

Abstract
The possibility of the release of smallpox virus into a predominantly nonimmunized population highlights the importance of understanding poxvirus biology. Poxviruses encode a conserved pathway that is required to oxidize disulfide bonds in nascent viral proteins that fold in the reducing environment of the eukaryotic host cytoplasm. We present the structure of the last enzyme of the vaccinia virus pathway, G4, which is almost identical in smallpox virus. G4 catalyzes the formation of disulfide bonds in proteins that are critical for virus maturation and host cell infection. G4 contains a thioredoxin fold and a Cys-X-X-Cys active site. In solution, G4 monomers and dimers are observed. In the crystal, G4 is found as a dimer that buries 4,500 Å2 in the interface and occludes the active site, which could protect the reactive disulfide from reduction in the cytoplasm. The structure serves as a model for drug design targeting viral disulfide bond formation.
Interest in poxviruses has increased in the last few years due to the possible existence of undeclared stocks of variola virus, the cause of smallpox. The lethality and ease of transmission makes the biology of the disease particularly important to understand (11). Although not as transmissible, the existence of a related monkeypox virus and other poxviruses that can infect humans also underscore the need to better understand this family of large DNA viruses (7).
Poxviruses replicate in the cytoplasm of infected cells, where noninfectious proviruses undergo a maturation step, which causes a morphological change visible by electron microscopy (1, 7). Upon maturation, the virus assumes its first infectious form, the intracellular mature virion (IMV). Some viral particles can exit the cell, acquiring an additional membrane and become cell-associated virions (CEV), or are released as extracellular enveloped virions (EEV) (7). The CEV/EEV forms are thought to mediate the spread of the virus within the host. The IMV form likely mediates host to host spread of the virus and can be released from the CEV/EEV by rupture of the fragile outermost CEV/EEV membrane, either mechanically or through antibody-dependent mechanisms (23).
In eukaryotic cells, disulfide bond formation is generally compartmentalized in organelles and catalyzed by proteins of the protein disulfide isomerase (PDI) family (6); in bacteria, disulfide bonds are formed in the periplasm by disulfide oxidoreductases of the DsbA family (41). Proteins of the PDI family can also isomerize or “shuffle” improperly paired disulfide bonds in eukaryotes; members of the DsbC and DsbG families perform this function in bacteria. Thermophilic archaea encode disulfide oxidoreductases that mediate disulfide bond formation in cytoplasmic proteins, which would stabilize the proteins in harsh growth environments (24). The IMV form of poxvirus has a number of surface proteins that contain disulfide bonds. Although many other viruses have surface proteins that contain disulfide bonds, those bonds form during transit through the endoplasmic reticulum of host cells. The proteins of the poxvirus IMV surface are unique in that they form in the reducing environment of the host cytoplasm.
Poxviruses encode three proteins—E10, A2.5, and G4—that form a pathway to make disulfide bonds in the host cell cytoplasm. E10 is similar to proteins of the EVR1/ALR family of eukaryotic proteins that utilize the oxidizing potential of flavin adenine dinucleotide (8, 36). The oxidizing potential is transferred from E10 through A2.5 to G4, and intermediate complexes have been detected between these proteins (38). Once oxidized, G4 can catalyze disulfide bond formation on a number of different poxvirus proteins. The deletion of any protein in this pathway blocks the maturation of the virus prior to formation of IMVs and therefore blocks infection (37, 39, 48). These essential enzymes are ideal targets for antiviral drugs (10). Since homologues of the proteins in the disulfide bond forming pathway have been found in other families of large DNA viruses (namely, asfarviruses, iridoviruses, and phycodnaviruses) (16), the requirement for disulfide bond formation may extend beyond the replication of poxviruses.
Here, we present the structure of G4, the essential disulfide oxidoreductase from vaccinia virus. Since vaccinia G4 is 98% identical in sequence to the G4 proteins from smallpox and monkeypox viruses, the structure of vaccinia G4 may help in developing therapeutics against other poxviruses. The G4 structure will also aid in understanding the biology and specificity of poxviral disulfide bond formation.
MATERIALS AND METHODS
Cloning and expression.
Viral DNA, isolated from vaccinia virus-infected cells, was used as a template for the amplification of g4l using the primers 5′-GGGGATCCATATGAAGAACGTACTGATTATTTTCGG-3′ and 5′-GGGGATCCGCGGCCGCTTATTCGGTAACAGGTGGCCAAACTC-3′. The PCR product, cut with NdeI and NotI, was cloned into the T7 promoter-driven vector, pNAN, a variant of the vector, pLM1, in which the NdeI site was removed and a polylinker containing a ribosome-binding site and restriction sites for NdeI, AscI, and NotI were introduced after the T7 promoter.
The protein was expressed in either BL21(DE3)-RIL cells for native protein expression or in BL21(DE3)-RIL-X cells (Stratagene, La Jolla, CA) for selenomethionine incorporation. Bacteria were grown to an optical density at 600 nm of 0.6 in LB (25) or selenomethionine media (Molecular Dimensions, Apopka, FL) supplemented with 50 mg of l-selenomethionine (Sigma, St. Louis, MO)/liter. Protein expression was induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 to 6 h. Cells were pelleted by centrifugation at 4,000 × g and resuspended in 100 to 150 ml of lysis buffer (100 mM TrisHCl [pH 8.0], 150 mM NaCl, 1% NP-40, 0.1% sodium deoxycholate, 0.1% sodium azide) with two to three Complete Protease tablets (Roche, Indianapolis, IN).
Since the majority of G4 appeared to be in inclusion bodies, we purified the inclusion bodies by multiple rounds of washing and homogenization with lysis buffer followed by the same buffer without detergent. Washed inclusion bodies were dissolved in 6 M guanidine-HCl with 2 mM dithiothreitol and subjected to ultracentrifugation at 45,000 × g for 30 min to remove the insoluble material. G4 was refolded in buffer containing 100 mM Tris-HCl (pH 8), 400 mM arginine-HCl, and 5 mM dithiothreitol. Some protein preparations were refolded without 5 mM dithiothreitol but with 5 mM reduced glutathione and 0.5 mM oxidized glutathione. After 24 h, the refolded protein was dialyzed at least twice against 10 mM Tris-HCl (pH 8.0). The protein was filtered, concentrated, and purified by size exclusion chromatography. G4 purity and extent of selenomethionine incorporation were confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and mass spectrometry. The G4 protein used for crystallization was pooled from the monomer fractions but possibly contained some dimers that formed upon concentrating the protein for crystallization.
Crystallization and structure determination.
Crystals of G4 were obtained by vapor diffusion in hanging drops over a reservoir of 100 mM citrate buffer (pH 4.6), 20% PEG1500, and 200 mM LiSO4. Larger, better-shaped crystals were obtained by spreading the drop on the cover, presumably by increasing the surface area for crystal nucleation. Crystallization was improved by the addition of either 5 mM dithiothreitol or 5 mM Tris(2-carboxyethyl) phosphine to the protein prior to crystallization. The crystals were frozen in 100 mM citrate buffer (pH 4.6), 30 to 35% PEG1500, 200 mM LiSO4 and 20% glycerol.
X-ray data were collected (Table 1) and then integrated and scaled using HKL2000 (30). Five of the nine selenium positions were found by SOLVE (44) using the two datasets from the same crystal. Density modification and building of an initial model was performed by RESOLVE (42, 43). The initial maps had discontinuous, fragmented Cα backbone electron density for some core secondary structural elements of two of the three molecules in the asymmetric unit and little density for the third molecule or for side chains. Iterative building in O (17) and refinement with CNS (2) and Refmac (28) improved the electron density maps and allowed tracing of the missing third molecule. The occupancies of the Se and Cɛ atoms of the selenomethionine residues were lowered to model radiation damage based on Fo-Fc difference electron densities for these atoms. Noncrystallographic symmetry restraints were used during refinement and each molecule was a separate translation/libration/screw (TLS) group for TLS refinement in Refmac (49). G4 is 124 residues in length; for the three molecules—A, B, and C—of the asymmetric unit, residues A1 to 49, A55 to 120, B1 to 121, C1 to 45 and C56 to 120 were modeled into density (19). Model geometry was analyzed with Procheck (20). LSQMAN was used for structural alignment between the monomers. The root-mean-square deviation between monomers A and B was 0.71 Å, and between monomers A and C it was 0.61 Å (106 Cα atoms for residues 1 to 43 and residues 58 to 120). Buried surface calculations were performed by using Areaimol (22). Surface complementarity was calculated with SC (21). Secondary structure assignment was calculated by using DSSP (18). Figures of protein models were generated with PyMOL (5). Sequence alignments were made by using Multalin (3). The structure of G4 has been deposited in the Research Collaboratory for Structural Bioinformatics protein data bank (RCSB pdb code 2G2Q).
TABLE 1.
Crystallographic statisticsa
| Dataset | SeMet | SeMet (remote) | SeMet (inflection) |
|---|---|---|---|
| Resolution (Å) | 50-2.50 (2.59-2.50) | 50-3.10 (3.21-3.10) | 50-3.15 (3.26-3.15) |
| X-ray source | 22-ID (SER-CAT) | 19-ID (SBC-CAT) | 19-ID (SBC-CAT) |
| Energy (eV) | 12,660 | 12,690 | 12,658 |
| Wavelength (Å) | 0.97931 | 0.97702 | 0.97948 |
| Completeness (%) | 99.1 (100) | 99.2 (99.9) | 99.1 (99.7) |
| Avg redundancy | 7.9 (7.8) | 7.7 (7.8) | 5.4 (5.5) |
| Rsym (%) | 8.2 (44.0) | 10.9 (47.3) | 11.1 (50.1) |
| <I>/<σI> | 19.3 (5.3) | 16.5 (3.7) | 13.7 (3.2) |
| No. of observed/unique reflections (anomalous pairs separate) | 189,234/23,962 | 104,503/13,560 | 72,666/13,383 |
| Dimensions of P41212 cell | |||
| a (Å) | 72.6 | 74.0 | |
| b (Å) | 72.6 | 74.0 | |
| c (Å) | 136.8 | 143.2 | |
| α, β, γ (°) | 90 | 90 | |
| RMS deviations | |||
| Bond lengths (Å) | 0.011 | ||
| Bond angles (o) | 1.298 | ||
| Ramachandran | |||
| Favored (%) | 92.4 | ||
| Allowed (%) | 7.6 | ||
| Mean B-value (Å2) for protein | 45.2 | ||
| Main chain | 44.2 | ||
| Side chain | 46.2 | ||
| No. of protein atoms (no H) | 2,757 | ||
| No. waters | 80 | ||
| No. SO4 | 2 | ||
| Mean B-value of all atoms | 45.2 | ||
| No. refl. refined/no. free | 12,338/754 | ||
| Rwork/Rfree (%) | 22.5 (24.1)/29.1 (30.2) |
Values in parentheses indicate the highest-resolution bin. Rsym=∑h∑i|Ih,i−〈Ih〉|/∑h∑i|Ih,i|, where Ih,i is the ith intensity measurement of reflection h, and 〈Ih〉 is the average intensity of that reflection. Rwork/Rfree = Σh|FP − Fc|/Σh|Fp|, where Fc is the calculated and Fp is the observed structure factor amplitude of reflection h for the working set and free set, respectively. refl., reflections.
RESULTS
The overall structure of G4.
The structure of G4 reveals a thioredoxin fold, despite having only ca. 25% sequence identity in the first 40 residues and less thereafter to proteins of known structure in this family (Fig. 1A and B). G4 has the N-terminal β-strand-α-helix-β-strand and the C-terminal β-strand-β-strand-α-helix that form the core of the thioredoxin fold and make a four-stranded β-sheet similar to glutaredoxin (26). In addition to the core fold, 26 residues connect strands β2 and β3 and include the α2 helix (Fig. 1C). The overall G4 structure resembles a hand, with the additional residues between β2 and β3 forming two upheld “fingers.” G4 contains a thioredoxin-like CXXC sequence motif located on a “knuckle,” between the β1-strand and the α1-helix (Fig. 1A). Alternating oxidation and reduction of the cysteines in the CXXC motif is the basis of disulfide reductase and oxidase activity in many thioredoxin family proteins. In the crystal, the cysteines of the CSIC motif of G4 are in a reduced state.
FIG. 1.

Structure of G4. (A) The “front” view shows G4 along the plane of the β-sheet of the thioredoxin fold, with the β-strands colored orange and the helices in blue. Indicated in red are the side chains of the two cysteines of the catalytic site of G4. By analogy to other proteins of this fold, the more N-terminal cysteine (Cys13), found in the loop between β1 and α1, forms disulfide bond intermediates with cysteines of substrate proteins. Helices and strands are labeled. (B) The “side” view is perpendicular to the β-sheet, allowing a different perspective of the sheet and extended “fingers” region. (C) Topology diagram of the secondary structural elements of G4, showing the core thioredoxin fold (box). The location of the cysteines are in red.
G4 dimers.
For each of the three G4 molecules A, B, and C of the asymmetric unit, another molecule makes extensive contact from the neighboring asymmetric unit that is crystallographically related by twofold rotational symmetry. Labeling the molecules of the neighboring asymmetric unit as A′, B′, and C′, dimers form between A and B′, B′ and A, and C and C′ (see Fig. S1 in the supplemental material).
Each G4 monomer has a surface area of ca. 8,500 Å2. In each G4 dimer, there is a buried surface area of ca. 4,500 Å2. By comparison, ca. 1,400 to 1,700 Å2 surface area is buried in forming a typical antibody-antigen complex (4). Each G4 monomer contributes ca. 2,250 Å2 to the dimer interface, which is about one-fourth of the total surface of the monomer.
In the “fingers” region of the G4 dimer, which extends beyond the thioredoxin core, the residues between β2 and α2 form an antiparallel β-sheet with β2 from the other molecule of the dimer, doubling the length of the β2-strand from each monomer through extensive hydrogen bonding. This adds a fifth strand to the β-sheet of each monomer (Fig. 2A). The five-stranded, extended β-sheet of one monomer wraps around the α2 helix of the other molecule, making numerous hydrophobic interactions provided by the side chains of the following residues: Leu33, Val35, Ile37, Phe40, Phe41, Leu66, Tyr69, Ile70, Phe74, Tyr76, Met83, Phe85, Phe94, and Trp119 (Fig. 2B).
FIG. 2.

G4 dimers. (A) Stereo view of the G4 dimer as a ribbon diagram. One monomer is colored by secondary structure as in Fig. 1, and the other is colored beige. The long β-strands that form due to the dimerization are shown. A gray line represents a loop between strand β2b and helix α2 that was not built into the model due to disorder in the crystal. The cysteines are colored red. (B) Stereo view of the interface of the dimer as a solvent-accessible surface representation of one molecule and a ribbon representation of the second molecule. The hydrophobic residues of the dimer are colored green in the surface representation and depicted as yellow sticks in the ribbon representation.
To evaluate the complementarity of the interacting surfaces of the two G4 molecules, we calculated the shape correlation statistic for the interface (21). The shape correlation is a measure of the geometric match between two interacting surfaces and is a function of the distance and matching curvature between points on both surfaces that form the interface. A perfect fit between two surfaces would yield a shape correlation of 1.0, with no separation between the surfaces. The interface between the two G4 molecules yields a shape correlation value of 0.71, which is in the range found for protein oligomers and protein-protein inhibitor interfaces and is higher than the values (0.64 to 0.68) typical for antibody-antigen complexes (21). In addition, no electron density for water molecules is present in the interface. Consistent with the dimer in the crystal, G4 appears to be capable of forming dimers in solution (Fig. 3; see also Fig. S2 in the supplemental material), although the protein used for crystallization was obtained by pooling the fractions from the monomeric peak. Some higher-order multimers can also been seen in Fig. 3, consistent with buried surfaces between dimers of ca. 1,100 to 1,300 Å2 that are seen in the crystal.
FIG. 3.

Size exclusion chromatogram of G4. This representative 280-nm absorption profile of G4 (molecular mass of 14.0 kDa), shown in blue, indicates two major populations of G4, consistent with a monomer-dimer equilibrium. The protein was run in phosphate-buffered saline buffer with the addition of 5 mM dithiothreitol. Immediately prior to the G4 run, molecular size standards (Stds) were run under the same conditions (orange peaks) with their molecular masses given at the top of the chromatogram.
Comparison of G4 sequences.
G4 is conserved in all sequenced poxviruses. Alignment of a number of G4 sequences, as shown in Fig. 4, indicates that the β2b strand and α2 helix extending beyond the core thioredoxin fold are conserved. Many of the hydrophobic residues that make contacts in the G4 dimer interface are also conserved, suggesting that all of the poxvirus G4 proteins are capable of forming dimers.
FIG. 4.

Alignment of vertebrate poxvirus G4 sequences. Differences in sequence are shown, and periods (.) indicate identical residues. Conserved hydrophobic residues that are in the dimer interface are boxed in green. Hydrophobic residues of the dimer interface that are less well conserved are in yellow boxes. In red boxes are the cysteines that mediate the oxidase function. The residue ranges of β-strands and α-helices observed in the G4 structure are shown above the alignment. The β2b strand and the α2 helix form the extended “finger” region. The conserved YGVWPP sequence close to the C terminus is underlined.
A feature of poxvirus G4 sequences is a conserved hexapeptide, YGVWPP, at the C terminus of G4 (9). In the G4 structure, Trp119 of the hexapeptide contributes hydrophobic contacts to the dimer interface. There is density to suggest that the Trp119 side chain can adopt an alternate conformation and may not be required for the dimer interaction. The electron density for the prolines following Trp119 suggest that they are disordered and do not play a structural role.
In the alignments, a completely conserved Gly-Lys-Pro sequence is adjacent to the catalytic site on the loop connecting the β1 strand and the α1 helix. The proximity of Lys10 to Asn68 on the α2 helix may contribute to stabilizing the dimer of G4. Gly9 and Pro11 may allow conformational flexibility for large movements of Lys10, which could be involved in the monomer-dimer transition or substrate binding.
Comparison to the binding site of other proteins with a thioredoxin fold.
To get an indication of the mode of binding substrates, we compared the structure of G4 to other proteins with thioredoxin folds. The structures of Escherichia coli thioredoxin bound to peptides from the proteins, NF-κB and Ref-1, have been determined by using nuclear magnetic resonance (31, 32). Figure 5A indicates that substrate peptides bind to thioredoxin by making numerous interactions with the loops at the top of the β sheet. Other proteins of this family are thought to bind peptides similarly, at a hydrophobic cleft next to the N-terminal, active cysteine, Cys13, of the CXXC motif (Fig. 5B). When G4 is oriented similarly, as shown in Fig. 5, the α2 helix and β strand of the “finger” region protrude beyond the substrate binding loops, thereby blocking the binding site seen in the other proteins. This suggests that G4 may not bind substrate as a dimer and indicates that dimerization may play a role in regulating its catalytic activity.
FIG. 5.

Comparison of substrate binding sites of thioredoxin-like proteins. (A) Ribbon diagrams of thioredoxin bound to a peptide from NF-κB (left) (PDB code 1MDI) and Ref-1 (center) (PDB code 1CQG). A similarly oriented G4 is shown (right), illustrating the extended “finger” region protruding through the peptide binding surface found in thioredoxin. (B) Ribbon diagrams of thioredoxin bound to a peptide from NF-κB (left) or Ref-1 (center) and a similarly oriented G4 (right). Surface representations are shown below each ribbon diagram in panel B, illustrating the hydrophobic cleft of the binding site on thioredoxin. The view of G4 shows the occlusion of the binding site due to the extended “fingers” region. The surfaces are colored from white to green representing increasing hydrophobicity.
By analogy to other thioredoxin-like proteins, Cys13 of G4 would contain the redox-active thiol, but the Sγ atom of Cys13 is almost entirely buried. In the dimer structure, the α2 helix contributes to occluding the Sγ, so that only 0.2 to 1.1Å2 is exposed to solvent. Although the cysteines are reduced in the structure, a disulfide bond could be protected from the solvent in a similar manner. Thus, the dimer may be capable of maintaining the oxidized state of G4 in the cytoplasm until it can react with substrate.
DISCUSSION
Together with the poxviral proteins E10 and A2.5, G4 constitutes a pathway that forms disulfide bonds in the cytoplasm of eukaryotic cells. A number of vaccinia virus proteins have been identified as substrates of G4. These include L1, a target of neutralizing antibodies and candidate for a DNA vaccine (13-15, 50), as well as A21, A28, L5, H2, and A16, proteins involved in membrane fusion during viral infection (33-35, 46, 47). The importance of this pathway is reinforced by the observation that deleting any of the three proteins in the disulfide bond-making pathway abrogates virus formation (37, 39, 48).
We observe the structure of G4 as a dimer in the crystal. In solution, a peak with the mobility of a dimer is always observed during size exclusion chromatography, although we isolated the monomer for crystallization (Fig. 3). For a monomer to exist in solution, the large hydrophobic surface that is buried in the dimer interface would need to be shielded from solvent. The positions of the α2 helices in the G4 dimer suggest a mechanism for such shielding. The monomer could form by binding its own α2 helix in the same position that is occupied by the α2 helix of its partner in the dimer, thus masking the hydrophobic surface. As modeled in Fig. 6, the “finger” region folds down in a similar position as the “finger” of the other molecule of the dimer. In the model of monomeric G4, the α2 helix swings away from the catalytic site and exposes the loops seen in other structures of thioredoxin-like proteins. A similar hydrophobic cleft would be created adjacent to the catalytic cysteine in monomeric G4. The Sγ of the reactive Cys13 also would become exposed to solvent when the α2 helix folds down in the monomer. The monomer may raise its “fingers” to form the dimer, which would protect the Cys13-Cys16 disulfide bond in the reducing cytoplasm.
FIG. 6.
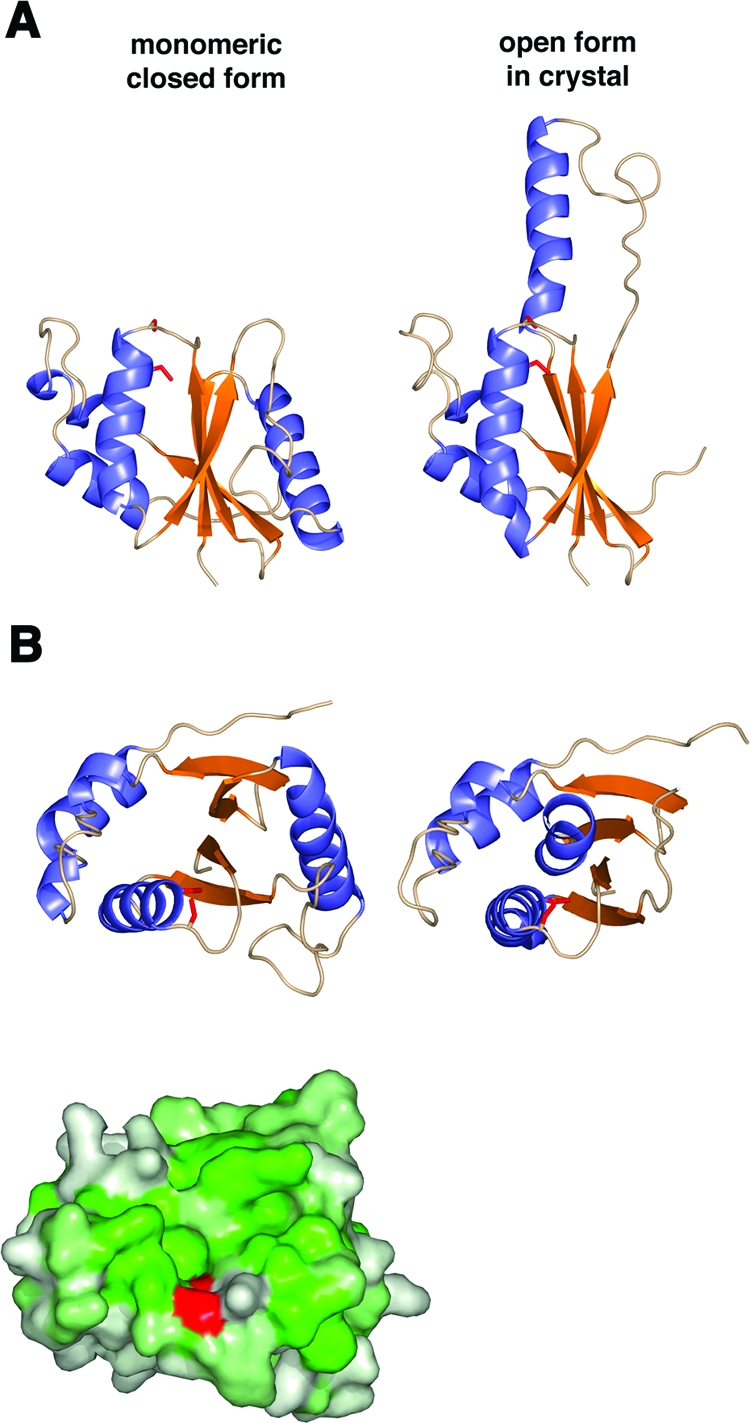
Model of monomeric G4. (A) A monomeric, closed model of G4 (left) was built based on the location of β2b and α2 in the dimer. For reference, the open form of the dimer in the crystal is shown on the right. (B) A view oriented from the top as in Fig. 5B shows the closed model (left) and open model (right) of G4 as a ribbon diagram (above). The same view of the closed model as a surface representation colored by hydrophobicity shows a similar hydrophobic cleft above the active-site cysteine, colored in red (below).
Alternatively, it is possible that the dimer interface is a surrogate for the binding of a substrate to the G4 monomer. The unfolded or partially folded substrate proteins may bind using similar interactions that occur in the dimer, through hydrophobic interactions or hydrogen bonding at the edge of the sheet. In the structure of L1, a substrate of G4, at least some disulfide bonds are formed prior to protein folding, since some disulfides are not solvent accessible in the final structure (40). G4 may function as a chaperone while disulfide bonds are formed. The complex between G4 bound to peptides from known substrates may also aid in designing drugs to block G4 activity.
The eukaryotic disulfide oxidoreductases also have disulfide isomerase activity. PDI contains multiple thioredoxin domains, and individual domains are sufficient for creating disulfide bonds, but multiple domains are required to isomerize improperly paired bonds (45). In bacteria, DsbC and DsbG both contain single thioredoxin domains that dimerize to provide isomerase activity (12, 27). PDI and the DsbC/DsbG proteins have similar “V”-shaped structures that expose a hydrophobic surface between two catalytic CXXC motifs. The hydrophobic surface is thought to hold the misfolded substrate proteins while the catalytic sites function as isomerases. Whether G4 can or cannot isomerize disulfide bonds is unknown, but the solvent-inaccessible environment of Cys13 suggests that G4 does not have isomerization activity as a dimer. The reducing environment of the cytoplasm itself may be capable of breaking improperly paired disulfide bonds.
The catalytic cysteines are located at the amino-terminal end of the G4 α1 helix and are found at the amino-terminal end of the structurally analogous helix in other thioredoxin-like proteins. This helix is thought to provide a macro-dipole that lowers the pKa of the catalytic cysteine and maintains it in an ionized, active form (29). In the model of G4, the carboxyl-terminal end of helix α2 is situated close to the catalytic site and would oppose the dipole of helix α1. The carbonyl oxygen of Asn68 at the carboxyl end of helix α2 is located 3.4 to 3.6 Å from the sulfur atom of Cys13 in the three molecules of the asymmetric unit. The location of the α2 helix further suggests that the G4 dimer is inactive. In the model of monomeric G4, α2 swings away from the catalytic site, allowing the α1 helix dipole to lower the pKa of Cys13, promoting its ionization and thus activating G4 (Fig. 6).
Part of the sequence noted to be conserved in G4 proteins was the YGVWPP sequence near the C terminus (9). Examination of the electron density map beyond Trp119 of the hexapeptide sequence indicates that the C terminus is disordered. The selective pressure for conservation of this region, throughout the poxvirus G4 proteins, does not appear to be structural. The hexapeptide sequence may perform a function in binding to A2.5 or in localization to the membrane, where G4 substrates are found. Due to the proximity of Trp119 to the dimer interface, A2.5 binding to the YGVWPP sequence may disrupt the dimer and be a means to regulate G4 activity.
Supplementary Material
Acknowledgments
We thank Scott Garman for help with the initial electron density maps, Apostolos Gittis for help with analyzing the model, and the other members of the Structural Biology Section for helpful discussions. We thank Bernard Moss and Tatiana Senkevich of the Laboratory of Viral Diseases, NIAID, for vaccinia virus DNA and for helpful discussions. We thank Mark Garfield for mass spectrometry and N-terminal protein sequencing and the staff of SBC-CAT and SER-CAT for assistance with data collection. The data were collected at the Structural Biology Center (SBC-CAT) and Southeast Regional Collaborative Access Team (SER-CAT) beam lines at the Advanced Photon Source, Argonne National Laboratory.
Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source beam lines was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38. This research was supported by the Division of Intramural Research of the NIAID, NIH.
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Bawden, A. L., K. J. Glassberg, J. Diggans, R. Shaw, W. Farmerie, and R. W. Moyer. 2000. Complete genomic sequence of the Amsacta moorei entomopoxvirus: analysis and comparison with other poxviruses. Virology 274:120-139. [DOI] [PubMed] [Google Scholar]
- 2.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54(Pt. 5):905-921. [DOI] [PubMed] [Google Scholar]
- 3.Corpet, F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16:10881-10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies, D. R., E. A. Padlan, and S. Sheriff. 1990. Antibody-antigen complexes. Annu. Rev. Biochem. 59:439-473. [DOI] [PubMed] [Google Scholar]
- 5.DeLano, W. L. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, Calif.
- 6.Ellgaard, L., and L. W. Ruddock. 2005. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6:28-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fields, B. N., D. M. Knipe, and P. M. Howley. 1996. Fields virology, 3rd ed. Lippincott-Raven Publishers, Philadelphia, Pa.
- 8.Gross, E., D. B. Kastner, C. A. Kaiser, and D. Fass. 2004. Structure of Ero1p, source of disulfide bonds for oxidative protein folding in the cell. Cell 117:601-610. [DOI] [PubMed] [Google Scholar]
- 9.Gvakharia, B. O., E. K. Koonin, and C. K. Mathews. 1996. Vaccinia virus G4L gene encodes a second glutaredoxin. Virology 226:408-411. [DOI] [PubMed] [Google Scholar]
- 10.Harrison, S. C., B. Alberts, E. Ehrenfeld, L. Enquist, H. Fineberg, S. L. McKnight, B. Moss, M. O'Donnell, H. Ploegh, S. L. Schmid, K. P. Walter, and J. Theriot. 2004. Discovery of antivirals against smallpox. Proc. Natl. Acad. Sci. USA 101:11178-11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henderson, D. A., T. V. Inglesby, J. G. Bartlett, M. S. Ascher, E. Eitzen, P. B. Jahrling, J. Hauer, M. Layton, J. McDade, M. T. Osterholm, T. O'Toole, G. Parker, T. Perl, P. K. Russell, K. Tonat, et al. 1999. Smallpox as a biological weapon: medical and public health management. JAMA 281:2127-2137. [DOI] [PubMed] [Google Scholar]
- 12.Heras, B., M. A. Edeling, H. J. Schirra, S. Raina, and J. L. Martin. 2004. Crystal structures of the DsbG disulfide isomerase reveal an unstable disulfide. Proc. Natl. Acad. Sci. USA 101:8876-8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hooper, J. W., D. M. Custer, C. S. Schmaljohn, and A. L. Schmaljohn. 2000. DNA vaccination with vaccinia virus L1R and A33R genes protects mice against a lethal poxvirus challenge. Virology 266:329-339. [DOI] [PubMed] [Google Scholar]
- 14.Hooper, J. W., D. M. Custer, and E. Thompson. 2003. Four-gene-combination DNA vaccine protects mice against a lethal vaccinia virus challenge and elicits appropriate antibody responses in nonhuman primates. Virology 306:181-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ichihashi, Y., and M. Oie. 1996. Neutralizing epitope on penetration protein of vaccinia virus. Virology 220:491-494. [DOI] [PubMed] [Google Scholar]
- 16.Iyer, L. M., L. Aravind, and E. V. Koonin. 2001. Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 75:11720-11734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones, T. A., J. Y. Zou, S. W. Cowan, and Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47(Pt. 2):110-119. [DOI] [PubMed] [Google Scholar]
- 18.Kabsch, W., and C. Sander. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577-2637. [DOI] [PubMed] [Google Scholar]
- 19.Kleywegt, G. J. 1996. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr. D Biol. Crystallogr. 52:842-857. [DOI] [PubMed] [Google Scholar]
- 20.Laskowski, R. A., M. M. W., Moss, D. S., and Thornton, J. M. 1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283-291. [Google Scholar]
- 21.Lawrence, M. C., and P. M. Colman. 1993. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234:946-950. [DOI] [PubMed] [Google Scholar]
- 22.Lee, B., and F. M. Richards. 1971. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55:379-400. [DOI] [PubMed] [Google Scholar]
- 23.Lustig, S., C. Fogg, J. C. Whitbeck, and B. Moss. 2004. Synergistic neutralizing activities of antibodies to outer membrane proteins of the two infectious forms of vaccinia virus in the presence of complement. Virology 328:30-35. [DOI] [PubMed] [Google Scholar]
- 24.Mallick, P., D. R. Boutz, D. Eisenberg, and T. O. Yeates. 2002. Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds. Proc. Natl. Acad. Sci. USA 99:9679-9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 26.Martin, J. L. 1995. Thioredoxin: a fold for all reasons. Structure 3:245-250. [DOI] [PubMed] [Google Scholar]
- 27.McCarthy, A. A., P. W. Haebel, A. Torronen, V. Rybin, E. N. Baker, and P. Metcalf. 2000. Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli. Nat. Struct. Biol. 7:196-199. [DOI] [PubMed] [Google Scholar]
- 28.Murshudov, G. N., A. A. Vagin, and E. J. Dodson. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53:240-255. [DOI] [PubMed] [Google Scholar]
- 29.Nelson, J. W., and T. E. Creighton. 1994. Reactivity and ionization of the active site cysteine residues of DsbA, a protein required for disulfide bond formation in vivo. Biochemistry 33:5974-5983. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307-326. [DOI] [PubMed] [Google Scholar]
- 31.Qin, J., G. M. Clore, W. M. Kennedy, J. R. Huth, and A. M. Gronenborn. 1995. Solution structure of human thioredoxin in a mixed disulfide intermediate complex with its target peptide from the transcription factor NF κB. Structure 3:289-297. [DOI] [PubMed] [Google Scholar]
- 32.Qin, J., G. M. Clore, W. P. Kennedy, J. Kuszewski, and A. M. Gronenborn. 1996. The solution structure of human thioredoxin complexed with its target from Ref-1 reveals peptide chain reversal. Structure 4:613-620. [DOI] [PubMed] [Google Scholar]
- 33.Senkevich, T. G., and B. Moss. 2005. Vaccinia virus H2 protein is an essential component of a complex involved in virus entry and cell-cell fusion. J. Virol. 79:4744-4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Senkevich, T. G., S. Ojeda, A. Townsley, G. E. Nelson, and B. Moss. 2005. Poxvirus multiprotein entry-fusion complex. Proc. Natl. Acad. Sci. USA 102:18572-18577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Senkevich, T. G., B. M. Ward, and B. Moss. 2004. Vaccinia virus entry into cells is dependent on a virion surface protein encoded by the A28L gene. J. Virol. 78:2357-2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Senkevich, T. G., A. S. Weisberg, and B. Moss. 2000. Vaccinia virus E10R protein is associated with the membranes of intracellular mature virions and has a role in morphogenesis. Virology 278:244-252. [DOI] [PubMed] [Google Scholar]
- 37.Senkevich, T. G., C. L. White, E. V. Koonin, and B. Moss. 2000. A viral member of the ERV1/ALR protein family participates in a cytoplasmic pathway of disulfide bond formation. Proc. Natl. Acad. Sci. USA 97:12068-12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Senkevich, T. G., C. L. White, E. V. Koonin, and B. Moss. 2002. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc. Natl. Acad. Sci. USA 99:6667-6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Senkevich, T. G., C. L. White, A. Weisberg, J. A. Granek, E. J. Wolffe, E. V. Koonin, and B. Moss. 2002. Expression of the vaccinia virus A2.5L redox protein is required for virion morphogenesis. Virology 300:296-303. [DOI] [PubMed] [Google Scholar]
- 40.Su, H. P., S. C. Garman, T. J. Allison, C. Fogg, B. Moss, and D. N. Garboczi. 2005. The 1.51-angstrom structure of the poxvirus L1 protein, a target of potent neutralizing antibodies. Proc. Natl. Acad. Sci. USA 102:4240-4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan, J. T., and J. C. Bardwell. 2004. Key players involved in bacterial disulfide-bond formation. Chembiochemistry 5:1479-1487. [DOI] [PubMed] [Google Scholar]
- 42.Terwilliger, T. C. 2003. Automated main-chain model building by template matching and iterative fragment extension. Acta Crystallogr. D Biol. Crystallogr. 59:38-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terwilliger, T. C. 2000. Maximum-likelihood density modification. Acta Crystallogr. D Biol. Crystallogr. 56(Pt. 8):965-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terwilliger, T. C., and J. Berendzen. 1999. Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55(Pt. 4):849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tian, G., S. Xiang, R. Noiva, W. J. Lennarz, and H. Schindelin. 2006. The crystal structure of yeast protein disulfide isomerase suggests cooperativity between its active sites. Cell 124:61-73. [DOI] [PubMed] [Google Scholar]
- 46.Townsley, A. C., T. G. Senkevich, and B. Moss. 2005. The product of the vaccinia virus L5R gene is a fourth membrane protein encoded by all poxviruses that is required for cell entry and cell-cell fusion. J. Virol. 79:10988-10998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Townsley, A. C., T. G. Senkevich, and B. Moss. 2005. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J. Virol. 79:9458-9469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.White, C. L., T. G. Senkevich, and B. Moss. 2002. Vaccinia virus G4L glutaredoxin is an essential intermediate of a cytoplasmic disulfide bond pathway required for virion assembly. J. Virol. 76:467-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winn, M. D., M. N. Isupov, and G. N. Murshudov. 2001. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57:122-133. [DOI] [PubMed] [Google Scholar]
- 50.Wolffe, E. J., S. Vijaya, and B. Moss. 1995. A myristylated membrane protein encoded by the vaccinia virus L1R open reading frame is the target of potent neutralizing monoclonal antibodies. Virology 211:53-63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.