPrep gradients as follows. Equal volumes of OptiPrep were added to NaCl-extracted or culture supernatant virus pellets, mixed, and transferred to Quickseal polyallomer tubes. Tubes were filled with 25% OptiPrep and centrifuged for 3 h at 354,000 × g in an NVT90 rotor. After centrifugation, virus fractions were collected, diluted in PBS, and centrifuged. Virus pellets from supernatants and cell lysates were combined, and the titer was determined on Vero cells as described previously (1).
Replication assays.
Viral replication was assessed in vitro as previously described (1). Studies were repeated at least three times, and results were analyzed by fitting a general linear model (analysis of variance) across groups and time points.
IL-12 ELISA.
Cells were seeded in 24-well plates and mock-infected or infected with a low MOI (0.1) of virus as described above. Supernatants from replicate wells (two to four wells) were harvested at multiple time points and tested for murine IL-12 (p70 heterodimer of p40 and p35 IL-12 subunits) using a commercially available enzyme-linked immunosorbent assay (ELISA) kit (Quantikine; R&D Systems, Minneapolis, MN). Statistical analysis was performed by t test across groups for each time point.
In vitro protein synthesis profiles.
Replicate cultures of D54-MG or SK-N-SH cells were mock infected or infected with a virus at a high MOI (40 to 50). Twelve hours postinfection (hpi), cultures were incubated in methionine-free DMEM (199V) supplemented for 1 h with 50 μCi 35S-labeled l-methionine (Amersham Bioscience, Piscataway, NJ). Cells were harvested, solubilized, run through a denaturing 15% (vol/vol) polyacrylamide gel cross-linked with N, N′-diallyltartardiamide to electrophoretically separate bands, and analyzed by autoradiography after transfer to nitrocellulose membranes.
US11 expression studies.
Vero monolayers in 12-well plates were infected at a high MOI (MOI of 50) and incubated with 300 μg/ml phosphonoacetic acid (PAA). US11 expression was examined at both 12 and 24 hpi. Culture medium was removed, and cells were lysed in disruption buffer (8 ml 20% sodium dodecyl sulfate, 4 ml β-mercaptoethanol, 4 ml 55% sucrose, 4 ml 1 M Tris HCl, pH 7.0, bromophenol blue). Cell lysates were transferred to microcentrifuge tubes, boiled, and loaded in a denaturing 12% (vol/vol) polyacrylamide gel to electrophoretically separate bands. Bands were transferred to nitrocellulose membranes and incubated in Tris-buffered saline-Tween 20 with a US11-specific mouse monoclonal antibody (*28S; a gift from B. Roizman, University of Chicago) overnight at 4°C. Membrane was washed and incubated with alkaline phosphatase-conjugated goat anti-mouse secondary antibody (1:3,000) (catalog no.170-6520; Bio-Rad Laboratories, Hercules, CA) for 1 h. US11 bands were visualized with 200 μl nitroblue tetrazolium (NBT; ICN Biomedical, Pasadena, CA) plus 200 μl 5-bromo-4-chloro-3-indolylphosphate (BCIP; Sigma, St. Louis, MO) in 20 ml alkaline phosphatase buffer.
Animal studies.
All mouse strains (CBA/J, SCID, and A/J) were obtained from the Frederick Cancer Research and Development Center, National Cancer Institute. All animal studies were conducted in accordance with guidelines for animal use and care established by the University of Alabama at Birmingham Animal Resource Program and the Institutional Animal Care and Use Committee (protocol no. 050607062).
(i) Survival studies.
Mice were injected intracerebrally with cells (via 5 μl of 5% methylcellulose) by drilling a small hole 2 mm anterior and 2 mm lateral to the bregma. A 30-gauge needle was inserted to a depth of 2.5 mm using a Kopf stereotaxic frame to implant cells in the caudate nucleus. SCID mice were injected with 1 × 106 D54-MG cells and inoculated stereotactically with virus 7 days later using the same coordinates. A/J mice were injected with 2 × 104 Neuro-2a cells intracranially and treated after 5 days. Virus treatments were administered (via 10 μl PBS) through the same burr hole. Incisions were closed with TissueMend II (Veterinary Product Laboratories, Phoenix, AZ), and all mice were maintained on a Tylenol-water mixture 2 days prior to and after surgery. Kaplan-Meier statistical analysis was used to determine any differences between survival curves of various cohorts.
(ii) Neurotoxicity studies.
The highly HSV-sensitive CBA/J mice (4 weeks old, female) were injected intracerebrally with graded doses of viruses in cohorts of 10 mice as described above, and deaths occurring within 30 days were recorded. HSV-1 (F) was injected for comparison. LD50 was determined by Spearman-Karber analysis.
RESULTS
Selection of novel Δγ134.5 HSV by in vivo and in vitro serial passage.
Parent recombinant viruses R3659 and M002 are strains with γ134.5 deleted but capable of replication in human glioma cells. Each virus was injected intratumorally into D54-MG flank tumor xenografts in SCID mice to generate in vivo-passaged viruses as described above (Fig. 1A). A total of 11 passage cycles were performed for both R3659 and M002 in D54-MG tumors. In parallel, in vitro-passaged R3659 and M002 viruses were selected by sequential passage in D54-MG cells in tissue culture, as described above (Fig. 1B). Throughout the subsequent text, passaged viruses are referred to as follows: vtD54-R3659 and vtD54-M002 refer to parent viruses selected in D54-MG glioma cells in culture (within an in vitro environment). Viruses vvD54-R3659 and vvD54-M002 refer to those passaged in D54-MG flank tumor xenografts (within an in vivo environment). Viruses recovered from final passages were grown to high titers, purified, and used for characterization studies.
FIG. 1.

Schematic of selection of novel Δγ134.5 HSV-1 by serial passage. (A) Selection by in vivo serial passage. (B) Selection by in vitro serial passage.
Replication of vtD54-M002 is 1 to 3 logs higher than that of the parent, nonpassaged M002, but replication of vvD54-M002 is no different from that of M002.
To determine whether mutants with differences in viral replication were selected and whether these mutations were cell line specific or generalized across different cell types, both multi- and single-step replication assays were performed. Multistep replication assays after D54-MG infection in vitro at low MOI with vvD54-M002 showed only slightly higher replication than M002 (P > 0.05) (Fig. 2A). A similar trend was observed after infection of a different human glioma cell line, U87-MG, but no change was seen in Neuro-2A cells or the highly permissive Vero or 4C8 murine glioma cells (not shown). In contrast, vtD54-M002 exhibited increased replication (approximately 2 logs higher) after infection of D54-MG cells compared to M002, though this difference did not reach statistical significance. The serially passaged R3659 viruses showed no difference in replication compared to the unpassaged parent virus in any of these cell lines (not shown). For the single-step replication assay, D54-MG cells were infected using a high MOI (20) and samples were harvested at multiple time points to determine viral recovery. Findings were similar to the multistep replication assays in that recovery of vvD54-M002 was no different from that of M002, whereas recovery of vtD54-M002 was higher and to wild-type levels but did not reach statistical significance (data not shown). Viral recovery was also measured after in vivo infection of D54-MG intracranial tumor xenografts in SCID mice with 1 × 107 PFU of each virus. Brain tumors were harvested every 2 days and homogenized, and viral titer was determined. At all time points, vtD54-M002 titers recovered from homogenates were 1 to 2 logs higher than M002 titers (Fig. 2B), but again did not reach statistical significance. By day 12, no titers were recovered from any of the homogenates. Lastly, we determined viral recovery after intratumoral injection of 1 × 107 PFU of each virus at various time points (2, 4, 7, 10, 14, and 21 days) from both D54-MG and Neuro-2A flank tumors. Recovery of vtD54-M002 was at most 1 log higher than that of the other viruses and was not statistically significant (not shown).
FIG. 2.

In vitro and in vivo replication of parent and passaged viruses. (A) D54-MG cells were cultured in 24-well plates in vitro, and replicate wells were infected (MOI = 0.1) with either HSV-1 (F), parent viruses (R3659 or M002), or a serially passaged virus. Samples were harvested 12, 24, 36, 48, and 72 hpi. (B) Replication time course after intratumoral injection of viruses in D54-MG brain tumors established intracranially in SCID mice.
IL-12 production differs from vvD54-M002- versus M002-infected cells.
Since M002 was engineered to express murine IL-12, we tested whether serial passage selected a mutant with altered IL-12 gene expression. After infection of D54-MG, U87-MG, 4C8, Neuro-2a, and Vero cells, supernatants were harvested at multiple time points to determine levels of murine IL-12 by ELISA, with control wells infected with R3659. As seen in Fig. 3A, a modest increase in IL-12 production was noted when D54-MG cells were infected with vvD54-M002 (late time points; P < 0.05). Conversely, IL-12 production was decreased after vvD54-M002 infection of Neuro-2a cells relative to M002 (late time points; P < 0.05) (Fig. 3B). However, no increase in recombinant IL-12 expression was noted from vvD54-M002-infected tissue culture monolayers of U87-MG, 4C8, or Vero cells (not shown). The vtD54-M002 virus did not exhibit statistically significant changes in IL-12 expression relative to M002 (not shown).
FIG. 3.

Recombinant IL-12 production after M002 and vvD54-M002 infection. IL-12 production after infection with viruses was determined using an ELISA kit (R&D Systems) specific for murine IL-12 p70 heterodimer. Shown are the levels of IL-12 detected after vvD54-M002 or M002 infection of (A) D54-MG cells (72 hpi, P = 0.0124; 84 hpi, P = 0.0093) and (B) Neuro-2a cells (48 hpi, P = 0.0152; 72 hpi, P = 0.0008).
The protein synthesis phenotype is restored by in vitro-passaged (vtD54-M002), but not in vivo-passaged (vvD54-M002) virus following infection of D54-MG and SK-N-SH cells.
Δγ134.5 HSV R3659 and M002, unlike HSV-1 (F), are unable to synthesize late viral proteins after infection of either D54-MG or SK-N-SH cells. To determine if the passaged viruses were able to overcome host translation shutoff, protein synthesis was assessed. The in vitro-passaged virus vtD54-M002 demonstrated the capacity to produce viral proteins after infection of the Δγ134.5 nonpermissive SK-N-SH cell line (Fig. 4A). The same was true for vtD54-M002 infection of D54-MG cells (Fig. 4B) and the heterologous human glioma U87 cells (not shown). In contrast, restoration of the protein synthesis phenotype was not observed in these cell lines following infection with vvD54-M002 (Fig. 4A and B) or with in vitro- or in vivo-passaged R3659 viruses (not shown). Relative to HSV (F)-infected cells, vtD54-M002 displays less robust production of viral proteins, suggesting an acquired mechanism of PKR evasion that is not as powerful as that seen with ICP34.5 expression, but which is still sufficient to continue viral protein production in even nonpermissive SK-N-SH cells.
FIG. 4.
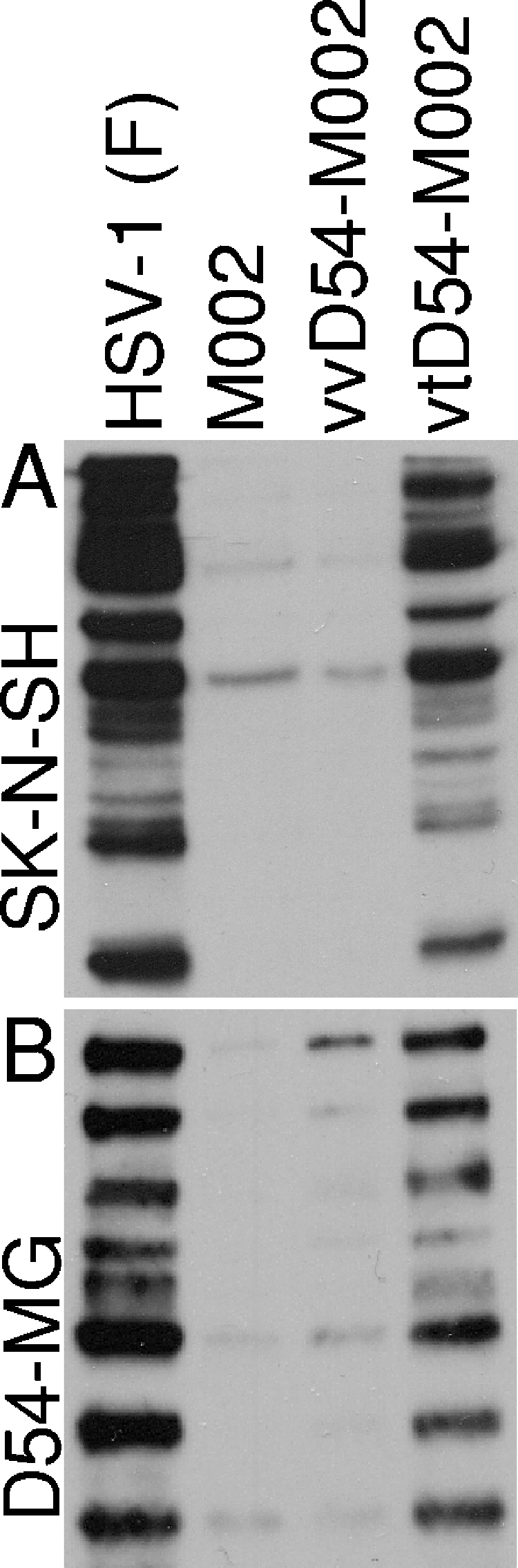
Protein synthesis profiles from infected D54-MG human glioma cells and SK-N-SH human neuroblastoma cells. An autoradiographic image of protein production from cells pulse-labeled with 35S-labeled l-methionine 12 hpi (MOI = 50) with either HSV-1 (F), parent viruses (M002 or R3659), or viruses serially passaged in vivo or in vitro in (A) SK-N-SH cells or (B) D54-MG cells.
Serial passage of R3659 and M002 selects mutants with altered neurotoxic potential depending on the environment (in vitro or in vivo) in which passage was performed.
To determine differences in neurotoxic potential, each passaged virus and its respective parent were administered in escalating doses by direct intracranial injection into HSV-sensitive CBA/J mice. Wild-type virus was injected at 50 and 100 PFU into five mice each as a positive control: HSV-1 (F), LD50 = 75 PFU. As with HSV-1 (F) inoculation, deaths consistent with HSV encephalitis after injection of other viruses generally occurred within 10 to 12 days. LD50 values (Spearman-Karber analysis) for the in vitro-passaged viruses vtD54-M002 and vtD54-R3659 were calculated to be approximately 5.1 × 105 PFU and 1.67 × 106 PFU, respectively, which were lower than the LD50 values of the nonpassaged parent viruses (M002 LD50 = 2 × 107 PFU and R3659 LD50 = 1.54 × 107 [and see Table 1 ]). Whereas the viruses passaged in D54-MG cells in vitro show lower LD50 values (i.e., more neurotoxicity) than the parent viruses, passage within flank tumor xenografts selected mutants with higher LD50 values (i.e., less neurotoxicity). At the highest dose tested (3 × 107 PFU), an LD50 value could not be calculated for either vvD54-R3659 or vvD54-M002 (Table 1). Thus, passage of these viruses in D54-MG flank tumor xenografts resulted in selection of mutants with a much safer neurovirulence profile.
TABLE 1.
Determination of neurovirulence of passaged viruses compared to parental viruses R3659 and M002
| Virus | LD50 (PFU) |
|---|---|
| R3659 | 1.54 × 107 |
| vtD54-R3659 | 1.67 × 106 |
| vvD54-R3659 | >3 × 107 |
| M002 | 2 × 107 |
| vtD54-M002 | 5.1 × 105 |
| vvD54-M002 | >3 × 107 |
Kinetics of US11 expression from vtD54-M002 is altered.
The increase in neurotoxic potential as well as the positive protein synthesis phenotype of the in vitro-passaged virus, vtD54-M002, raised the question of whether its phenotype was due to changes in US11 gene expression, as reported previously for an in vitro-selected Δγ134.5 HSV mutant in SK-N-SH cells (4). US11 expression kinetics was examined in vtD54-M002-infected cells in the presence or absence of phosphonoacetic acid, and lysates were analyzed by Western blotting using the US11-specific *28S monoclonal antibody. PAA inhibits HSV DNA synthesis and the activity of viral DNA polymerase and, thereby, halts viral replication such that transcription of the α (immediate-early), β (early), and γ1 (early-late) genes is permitted, but γ2 (true late) proteins will not be produced (13). As shown in Fig. 5, at 12 hpi, US11 expression kinetics has shifted to an earlier time point with vtD54-M002 infection compared to M002 infection. Recombinant HSV-1 R5104, engineered to express US11 as an early gene, was used as a positive control (6). Since the US11 gene product is virion associated (23), small amounts were detected from lysates of HSV-1 (F)-, R3659-, and M002-infected cells. In contrast, kinetics of US11 expression appeared unaltered in lysates from vvD54-M002-infected cells (Fig. 5). Neither vtD54-R3659 nor vvD54-R3659 exhibited altered US11 expression kinetics (not shown).
FIG. 5.

Expression kinetics of US11 from infected Vero cells. Shown is US11 expression 12 hpi from Vero cells in the absence (−) or presence (+) of 300 μg/ml PAA infected. Cells were infected with R5104 (early US11 expression), HSV-1 (F), M002, or the in vivo- and in vitro-passaged M002 viruses. The arrow indicates US11.
The vvD54-M002 virus improves survival of tumor-bearing animals in two experimental murine brain tumor models.
In vivo-passaged viruses were evaluated relative to the nonpassaged parent viruses as treatments for brain tumors in mice. Intracranial D54-MG tumor xenografts in SCID mice were treated with 1 × 107 PFU of virus or saline control. The study was repeated twice, and combined results are presented in Fig. 6. Median survival for control mice (saline treated) was 28 days and was improved minimally by treatment with R3659 (32 days; P = 0.0518). As demonstrated previously (21), intratumoral administration of M002 significantly improved survival (median = 41 days) as compared to saline- or R3659-treated controls (P ≤ <0.0001 versus saline; P = 0.0020 versus R3659). However, vvD54-M002 dramatically improved survival of mice compared to treatment with M002, with a median survival of 57 days (P = 0.0030). Additionally, there were no long-term survivors in the M002-treated cohort from either experiment, whereas the vvD54-M002-treated cohort had three apparent cures 150 days following tumor implantation, the experiment endpoint.
FIG. 6.

Survival of mice bearing human D54-MG brain tumors after vvD54-M002 treatment. Immunocompromised SCID mice bearing D54-MG brain tumors were treated with either saline (closed circles) as a control or 1 × 107 PFU of either R3659 (closed diamonds), M002 (closed squares), or vvD54-M002 (open squares) after 7 days. The survival graph shows two separate studies combined. Cohorts were compared using Kaplan-Meier statistical analysis.
The other passaged viruses did not demonstrate enhanced antitumor activity in the D54-MG xenograft brain tumor model.
Given that vtD54-M002 had regained the wild-type protein synthesis phenotype and increased replication in vitro, we predicted that this would translate to improved tumor killing and survival of tumor-bearing mice. However, when tested at equivalent doses, neither the in vitro-passaged vtD54-M002, nor either in vitro- or in vivo-passaged R3659 viruses (vtD54-R3659 and vvD54-R3659), conferred increased survival to SCID mice bearing intracranial D54-MG tumors relative to the parent viruses (not shown).
The vvD54-M002 virus demonstrated therapeutic efficacy in A/J mice bearing syngeneic Neuro-2a neuroblastoma brain tumors.
We next determined whether prolonged survival following vvD54-M002 treatment of tumors was applicable to other, nonhuman and nonglioma models of malignant brain tumors. For these studies, the murine Neuro-2a neuroblastoma cell line was used to establish intracranial tumors in the syngeneic A/J strain mice. Five days after implantation, either R3659, M002, or vvD54-M002 (at equivalent doses to those given above) was directly injected into the tumors. The results of two separate experiments were combined, and the data are shown in Fig. 7. In this model, untreated mice succumb much more quickly to the implanted tumors, with a median survival of only 10 days for the saline cohort. Mice treated with vvD54-M002 had a median survival of 14 days, which was significantly improved over treatment with M002 (P = 0.0193). Moreover, the vvD54-M002 treatment cohort produced two apparent cures (surviving >155 days post-tumor implantation), whereas the M002 cohort had no long-term survivors.
FIG. 7.

Survival of mice bearing Neuro-2a brain tumors after vvD54-M002 treatment. Immunocompetent A/J mice bearing Neuro-2a brain tumors were treated with either saline (closed circles) as a control or 1 × 107 PFU of either R3659 (closed diamonds), M002 (closed squares), or vvD54-M002 (open squares) after 5 days. The survival graph shows two separate studies combined. Cohorts were compared using Kaplan-Meier statistical analysis.
Treatment of SCID mice bearing D54 human glioma brain tumors with vvD54-M002 significantly enhanced survival and resulted in a 5% apparent cure rate, with mice showing no signs of tumor burden. There were no long-term survivors after M002 treatment. Importantly, the improvement in survival time was translatable to other nonhuman and nonglioma-derived tumor types. In the murine Neuro-2a neuroblastoma model, 7% of vvD54-M002-treated mice were long-term survivors. In contrast, all M002-treated animals eventually succumbed to their tumors. These results confirm that serial passage of a cytokine-expressing Δγ134.5 HSV strain, like M002, through xenografted human glioma-derived tumors selects for mutants that mediate improved survival in murine brain tumor models.
DISCUSSION
The antitumor benefits of Δγ134.5 HSV-1, demonstrated through numerous preclinical studies (2, 3, 12, 16), and their favorable safety profiles, demonstrated in separate clinical trials of two different constructs (15, 22), have validated the promise of oncolytic HSV-1 vectors for the treatment of malignant glioma. The focus of new generation viruses has now shifted towards augmenting the intrinsic oncolytic abilities of a Δγ134.5 HSV-1 virus, while maintaining its safety profile, to enhance delivery and expression of therapeutic genes. To this end, we hypothesized that serial passage of a virus through human glioma cells, either in vitro or in vivo, would select mutants with an enhanced therapeutic profile. Previous studies have shown in vitro passage can increase the oncolytic potential of HSV (19). Greater glioma cell tropism has also been reported for vesicular stomatitis virus after passage in vitro (27). Here we report for the first time that in vivo intratumoral passage can augment the efficacy of an oncolytic virus engineered to express a foreign gene. An unanticipated result of these studies is that previously defined mechanisms (restoration of the wild-type protein synthesis phenotype and improved replication) that have mediated antitumor efficacy in other tumor models did not improve survival of tumor-bearing animals in the models evaluated here.
To maximize opportunities for selecting mutants and to determine the potential impact of foreign gene expression on the selection process, both R3659 and M002 were passaged in D54-MG cells in vitro and flank tumors in vivo. D54-MG is capable of type I interferon secretion and would be expected to exert selective pressures involving interferon antiviral responses. We hypothesized passage of these viruses through in vivo-xenografted human glioma tumors would subject the viruses to intratumoral pressures not encountered in cell culture, including viral spread through the three-dimensional tumor stroma and the host's innate immune responses to viral infection. Additionally, the in vivo tumor microenvironment imposes selective pressures related to regional heterogeneities, including hypoxia, necrosis, angiogenesis, and variable tumor cell mitotic rates. Thus, in vivo passage using a human tumor type provides the more relevant environment in which to select tumor-adapted mutants. Finally, to avoid selection of mutants adapted to replication in normal brain, we passaged the viruses in flank xenografts.
Selectivity for neoplastic cells alone and minimal toxicity to the adjacent normal brain are of paramount importance for oncolytic HSV therapy of malignant glioma. In vitro passage restored some of the neurotoxic potential initially eliminated with deletion of the γ134.5 genes, but to levels still well below that of HSV-1 (F). Similarly, Cassady et al. found at least one of the in vitro SK-N-SH-passaged Δγ134.5 R3616 variants had partially reacquired neurovirulence, but was still significantly reduced compared to HSV-1 (F) (4). The increased neurotoxicity of vtD54-M002 suggests it is able to replicate in normal brain, although not to the same level of efficiency as HSV-1 (F). This phenotype may account in part for the greater viral recovery from SCID mice bearing D54-MG tumors. Interestingly, we found that vvD54-R3659 and vvD54-M002 were less neurovirulent than M002; indeed, an LD50 was not achieved at the highest dose tested for both in vivo-passaged viruses. The benefit of this difference is twofold: increased safety at a particular dose and the ability to administer higher doses if required to attain maximum tumor cell killing without increasing the risk of HSV-induced encephalitis.
A potential concern was that the in vivo-passaged variant with an attenuated neurovirulence profile may show a corresponding loss of efficient tumor killing. Conversely, an increase in efficacy may be seen at the expense of safety with the in vitro-passaged viruses. In contrast, treatment with the safer vvD54-M002 virus demonstrated a greater improvement in survival. Importantly, we have demonstrated that a lack of (i) restoration of viral protein synthesis, (ii) early US11 expression, and (iii) significant improvement in replicative abilities in vitro and in vivo did not preclude the ability of vvD54-M002 from improving survival of brain tumor-bearing mice. Moreover, though we note both early US11 expression and slightly greater viral recovery of vtD54-M002 from infected cells in vitro—similar to wild-type HSV-1 (F) replication levels—and brain tumors in vivo, this did not translate to greater antiglioma effects. Previous studies show a shift in US11 expression from late in infection (under control of its native γ2 promoter) to much earlier (under control of the immediate-early α47 promoter), permitting it to preclude PKR activation and eIF-2α phosphorylation, resulting in continued viral protein synthesis in U373-MG cells (5, 11, 19). Late viral protein synthesis after infection of prostate carcinoma PC3 cells and subsequent efficacy in treatment of the PC3 murine model of prostate carcinoma are attributed to the new kinetics of US11 expression (25). However, our results are not necessarily at odds with these data; improvement of SUP-1 tumor killing in vivo is demonstrated in PC3 tumors, whereas we have demonstrated improved survival rates with vvD54-M002 in two brain tumor models, a human malignant glioma and a murine neuroblastoma. Just as we have seen the selective pressures between in vitro and in vivo serial passage differ, the properties considered beneficial of Δγ134.5 mutant HSV-1 oncolytic vectors may differ between brain and prostate tumors. Indeed, since early US11 expression and a resultant ability to produce late viral proteins may in part account for the increased neurotoxicity of vtD54-M002, testing these viruses in tumors outside the central nervous system may show benefit since the risk of HSV-induced encephalitis would be greatly reduced. Conversely, it is unclear whether SUP-1 demonstrated greater antiglioma activity, since the virus was tested in a human glioma cell line in vitro but not in an in vivo model of human glioma.
While the modest increases in vvD54-M002 replication in D54-MG cells as compared to the parent M002 may be responsible for the slightly increased IL-12 production observed in these cells, there are at least two other possibilities. First, these differences may be due to a global change in viral protein production from the serially passaged virus. This is unlikely in that protein synthesis profiles were not different between the parent M002 and vvD54-M002 viruses. A more likely explanation is that mutations were selected which affected foreign gene expression, specifically, expression of the murine IL-12 gene. Such mutants would include those with mutations in the promoter or enhancer regions that directly affected mRNA synthesis or those which affected mRNA or protein stability. By passage in human D54-MG tumors, which are somewhat resistant to HSV infection, more optimal foreign gene expression may have been achieved. The relative IL-12 levels from vvD54-M002- versus M002-infected cells depended on the cell lines infected (human D54-MG versus murine Neuro-2A). It is difficult to predict how the changes seen in vitro might affect the antitumor activity of the viruses within the in vivo environment; a tumor treated with these oncolytic viruses would be expected to have a complex set of interactions between infectious virions, actively growing, infected and lysed tumor cells, ongoing angiogenesis, recruited immune cells, and the host cellular responses to all these factors. Studies are in progress to examine promoter regions for mutations, which may be cell line specific, that account for enhanced IL-12 expression in D54-MG cells but reduced expression in Neuro-2a cells and their significance to the antitumor benefits demonstrated in vivo. Since in vivo passage of M002, but not R3659, selected a mutant demonstrating greater improvements in survival, it is unlikely mutations in surface glycoproteins improving viral entry account for the change, and this makes involvement of the foreign IL-12 gene insert important to delineate. Studies are under way to assess changes in viral spread or secondary effects of virus-host interaction, such as innate immune responses and angiogenesis induction, which may account for the change in phenotype.
Finally, the fact that a more effective mutant was selected from our virus encoding IL-12, and not in a virus expressing only native viral proteins, suggests in vivo passage may be particularly relevant for oncolytic viruses engineered to express foreign genes. While viruses have evolved towards optimal efficiency over the course of their existence, subtle changes in viral genotype may improve the efficacy of oncolytic viruses expressing foreign gene products, contributing to a new equilibrium between the engineered virus and its replication milieu. In vivo passage allows the virus and its milieu to select these changes and could represent a new paradigm for development of optimized oncolytic vectors expressing foreign proteins prior to use in clinical trials. Studies are under way to elucidate the mutation(s) responsible for the acquired phenotype of vvD54-M002, which will be vital for construction of future generations of therapeutic HSVs.
Acknowledgments
We thank Bernard Roizman, University of Chicago, for helpful discussions throughout the course of this work and for providing viruses and the US11 antibody. We thank Huey Nguyen and Jennifer Coleman for excellent technical assistance.
These studies were initiated and supported by NCI P01 CA 71933 (R.J.W., J.N.P., G.Y.G., and J.M.M.), the Brain Tumor Society's Neal P. Levitan Leadership Chair of Research Award (J.N.P.), and the Ruth L. Kirschstein NRSA Fellowship 1 F31 NS050924-01 (A.C.S.). We acknowledge the Medical Scientist Training Program and Department of Physiology and Biophysics, University of Alabama, for their support of A.C.S.
REFERENCES
- 1.Andreansky, S., B. He, J. van Cott, J. McGhee, J. M. Markert, G. Y. Gillespie, B. Roizman, and R. J. Whitley. 1998. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine interleukins. Gene Ther. 5:121-130. [DOI] [PubMed] [Google Scholar]
- 2.Andreansky, S., L. Soroceanu, E. R. Flotte, J. Chou, J. M. Markert, G. Y. Gillespie, B. Roizman, and R. J. Whitley. 1997. Evaluation of genetically engineered herpes simplex viruses as oncolytic agents for human malignant brain tumors. Cancer Res. 57:1502-1509. [PubMed] [Google Scholar]
- 3.Andreansky, S. S., B. He, G. Y. Gillespie, L. Soroceanu, J. Markert, J. Chou, B. Roizman, and R. J. Whitley. 1996. The application of genetically engineered herpes simplex viruses to the treatment of experimental brain tumors. Proc. Natl. Acad. Sci. USA 93:11313-11318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cassady, K. A., M. Gross, G. Y. Gillespie, and B. Roizman. 2002. Second-site mutation outside of the US10-12 domain of Δγ134.5 herpes simplex virus 1 recombinant blocks the shutoff of protein synthesis induced by activated protein kinase R and partially restores neurovirulence. J. Virol. 76:942-949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cassady, K. A., M. Gross, and B. Roizman. 1998. The herpes simplex virus US11 protein effectively compensates for the γ134.5 gene if present before activation of protein kinase R by precluding its phosphorylation and that of the α subunit of eukaryotic translation initiation factor 2. J. Virol. 72:8620-8626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cassady, K. A., M. Gross, and B. Roizman. 1998. The second-site mutation in the herpes simplex virus recombinants lacking the γ134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2α. J. Virol. 72:7005-7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chou, J., E. R. Kern, R. J. Whitley, and B. Roizman. 1990. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 250:1262-1266. [DOI] [PubMed] [Google Scholar]
- 8.Ejercito, P. M., E. D. Kieff, and B. Roizman. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol. 2:357-364. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein, D. J., and S. K. Weller. 1988. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology 166:41-51. [DOI] [PubMed] [Google Scholar]
- 10.Goldstein, D. J., and S. K. Weller. 1988. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J. Virol. 62:196-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He, B., J. Chou, R. Brandimarti, I. Mohr, Y. Gluzman, and B. Roizman. 1997. Suppression of the phenotype of γ134.5− herpes simplex virus 1: failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the α47 gene. J. Virol. 71:6049-6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hellums, E. K., J. M. Markert, J. N. Parker, B. He, B. Perbal, B. Roizman, R. J. Whitley, C. P. Langford, S. Bharara, and G. Y. Gillespie. 2005. Increased efficacy of an interleukin-12-secreting herpes simplex virus in a syngeneic intracranial murine glioma model. Neuro-Oncol. 7:213-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honess, R. W., and D. H. Watson. 1977. Herpes simplex virus resistance and sensitivity to phosphonoacetic acid. J. Virol. 21:584-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lagunoff, M., and B. Roizman. 1995. The regulation of synthesis and properties of the protein product of open reading frame P of the herpes simplex virus 1 genome. J. Virol. 69:3615-3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Markert, J. M., M. D. Medlock, S. D. Rabkin, G. Y. Gillespie, T. Todo, W. D. Hunter, C. A. Palmer, F. Feigenbaum, C. Tornatore, F. Tufaro, and R. L. Martuza. 2000. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 7:867-874. [DOI] [PubMed] [Google Scholar]
- 16.Martuza, R. L., A. Malick, J. M. Markert, K. L. Ruffner, and D. M. Coen. 1991. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 252:854-856. [DOI] [PubMed] [Google Scholar]
- 17.Mineta, T., S. D. Rabkin, and R. L. Martuza. 1994. Treatment of malignant gliomas using ganciclovir-hypersensitive, ribonucleotide reductase-deficient herpes simplex viral mutant. Cancer Res. 54:3963-3966. [PubMed] [Google Scholar]
- 18.Mineta, T., S. D. Rabkin, T. Yazaki, W. D. Hunter, and R. L. Martuza. 1995. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1:938-943. [DOI] [PubMed] [Google Scholar]
- 19.Mohr, I., and Y. Gluzman. 1996. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J. 15:4759-4766. [PMC free article] [PubMed] [Google Scholar]
- 20.Mohr, I., D. Sternberg, S. Ward, D. Leib, M. Mulvey, and Y. Gluzman. 2001. A herpes simplex virus type 1 γ34.5 second-site suppressor mutant that exhibits enhanced growth in cultured glioblastoma cells is severely attenuated in animals. J. Virol. 75:5189-5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parker, J. N., G. Y. Gillespie, C. E. Love, S. Randall, R. J. Whitley, and J. M. Markert. 2000. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 97:2208-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rampling, R., G. Cruickshank, V. Papanastassiou, J. Nicoll, D. Hadley, D. Brennan, R. Petty, A. MacLean, J. Harland, E. McKie, R. Mabbs, and M. Brown. 2000. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant glioma. Gene Ther. 7:859-866. [DOI] [PubMed] [Google Scholar]
- 23.Roller, R. J., and B. Roizman. 1992. The herpes simplex virus 1 RNA binding protein US11 is a virion component and associates with ribosomal 60S subunits. J. Virol. 66:3624-3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shah, A. C., D. Benos, G. Y. Gillespie, and J. M. Markert. 2003. Oncolytic viruses: clinical applications as vectors for the treatment of malignant gliomas. J. Neurooncol. 65:203-226. [DOI] [PubMed] [Google Scholar]
- 25.Taneja, S., J. MacGregor, S. Markus, S. Ha, and I. Mohr. 2001. Enhanced antitumor efficacy of a herpes simplex virus mutant isolated by genetic selection in cancer cells. Proc. Natl. Acad. Sci. USA 98:8804-8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valyi-Nagy, T., M. U. Fareed, J. S. O'Keefe, R. M. Gesser, A. R. MacLean, S. M. Brown, J. G. Spivack, and N. W. Fraser. 1994. The herpes simplex virus type 1 strain 17+ gamma 34.5 deletion mutant 1716 is avirulent in SCID mice. J. Gen. Virol. 75:2059-2063. [DOI] [PubMed] [Google Scholar]
- 27.Wollmann, G., P. Tattersall, and A. N. van den Pol. 2005. Targeting human glioblastoma cells: comparison of nine viruses with oncolytic potential. J. Virol. 79:6005-6022. [DOI] [PMC free article] [PubMed] [Google Scholar]