

Abstract
Currently, little is known about the viral kinetics of influenza A during infection within an individual. We utilize a series of mathematical models of increasing complexity, which incorporate target cell limitation and the innate interferon response, to examine influenza A virus kinetics in the upper respiratory tracts of experimentally infected adults. The models were fit to data from an experimental H1N1 influenza A/Hong Kong/123/77 infection and suggest that it is important to include the eclipse phase of the viral life cycle in viral dynamic models. Doing so, we estimate that after a delay of ∼6 h, infected cells begin producing influenza virus and continue to do so for ∼5 h. The average lifetime of infected cells is ∼11 h, and the half-life of free infectious virus is ∼3 h. We calculated the basic reproductive number, R0, which indicated that a single infected cell could produce ∼22 new productive infections. This suggests that antiviral treatments have a large hurdle to overcome in moderating symptoms and limiting infectiousness and that treatment has to be initiated as early as possible. For about 50% of patients, the curve of viral titer versus time has two peaks. This bimodal behavior can be explained by incorporating the antiviral effects of interferon into the model. Our model also compared well to an additional data set on viral titer after experimental infection and treatment with the neuraminidase inhibitor zanamivir, which suggests that such models may prove useful in estimating the efficacies of different antiviral therapies for influenza A infection.
Outbreaks of human disease caused by influenza virus can be traced through history as far back as 430 BC (21). Worldwide influenza outbreaks can be very costly in terms of human life, with over 20 million lives lost during the 1918 “Spanish flu” pandemic (6). During a “normal” influenza season in the United States, there are an estimated 20 to 50 million cases of influenza and influenza-like illnesses, which result in over 100,000 hospitalizations (43) and over 36,000 influenza-related cardiopulmonary deaths (46) and incur 1 to 3 billion U.S. dollars in direct medical costs and 10 to 15 billion U.S. dollars in indirect costs (32a).
Since influenza virus was first isolated in 1933 (44), overwhelming attention has been given to its structure, its genome, the immune response elicited against it, vaccines to protect against it, and its epidemiology. Information concerning the kinetics of influenza virus during an infection within an individual, however, is limited. The standard pattern of an influenza A virus infection in adults is characterized by an exponential growth of virus titer, which peaks 2 to 3 days postinfection (DPI), followed by an exponential decrease until it is undetectable after 6 to 8 DPI (51).
Innate and adaptive immune responses are important in modulating virus replication in the respiratory tract. For example, extended periods of replication (up to 21 days [d]) occur in young children experiencing initial infections, in infections due to novel viruses in susceptible persons (e.g., avian A [H5N1]), and in highly immunocompromised hosts in whom shedding may sometimes last weeks or months. In one study of experimentally infected adults, nasal wash neutralizing antibodies against influenza virus did not begin to increase until around 7 DPI and did not reach high titers until approximately 2 to 4 weeks postinfection (38). In another study, Ennis et al. report that cytotoxic T lymphocytes were not detected until 6 to 14 DPI and disappeared by day 21 (10). Thus, both the cytotoxic T-lymphocyte-mediated and the antibody-mediated immune responses tend to be detected after peak viral replication. Unlike the slow adaptive immune response, innate responses are detected early and are thought to provide the first line of defense against influenza replication (42). Interferons (IFNs), particularly of type I (IFN-α/β), tumor necrosis factor alpha, and other cytokines, such as interleukin-6, become elevated early after infection (12, 13, 20, 42, 48). Type I IFNs are produced by infected epithelial cells (19) and other host cells, such as macrophages, monocytes, and dendritic cells (3, 19), in response to the presence of viral double-stranded RNA (45). High IFN titers are detected 1 day after virus shedding begins and generally peak simultaneously with, or up to 1 day after, virus titer peak (12, 13, 38). Even though a general outline of the immune response to influenza A virus infection has been described, it does not account for the extremely rapid kinetics of influenza virus infection; clearly, we do not quantitatively understand the complex interplay between viral dynamics and the host response during infection.
Mathematical models have proven to be useful tools in the analysis of viral infections. For example, the dynamics of human immunodeficiency virus (HIV) infection in vivo were poorly understood until simple mathematical models were developed. These models examined the kinetics of viral load decline in patients treated with potent antiviral drug therapy and were able to estimate the rate of HIV replication, the number of virus particles produced and cleared daily, and the average life span of productively infected CD4+ T cells (17, 35, 36). Similar models have been used to study the action of IFN-α and ribavirin on hepatitis C virus (HCV) kinetics (8, 29, 30) as well as the effects of antiviral drugs, such as adefovir dipivoxil (47), lamivudine (32), and lamivudine/famciclovir (23), on hepatitis B virus (HBV) kinetics. These models took advantage of the fact that HIV, HCV, and HBV produced prolonged, chronic disease, with the virus population at or near steady state for long periods of time. Perturbation of the steady state with antiviral agents provided insight into viral dynamics.
In contrast, the extremely fast and relatively short duration of replication of influenza A virus in immunocompetent adults invites the search for alternative views of influenza virus and immune system dynamics. To date, there are only three models of influenza virus dynamics within a single infected host. One study used a compartmental model to describe the dynamics of influenza virus within infected mice (22). The compartments, however, did not correspond to any immune populations or factors, so changes in viral kinetics could not be connected with specific immune effects. Another model studied the viral dynamics in human infections and was composed of a system of differential equations representing 12 immune populations involving more than 60 parameters (2). Finally, the third and most recent model used cellular automaton simulations to include spatial effects and to visualize the spread of the infection in lung epithelial tissue (1).
In an effort to better understand influenza A virus kinetics in humans, we applied simple models, similar to those used to study HIV, HCV, and HBV (17, 23, 30, 32, 34, 36, 47), in which target cell limitation is important, to data derived from experimentally infected volunteers. We also used a more complex model that included the antiviral effects of IFN. One limitation of our study is that the data we evaluate are derived from experimentally infected subjects given intranasal challenge, which generally results in upper respiratory tract infection. In contrast, natural influenza infection most likely involves lower respiratory tract, i.e., tracheobronchial, viral replication.
MATERIALS AND METHODS
Study data.
The data examined here came from an experimental infection study of H1N1 influenza virus (28). Briefly, six serosusceptible adult volunteers were experimentally infected intranasally with 104.2 50% tissue culture infective doses (TCID50) of cloned wild-type influenza A/Hong Kong/123/77. Prescreening of the volunteers ensured that each had no recent influenza infections related to the challenge strain (hemagglutinin inhibition antibody titers of ≤1:8 and neuraminidase inhibition antibody titers of ≤1:2). As a result of the experimental infections, five of the six volunteers developed fever or systemic symptoms, and all were infected as determined by virus recovery. Nasal washes were collected daily for the first week of infection and serially cultured in 10-fold dilutions to determine infectious virus titers. The daily viral titers are presented in Table 1.
TABLE 1.
Results of nasal washes taken daily from six volunteers inoculated with 104.2 TCID50 of cloned wild-type influenza A/Hong Kong/123/77 (H1N1)a
| Patient | Result for day no.:
|
|||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | |
| 1 | 2.0 | 5.5 | 4.0 | 5.5 | 3.0 | ≤0.5 | ≤0.5 | ≤0.5 |
| 2 | 1.0 | 6.0 | 3.0 | 1.5 | 3.5 | 1.3 | ≤0.5 | ≤0.5 |
| 3 | 2.5 | 5.0 | 5.0 | 3.0 | 5.5 | 3.5 | ≤0.5 | ≤0.5 |
| 4 | 3.5 | 5.5 | 6.5 | 5.5 | 3.5 | 4.0 | ≤0.5 | ≤0.5 |
| 5 | 2.5 | 3.0 | 6.5 | 6.5 | 2.0 | 0.8 | ≤0.5 | ≤0.5 |
| 6 | 4.0 | 5.0 | 5.5 | 7.5 | 5.5 | 1.3 | ≤0.5 | ≤0.5 |
| Avg | 2.6 | 5.0 | 5.1 | 4.9 | 3.8 | 1.9 | ≤0.5 | ≤0.5 |
| SEM | 0.4 | 0.4 | 0.7 | 1.7 | 0.7 | 0.8 | NA | NA |
The data are given in log10 TCID50/ml of nasal wash. Data are provided by R. Murphy from the study presented in reference 28. NA, not applicable.
Fitting data to models.
Experimental viral titer is given in TCID50/ml of nasal wash. Particles deposited in the trachea and initial bronchial divisions are cleared with a half time of ∼30 min, and it takes up to 24 h to clear the airways down to approximately the 16th division, i.e., the end of the terminal bronchioles (4, 31). We took concentration of virus measured by TCID50/ml of nasal wash to be proportional to concentration of free virions at the site of infection at the time of nasal wash, thus ignoring any transport delay, since its precise length was unknown. Also, the time resolution of the data, with daily measurements, is such that small delays would have little impact on our results. The models were formulated in terms of ordinary or delay differential equations and were numerically solved using Berkeley Madonna (24). The Runge-Kutta 4 or the Euler method of integration (for models with fixed delays) was employed with a step size of 0.0003. Madonna's “curve fitter” option was used to establish a set of initial parameter estimates. The curve-fitting method uses nonlinear least-squares regression that minimizes the sum of the squared residuals between the experimental and predicted values of log10 TCID50/ml of daily nasal wash for each patient. Because no uncertainty was provided for experimental viral titer, we weighted the data points equally in our fitting procedure. The set of parameter estimates derived from Madonna was used as initial guesses for a more sophisticated subroutine, DNLS1, from the Common Los Alamos Software Library, which is based on a finite-difference, Levenberg-Marquardt algorithm for solving nonlinear least-squares problems and a more sophisticated ordinary equation solver that uses an implicit Adam's method or, if the equations are stiff, Gear's method. We examined the dependence of the best-fit estimates of parameters on the initial parameter guesses by systematically exploring all possible combinations of twofold increases over the initial estimates. Finally, for each best-fit parameter estimate, we provide a 95% confidence interval (CI), which was computed from 200 bootstrap replicates, using the method of Efron and Tibshirani (9).
For some parameters, there was large interpatient variation in best-fit value. Moreover, because parameter values were constrained to be positive, their distributions were skewed to the right. For these reasons, we decided to compute the geometric mean along with the geometric 95% confidence interval rather than the arithmetic analogues. Specifically, we took the log10 value of the six patient-specific parameter estimates and then calculated the mean and 95% confidence interval (mean ± 1.96 times the standard error of the mean) on the log scale. We then raised the mean and end points of the confidence interval to the power of 10 and thus obtained the overall geometric mean and the geometric 95% confidence interval for the parameters shown in Tables 2 and 3.
TABLE 2.
Patient-specific best-fit parameter values for the target cell-limited model with no delay (equations 1 through 3)a
| Patient | V0 (TCID50/ml) | β [(TCID50/ml)−1 · d−1] | p (TCID50/ml · d−1) | c (d−1) | t1/2 (h) | δ (d−1) | 1/δ (h) | R0 | SSR (TCID50/ml)2 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3.5 × 10−1 | 3.4 × 10−5 | 7.9 × 10−3 | 3.3 | 5.0 | 3.4 | 7.1 | 9.6 | 4.6 |
| 2 | 1.4 × 10−3 | 1.6 × 10−4 | 4.1 × 10−3 | 2.1 | 7.7 | 11.2 | 2.1 | 11.2 | 7.0 |
| 3 | 1.0 × 10−2 | 1.3 × 10−4 | 3.2 × 10−3 | 2.1 | 7.9 | 2.1 | 11.4 | 37.7 | 8.3 |
| 4 | 9.1 × 10−1 | 6.3 × 10−6 | 4.2 × 10−2 | 3.1 | 5.4 | 2.8 | 8.7 | 12.4 | 4.0 |
| 5 | 4.3 × 10−1 | 2.3 × 10−5 | 1.0 × 10−2 | 4.2 | 4.0 | 5.1 | 4.7 | 4.4 | 9.0 |
| 6 | 3.3 × 10−1 | 3.8 × 10−6 | 7.1 × 10−2 | 3.6 | 4.6 | 3.6 | 6.7 | 8.3 | 13.6 |
| Avg | 9.3 × 10−2 | 2.7 × 10−5 | 1.2 × 10−2 | 3.0 | 5.6 | 4.0 | 6.0 | 11.1 | 7.1 |
| 95% CI | 1.4 × 10−2-6.1 × 10−1 | 8.8 × 10−6-8.3 × 10−5 | 4.8 × 10−3-3.0 × 10−2 | 2.4-3.6 | 4.6-6.9 | 2.6-6.1 | 3.9-9.2 | 6.6-18.5 | 5.1-9.9 |
For each patient, the best-fit initial virus titer (V0), infection rate constant (β), average rate of increase of viral titer per infected cell (p), viral clearance rate (c), half-life of free virus (t1/2), infected-cell lifespan (1/δ), basic reproductive number (R0), and sum of square residuals (SSR) are given along with their geometric average and geometric 95% CI. The parameter T0 was held fixed at a value of 4 × 108 cells.
TABLE 3.
Patient-specific best-fit parameter values for the target cell-limited model with a delay (equations 5 through 8)a
| Patient | V0 (TCID50/ml) | β [(TCID50/ml)−1 × d−1] | k (d−1) | 1/k (h) | p (TCID50/ml × d−1) | c (d−1) | t1/2 (h) | δ (d−1) | 1/δ (h) | 〈t〉 (h) | R0 | SSR (TCID50/ml)2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 4.3 × 10−2 | 4.9 × 10−5 | 3.9 | 6.2 | 2.8 × 10−2 | 4.3 | 3.9 | 4.2 | 5.7 | 11.9 | 30.4 | 4.3 |
| 2 | 3.1 × 10−7 | 1.1 × 10−3 | 2.0 | 12.1 | 2.1 × 10−2 | 11.0 | 1.5 | 10.9 | 2.2 | 14.3 | 75.0 | 6.5 |
| 3 | 7.0 × 10−1 | 1.7 × 10−4 | 4.9 | 4.9 | 3.0 × 10−3 | 2.2 | 7.5 | 2.3 | 10.3 | 15.2 | 39.6 | 8.0 |
| 4 | 4.9 | 5.3 × 10−6 | 4.0 | 6.0 | 1.3 × 10−1 | 3.8 | 4.4 | 3.8 | 6.4 | 12.4 | 19.1 | 2.9 |
| 5 | 1.7 | 2.7 × 10−6 | 6.0 | 4.0 | 5.9 × 10−1 | 13.5 | 1.2 | 13.5 | 1.8 | 5.8 | 3.5 | 6.6 |
| 6 | 2.4 | 8.4 × 10−6 | 4.4 | 5.5 | 7.1 × 10−2 | 3.7 | 4.5 | 3.8 | 6.3 | 11.8 | 16.6 | 11.8 |
| Avg | 7.5 × 10−2 | 3.2 × 10−5 | 4.0 | 6.0 | 4.6 × 10−2 | 5.2 | 3.2 | 5.2 | 4.6 | 11.4 | 21.5 | 6.1 |
| 95% CI | 7.6 × 10−4-7.5 | 6.0 × 10−6-1.7 × 10−4 | 3.0-5.2 | 4.6-7.9 | 1.2 × 10−2-1.7 × 10−1 | 3.1-8.7 | 1.9-5.3 | 3.2-8.6 | 2.8-7.5 | 8.8-14.7 | 10.1-46.1 | 4.3-8.7 |
For each patient, the best-fit initial virus titer (V0), infection rate constant (β), transition time to I2 (1/k), rate of increase of viral titer per infected cell (p), viral clearance rate (c), half-life of free virus (t1/2), infected cell life span (1/δ), average lifetime of infected cells (〈t〉, calculated as k−1 + δ−1, the sum of the lifetimes of cells while infected and not producing virus and while producing virus), basic reproductive number (R0), and sum of square residuals (SSR) are given along with their geometric average and geometric 95% CI. The parameter T0 was held fixed at a value of 4 × 108 cells.
In fitting the model with a delay from time of infection to viral production for patient 5, we fixed the parameter k to 6 d−1 to keep it and the parameters c and δ in a biologically realistic range. This led to a fit visually indistinguishable from the best fit and a sum of squared residuals a few percent higher than the minimum.
RESULTS
A target cell-limited model.
In the simplest model, influenza A virus infection is limited by the availability of susceptible target (epithelial) cells rather than the effects of the immune response. A model of acute viral infection that incorporates target cell limitation can be described by the following differential equations:
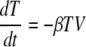 |
(1) |
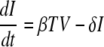 |
(2) |
 |
(3) |
where T is the number of uninfected target cells, I is the number of productively infected cells, and V is the infectious-viral titer expressed in TCID50/ml of nasal wash. We assume that infection is initiated by the introduction of virus into the upper respiratory tract at a concentration equivalent to V0 TCID50/ml of nasal wash. Susceptible cells become infected by virus at rate βTV, where β is the rate constant characterizing infection.
Virally infected cells, I, by shedding virus increase viral titers at an average rate of p per cell and die at a rate of δ per cell, where 1/δ is the average life span of a productively infected cell. Free virus is cleared at a rate of c per day. The effects of immune responses are not explicitly described in this simple model, but they are implicitly included in the death rate of infected cells (δ) and the clearance rate of virus (c). The reduction in viral titer due to binding and infection of target cells at rate βTV makes little impact on the amount of free virus and was neglected. The mechanism of virion clearance is unknown and may involve mucociliary clearance as well as binding of virions to cells and to respiratory secretions, such as mucins.
A variant of the model, incorporating the logistic term rT (1 − T/T0) into equation 1 to represent the potential regeneration of target cells as infection proceeds as well as their natural death, was tested. We found that including this term did not lead to an improvement of the fit of the model to the data. Further, it is only around 3 to 5 days after the onset of symptoms (5 to 7 days after infection) that mitoses are detected in the basal cell layer and regeneration of the epithelium begins (51). Complete resolution of the epithelial necrosis probably takes up to 1 month (51), and thus it is not surprising that this term has little effect on the fit of the model to viral titer data collected over the first week of infection. In all further analyses, this term was omitted from the model.
We were fortunate to obtain a data set from individuals experimentally infected with H1N1 influenza A virus (28), and these data were analyzed with the viral kinetic model given by equations 1 to 3 as well as refinements discussed below. Estimates of the parameters were obtained by using nonlinear least-squares regression to fit the model to the longitudinal viral titers for each subject in the study. The initial number of target cells in an adult, T0, is approximately 4 × 108 cells, which was calculated from the area of epithelial cells lining the nasal turbinates of the upper respiratory tract, 160 cm2 (26), and the surface area per epithelial cell, 2 × 10−11 to 4 × 10−11 m2/cell (11). Thus, T0 was fixed at 4 × 108 cells, and the remaining parameter values were estimated. Because T0 refers to the total number of target cells in the upper respiratory tract, I, the number of infected cells, will also be the total number in the upper respiratory tract.
As mentioned above, there is no simple mapping between number of free infectious virions and experimental infectious viral titer given in TCID50/ml of nasal wash. Thus, the parameter β, which is in units of (TCID50/ml)−1 · d−1, the parameter p, which is in units of TCID50/ml · d−1, and the initial number of virions, V0, which is in units of TCID50/ml of nasal wash, cannot be expressed in values that are more biologically meaningful. Although there are many more noninfectious virions released per cell than infectious ones, these were not considered in our analyses.
The parameter estimates obtained from fitting the model to the experimental data for each patient are given, along with their geometric mean and 95% confidence intervals, in Table 2. The model fit to the data using these best-fit parameter estimates is shown in Fig. 1.
FIG. 1.

Fits of the target cell-limited model without delay (equations 1 to 3) (solid lines) and with delay (equations 5 to 8) (dashed lines) to experimental data (filled squares) from H1N1 experimental influenza virus infections (28). The graphs present viral titers in TCID50/ml of nasal wash (black) and fractions of target cells remaining (blue) over the courses of the infections. The horizontal dotted lines mark the limit of detection for viral titer.
The estimate of the average lifetime of productively infected epithelial cells (1/δ) was approximately 6 h, which is shorter than a previously published estimate of 24 h (52). Our lower estimate of the average lifetime of infected cells may be due to the lack of a delay between infection and virus production (virus production typically begins 6 to 8 h after infection [41]) or experimental inaccuracies in the previously published estimate. The estimated average half-life of free infectious virus [t1/2 = ln(2)/c] ranged from 4.0 to 7.9 h, with a mean of 5.6 h (95% CI, 4.6 to 6.9 h) (Table 2).
The basic reproductive number, R0.
The rate at which virus infections establish themselves is frequently analyzed in terms of the basic reproductive number, R0. This number represents the average number of second-generation infections produced by a single infected cell placed in a population of entirely susceptible cells. If R0 is greater than 1, then an infection can be established, whereas an infection rapidly dies out if R0 is less than 1. For the model given by equations 1 to 3, R0 can be computed from the following formula (32):
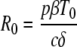 |
(4) |
Using equation 4 and the patient-specific parameter estimates, we calculated R0 for each patient; the mean was 11.1, with a 95% CI of 6.6 to 18.5 (Table 2). Thus, experimental influenza A virus infection is predicted to spread rapidly through the upper respiratory tract.
A target cell-limited model with delayed virus production.
After influenza A virus infects epithelial cells, progeny virions are usually not detected for 6 to 8 h (41). This delay in the production of free virus was modeled by defining two separate populations of infected epithelial cells: one population (I1) is infected but not yet producing virus; the second population (I2) is actively producing virus.
Equations 5 to 8 represent this delay model.
 |
(5) |
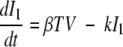 |
(6) |
 |
(7) |
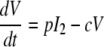 |
(8) |
where 1/k is the average transition time from I1 to I2. The separation of infected cells into two populations is similar to that in a model proposed earlier for HIV infection (35) and increases the realism of the model, as delays in the production of virus after the time of initial infection are part of the viral life cycle (the eclipse phase). However, this increase in realism occurs at the expense of adding one additional parameter, which is not justifiable on statistical grounds (F test). Nonetheless, we prefer this model for biological reasons (see Discussion).
Fitting the experimental data to this model (Fig. 1) gave the patient-specific and mean parameter estimates shown in Table 3. The geometric mean transition time from I1 to I2, 1/k, was 6.0 h (95% CI, 4.6 to 7.9 h) (Table 3). The geometric mean of the estimates of the average lifetime of virus-producing infected cells (1/δ) was 4.6 h (95% CI, 2.8 to 7.5 h). Putting both stages of infection together and then taking the geometric mean gave an average life span of infected epithelial cells (〈t〉) of 11.4 h (95% CI, 8.8 to 14.7 h) (Table 3).
In addition to a longer estimated life span, the estimate of viral production rate and virion clearance changed under equations 5 to 8. The new estimate of the free virion half-life (t1/2) is 3.2 h (95% CI, 1.9 to 5.3 h) (Table 3). This estimate is similar to the half-lives found for HIV and HCV (30, 36, 37, 53).
Overall, the changes in parameter estimates in the delay model resulted in a higher R0 value of 22 (also computed using equation 4), with a 95% CI of 10 to 46. This increase in the estimated R0 value, relative to that in the nondelay model, is due to the inclusion of a delay prior to production of free virus in this model. The delay leads to a longer viral generation time, and hence, a larger number of infections per generation are required to produce a rate of exponential growth similar to that in the nondelay model.
Damage to the epithelial cells during influenza infection is difficult to estimate. One study suggested that at the peak of infection, about 30 to 50% of the epithelial cells in the upper respiratory tract are destroyed (2). By numerically integrating the loss of productively infected cells (dD/dt = δI2) up to the time of the virus titer peak predicted by the model's best fit, we find that among the six patients, the fraction of dead cells at viral titer peak ranged from 37% (patient 2) to 66% (patient 3).
The model also provides us with a way of extracting the number of productively infected cells at viral titer peak. Viral titer peak occurs when the change in the virus population with respect to time is equal to zero. In our delay model (equations 5 to 8), this condition is met when pI2 is equal to cV. Thus, at viral titer peak, the level of productively infected target cells is given by I2 = cV/p. Using estimates of peak virus titer, viral production rate (p), and clearance rate of free virus (c), we find that, in the six volunteers, the proportion of productively infected cells (I2) at viral titer peak predicted by the model's best-fit parameters ranged from 8% (patient 5) to 30% (patient 3).
In both our simple (equations 1 to 3) and delay (equations 5 to 8) models, infection is cleared without the action of interferon or the immune system. The resolution of infection in our model is a consequence of target cell limitation. Figure 1 shows, for each patient's best-fit parameter values, the predicted fraction of remaining targets available for infection as a function of time. The figure shows that near viral titer peak, the vast majority of target cells have been depleted. While this would seem to exclude the possibility of the infection lasting as late as days 6 to 8, as is sometimes observed (51), Fig. 1 shows that despite the few remaining target cells past the viral titer peak, our model can indeed sustain infection as late as days 6 to 8.
Models incorporating an interferon response.
Type I IFNs are induced by influenza virus infection and act to inhibit viral replication within the infected cell and also induce an antiviral state in surrounding cells (45). During influenza A virus infections, IFNs and IFN activity are generally detected by 24 h postinfection (39), and peak nasal wash IFN titers are found at the time of or up to 1 day after nasal virus titer peak (12, 13, 38). Interferon, through induction of Mx and PKR proteins, has been shown to affect influenza infections by interfering with synthesis and/or translation of viral RNA (40, 49). In turn, influenza NS1 protein can counteract the antiviral effects of IFN through several mechanisms (50), but nonetheless, human plasmacytoid dendritic cells produce high levels of IFN-α after infection with influenza (7).
Because of the importance of IFN in the response to influenza infection, we explore through modeling the effects of including an IFN response on the kinetics of viral infection. The net effect of IFN on viral replication can be modeled by increasing 1/k or the transition time from the nonproductive state I1 to the productive state I2 and/or by lowering the viral production rate, p, as has been done in modeling HCV infections (30). We let F be the quantity of IFN. While it is known that infected epithelial cells produce IFN-α, it can also be produced by monocytes, macrophages, and plasmacytoid dendritic cells (7, 19). Here, we assumed that IFN is secreted from virus-producing cells, I2, at a rate of s per cell, beginning τ time units after cells begin producing virus and that this amount is proportional to the amount made collectively by infected cells, monocytes, macrophages, and plasmacytoid dendritic cells. We also assume that IFN is lost (either by binding to cellular IFN receptors, which are then internalized, or through degradation) according to rate constant α, so that
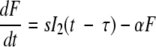 |
(9) |
We assume that IFN may affect the rate at which infected target cells move into the virion producing state, k, and/or the rate of viral production, p, according to the following equations:
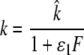 |
(10) |
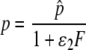 |
(11) |
where the parameters k̂ and p̂ correspond to the values of these parameters in the absence of IFN. We tested models where IFN affected k only, p only, or both k and p. The available data do not allow us to determine which model formulation may be more appropriate since decreasing either k or p reduces viral production. We set s to 1 without loss of generality, as this changes only the units in which IFN is measured. Nonetheless, the model that includes IFN has more parameters than data points and hence cannot be supported statistically, nor can parameters be estimated uniquely. Despite the limitation of overfitting the data, we used nonlinear regression to see how well this model would agree with the data. Preliminary data fitting suggested that a half-day lag in IFN response was better than a full day or no delay. Thus, the delay τ was set at 0.5 days. More-sophisticated models and methods of analysis could be envisioned but were not pursued due to the lack of data on IFN levels in these patients. However, as described below, including IFN in the model allowed us to explain the occurrence of bimodal virus titer curves.
Bimodal virus titer curves.
An interesting feature observed in the experimental influenza A viral kinetic data examined here was the presence of two virus titer peaks in some patients. This phenomenon of bimodal virus titer peaks was also observed in another study, where the average virus titer of 15 infected individuals had a bimodal peak (18). In studies where viral titers were given for each patient, the bimodal virus peaks were present in 6 of 12 patients (27, 28) and hence may be biologically relevant. The data show that virus titer can drop 1 to 2 logs after the initial peak and then rebound to a similar peak before decreasing again.
The simple target cell-limited models described above could not produce bimodal virus titer peaks to match the data for patients 1 to 3, in whom double peaks were observed. Using the interferon model, however, we were able to produce bimodal virus titer peaks (Fig. 2). Because of the delay in IFN production from infected cells, IFN levels peaked after viral titer peak (Fig. 2). As the infection waned due to target cell limitation, the infected cell and IFN levels fell. The fall in IFN allowed viral production to increase in the remaining infected cells, which in some patients created a second viral titer peak. Because we overfit the data, the “best-fit” parameter values may not be reliable and need to be interpreted with caution. Thus, we have not included them here, but they will be made available upon request.
FIG. 2.

Fits of the target cell-limited model with delay that incorporates interferon (equations 5 to 8 and 9 to 11) to experimental data (filled squares) from H1N1 experimental influenza virus infections (28). The graphs present the viral titers (black), the IFN concentrations (red), and the fractions of target cells remaining (blue) over the courses of the infections. The horizontal dotted lines mark the limit of detection for viral titer.
NI study.
The rather large values of R0 found by our models and the rapid viral kinetics suggest that antiviral treatment needs to be given before or very early after infection. This interpretation is well supported by studies where a neuraminidase inhibitor (NI), such as zanamivir or oseltamivir, was used prophylactically or as treatment after natural and experimental infections (14, 15, 16). Antiviral treatment 1 to 2 DPI generally resulted in viral clearance 2 to 3 days earlier and cessation of symptoms 1 day earlier than in placebo controls. Since NIs prevent new virions from budding off an infected cell, the use of an NI was incorporated into our models by reducing viral production rate.
In order to assess whether the type of simple model we developed here can be used to understand the effects of a neuraminidase inhibitor, we compare (in Fig. 3) the predictions of a simulation of the delay model with experimental data taken from reference 15. In this study, adult volunteers were inoculated intranasally with influenza A/Texas/91 (H1N1) (15). Zanamivir was then given early, at either 26 h or 32 h postinfection, which we modeled as being given at 29 h, or late, at 50 h postinfection. Assuming that the NI reduced viral production by 97% gave reasonable agreement with the data (Fig. 3). For the placebo group, we assumed that there was no reduction in viral production.
FIG. 3.

Course of influenza virus infections with and without the neuraminidase inhibitor zanamivir given intranasally. The average virus titers for 26 volunteers given placebo (solid triangles), 31 volunteers given an NI early (26 or 32 h) (filled circles), and 12 volunteers given an NI delayed (50 h) (filled squares) following experimental infection are shown. The predicted virus titers using the target cell-limited model with delay (solid line) are shown for the placebo group, the early-treatment group, and the delayed-treatment group. The horizontal dotted line marks the limit of detection for viral titer. The parameter values used to describe the infections before therapy (placebo group) are V0 = 0.25 TCID50/ml, β = 1.4 × 10−2 (TCID50/ml)−1 · d−1, k = 3.2 d−1, p = 2.7 × 10−5TCID50/ml · d−1, c = 3.2 d−1, and δ = 3.2 d−1. Those parameter values are held constant for the treatment groups, except for p, which was set to 0.03p from time of drug administration onwards, namely, from 1.2 d and 2.08 d for the early- and delayed-treatment groups, respectively. Prophylactic use of an NI was modeled as a reduction in viral production rate by 97% at time of infection (dashed line). Experimental data were taken from Hayden et al. (15).
Lastly, we modeled prophylactic use of NI by reducing viral production by 97% immediately at the time of infection. As shown in Fig. 3, virus is predicted to be cleared before an infection can become established, and this is consistent with studies in which preinoculation of intranasal zanamivir once daily was highly protective against infection and illness (14).
DISCUSSION
Using simple models for the kinetics of influenza A virus infection, we have been able to fit experimentally derived viral titer data and estimate parameters characterizing viral production and clearance in experimentally infected individuals. Tables 2 and 3 present the patient-specific and geometric mean parameter estimates that correspond to the best fits of the models with and without delay to the data. These estimates were sensitive to choice of initial parameter guesses, which suggests that there may be other parameter estimates that can also fit the data reasonably well. Examining the surface generated by plotting the sum of squared residuals versus various pairs of parameters, we find that rather than having a deep, well-defined minimum, the surface tends to be flat at the minimum, thus explaining how different initial parameter guesses can give rise to different parameter estimates. Nonetheless, the estimates we found generally were consistent among patients, had reasonably tight 95% confidence intervals, and generally fell within experimentally determined ranges. Studies with more-frequent sampling as well as studies that independently estimated some parameters, such as infected cell life span, would be useful in confirming the parameter estimates we report here.
We studied both a version of the standard target cell-limited model of viral infection developed for the characterization of HIV infection and a more accurate variant that incorporated a delay from time of cell infection to beginning of viral production. From a statistical standpoint, the improvement of the fit with the delay model (which had one more parameter) was not great enough to justify adding an additional parameter. Nevertheless, the target cell-limited model with a delay in viral production is more realistic than the model without the delay, and it yielded more-reasonable parameter estimates. The estimate of the life span of infected cells in the model without delay was 6 h, which is not realistic. Incorporating the eclipse phase of the viral life cycle lengthened the total life span to close to 12 h. Thus, we prefer the model with delay and below discuss only the results of this model.
The delay model suggests that influenza virus is produced by an infected cell for a period of about 5 h, that it takes about 6 h between infection of target cells and virion release, and that infected cells have an average life span of about 12 h (Table 3). If cells stop producing virus without dying, their life spans would be longer, as our model tracks the states of cells only from time of infection to loss of viral production. Such a noncytolytic loss of viral production might be caused by IFN or other antiviral cytokines.
Estimates of the average lifetime of infected cells derived from our model differed from experimental values. While one report suggests that cells infected with influenza virus have an average lifetime of ∼24 h (52), our target cell-limited model with delay yielded an estimated average lifetime of infected cells of ∼12 h. Whether this discrepancy is real needs to be examined with more-refined experiments in which the half-life of infected cells is more precisely defined, especially since the experimental estimates (52) were derived over 30 years ago.
Our data and models allowed us to estimate the basic reproductive number, R0, for influenza A virus infection of susceptible nasal epithelial cells within an individual. The estimated R0 was about 22 in the target cell-limited model with delay. This high value of R0 suggests that initial infection spreads rapidly and would be difficult to extinguish.
As we illustrated with our models (Fig. 3), treatment of influenza infection with NIs reduces viral load and hence should reduce period of symptomatic disease. Further, prophylactic use with a highly effective NI is predicted to prevent infection. Hayden et al. (15) observed that zanamivir (GG167) administered prophylactically as intranasal drops is highly protective against infection, as only 3 of 44 volunteers receiving zanamivir became infected, whereas 24 of 33 volunteers receiving placebo became infected. A similar study with oseltamivir showed that prophylactic therapy protected 8 of 21 volunteers from infection, whereas 8 of 12 volunteers receiving placebo became infected (16).
In light of the fact that adaptive immune responses in primary influenza infection are not detected until 6 to 8 DPI (10, 38), rapid innate immune response may play a major role in resolving influenza virus infection. However, the first line of defense to virus is probably physical clearance due to mucociliary action. The half-life of free virus in the target cell-limited model with delay averaged 3.2 h (Table 3). If mucociliary clearance is the major explanation for viral clearance, then clearance rate of free virus, c, would be expected to be lower in individuals such as smokers and people with emphysema, asthma, and cystic fibrosis, all of whom have decreased capacity for mucociliary clearance (33). In our model, a decrease in the parameter c, which reflects decreased physical clearance, results in a faster increase of virus titer and a higher peak titer during influenza infection and hence may be correlated with the increased seriousness of influenza infections noted for people with decreased capacity for mucociliary clearance.
The target cell-limited models we described can easily fit virus titer data when there is only one peak. However, in approximately 50% of individuals studied here and in another study, we observed bimodal virus titer profiles, which could be explained by the incorporation of an IFN response. Our model with IFN generally produced a second virus titer peak 36 to 48 h after the initial peak. When the second peak appears later than 48 h after the initial peak, more-complex models that incorporate explicit immune responses along with the possible generation of escape mutants may be needed. Other possible explanations could also account for the second virus titer peak. The extension of active viral replication to a previously uninvolved site in the upper respiratory tract, e.g., sinus or nasal passages, could result in a second peak. If the free virus migrates into a new area, then the number of susceptible target cells increases and allows the virus to undergo another surge in viral titer. Also, the types of cells infected in the nasal mucosa differ by virus type (avian versus human influenza viruses) and likely over time with human influenza viruses (25). The sequential infection of different cell types could also produce viral titer peaks at different times.
To increase our understanding of the control of influenza infection in humans, additional data in which antigen-specific T cells, antibody titers, NK cells, IFN levels, and viral titers are measured frequently (maybe more often then daily) need to be collected. Also, it is important to keep in mind that the experimental data used in our analysis are derived from upper respiratory viral infection, and hence, parameters characterizing the kinetics of natural infection in the lower respiratory tract may be different from those derived here.
In summary, we have shown that simple target cell-limited models of virus infection, as previously developed to study HIV, can be applied to improve our understanding of influenza virus infection. We estimated, using our model with delay from time of infection to viral production, that during upper respiratory tract infection, influenza virus initially spreads rapidly with 1 cell, on average, infecting ∼22 others. The infection slows as target cells are consumed, and by the time of the virus peak at days 2 to 3, the vast majority of the initial target cells have been destroyed. Thus, influenza A infection could be self-limiting.
In the case of therapy for hepatitis C virus infection, simple target cell-limited models have been used to estimate the antiviral efficacy of interferon (30) and the effects of ribavirin (8). We believe that models of the type introduced here, once more fully validated, can be used to estimate the efficacies of established antivirals, such as oseltamivir and zanamivir, as well as new agents for the treatment of influenza. We illustrated this by comparing model predictions with data in which the neuraminidase inhibitor zanamivir was used to treat experimentally infected adult volunteers.
Acknowledgments
We thank Brian R. Murphy for kindly providing the viral kinetics data that were analyzed in this study.
This work was done under the auspices of the U. S. Department of Energy under contract W-7405-ENG-36 and supported by NIH grants AI28433 and RR06555 and contract N01-AI-50020.
REFERENCES
- 1.Beauchemin, C., J. Samuel, and J. Tuszynski. 2005. A simple cellular automaton model for influenza A viral infections. J. Theor. Biol. 232:223-234. [DOI] [PubMed] [Google Scholar]
- 2.Bocharov, G. A., and A. A. Romanyukha. 1994. Mathematical model of antiviral immune response. III. Influenza A virus infection. J. Theor. Biol. 167:323-360. [DOI] [PubMed] [Google Scholar]
- 3.Cella, M., F. Facchetti, A. Lanzavecchia, and M. Colonna. 2000. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat. Immunol. 1:305-310. [DOI] [PubMed] [Google Scholar]
- 4.Clarke, S. 1983. Physical defenses of the respiratory tract. Eur. J. Respir. Dis. Suppl. 126:27-30. [Google Scholar]
- 5.Reference deleted.
- 6.Crosby, A. W., Jr. 1976. >Epidemic and peace, 1918, p. 145-170. Greenwood Press, Westport, Conn.
- 7.Diebold, S. S., T. Kaisho, H. Hemmi, S. Akira, and C. Reis e Sousa. 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529-1531. [DOI] [PubMed] [Google Scholar]
- 8.Dixit, N., J. Layden-Almer, T. Layden, and A. Perelson. 2004. Modelling how ribavirin improves interferon response rates in hepatitis C virus infection. Nature 432:922-924. [DOI] [PubMed] [Google Scholar]
- 9.Efron, B., and R. Tibshirani. 1986. Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Stat. Sci. 1:54-75. [Google Scholar]
- 10.Ennis, F. A., A. H. Rook, Y. H. Qi, G. C. Schild, D. Riley, R. Pratt, and C. W.Potter. 1981. HLA restricted virus-specific cytotoxic T-lymphocyte responses to live and inactivated influenza vaccines. Lancet ii:887-891. [DOI] [PubMed] [Google Scholar]
- 11.Fedoseev, G. B., and S. S. Geharev. 1989. Basic defense mechanisms of bronchio-lung system, p. 112-144. In N. V. Putov (ed.), General pulmonology, vol. 1. Medicina, Moscow, Russia. [Google Scholar]
- 12.Fritz, R. S., F. G. Hayden, D. P. Calfee, L. M. R. Cass, A. W. Peng, W. G. Alvord, W. Strober, and S. E. Straus. 1999. Nasal cytokine and chemokine responses in experimental influenza A virus infection: results of a placebo-controlled trial of intravenous zanamivir treatment. J Infect. Dis. 180:586-593. [DOI] [PubMed] [Google Scholar]
- 13.Hayden, F. G., R. Fritz, M. C. Lobo, W. Alvord, W. Strober, and S. E. Straus. 1998. Local and systemic cytokine responses during experimental human influenza A virus infection. Relation to symptom formation and host defense. J. Clin. Investig. 101:643-649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayden, F. G., L. V. Gubareva, A. S. Monto, T. C. Klein, M. J. Elliot, J. M. Hamond, S. J. Sharp, and M. J. Ossi. 2000. Inhaled zanamivir for the prevention of influenza in families. Zanamivir family study group. N. Engl. J. Med. 343:1282-1290. [DOI] [PubMed] [Google Scholar]
- 15.Hayden, F. G., J. J. Treanor, R. F. Betts, M. Lobo, J. D. Esinhart, and E. K. Hussey. 1996. Safety and efficacy of the neuraminidase inhibitor GG167 in experimental human influenza. JAMA 275:295-299. [PubMed] [Google Scholar]
- 16.Hayden, F. G., J. J. Treanor, R. S. Fritz, M. Lobo, R. F. Betts, M. Miller, N. Kinnersley, R. G. Mills, P. Ward, and S. E. Straus. 1999. Use of the oral neuraminidase inhibitor oseltamivir in experimental human influenza: randomized controlled trials for prevention and treatment. JAMA 282:1240-1246. [DOI] [PubMed] [Google Scholar]
- 17.Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, and M. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123-126. [DOI] [PubMed] [Google Scholar]
- 18.Jao, R. L., E. F. Wheelock, and G. G. Jackson. 1970. Production of interferon in volunteers infected with Asian influenza. J Infect. Dis. 121:419-426. [DOI] [PubMed] [Google Scholar]
- 19.Julkunen, I., K. Melen, M. Nyqvist, J. Pirhonen, T. Sareneva, and S. Matikainen. 2000. Inflammatory responses in influenza A virus infection. Vaccine 19(Suppl. 1):S32-S37. [DOI] [PubMed] [Google Scholar]
- 20.Kaiser, L., R. S. Fritz, S. E. Straus, L. Gubareva, and F. G. Hayden. 2001. Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J. Med. Virol. 64:262-268. [DOI] [PubMed] [Google Scholar]
- 21.Langmuir, A. D., T. D. Worthen, J. Solomon, C. G. Ray, and E. Petersen. 1985. The thucydides syndrome. A new hypothesis for the cause of the plague of Athens. N. Engl. J. Med. 313:1027-1030. [DOI] [PubMed] [Google Scholar]
- 22.Larson, E. W., J. W. Dominik, A. H. Rowberg, and G. A. Higbee. 1976. Influenza virus population dynamics in the respiratory tract of experimentally infected mice. Infect. Immun. 13:438-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewin, S. R., R. M. Ribeiro, T. Walters, G. K. Lau, S. Bowden, S. Locarnini, and A. S. Perelson. 2001. Analysis of hepatitis B viral load decline under potent therapy: complex decay profiles observed. Hepatology 34:1012-1020. [DOI] [PubMed] [Google Scholar]
- 24.Macey, R. I., and G. F. Oster. 2001. Berkeley Madonna, version 8.0. University of California at Berkeley, Berkeley, Calif.
- 25.Matrosovich, M. N., T. Y. Matrosovich, T. Gray, N. A. Roberts, and H.-D. Klenk. 2004. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc. Natl. Acad. Sci. USA 101:4620-4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Menache, M. G., L. M. Hanna, E. A. Gross, S. R. Lou, S. J. Zinreich, D. A. Leopold, A. M. Jarabek, and F. J. Miller. 1997. Upper respiratory tract surface areas and volumes of laboratory animals and humans: considerations for dosimetry models. J. Toxicol. Environ. Health 50:475-506. [DOI] [PubMed] [Google Scholar]
- 27.Murphy, B. R., R. M. Chanock, R. G. Douglas, R. F. Betts, D. H. Waterman, H. Holley, Jr., D. L. Hoover, S. Suwanagool, D. R. Nalin, and M. M. Levine. 1980. Temperature-sensitive mutants of influenza A virus: evaluation of the Alaska/77-ts-1A2 temperature-sensitive recombinant virus in seronegative adult volunteers. Arch. Virol. 65:169-173. [DOI] [PubMed] [Google Scholar]
- 28.Murphy, B. R., M. B. Rennels, R. Douglas, Jr., R. F. Betts, R. B. Couch, T. Cate, Jr., R. M. Chanock, A. P. Kendal, H. F. Maassab, S. Suwanagool, S. B. Sotman, L. A. Cisneros, W. C. Anthony, D. R. Nalin, and M. M. Levine. 1980. Evaluation of influenza A/Hong Kong/123/77 (H1N1) ts-1A2 and cold-adapted recombinant viruses in seronegative adult volunteers. Infect. Immun. 29:348-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neumann, A. U., N. P. Lam, H. Dahari, M. Davidian, T. E. Wiley, B. Mika, A. Perelson, and T. J. Layden. 2000. Differences in viral dynamics between genotypes 1 and 2 of hepatitis C virus. J. Infect. Dis. 182:28-35. [DOI] [PubMed] [Google Scholar]
- 30.Neumann, A. U., N. P. Lam, H. Dahari, D. R. Gretch, T. E. Wiley, T. J. Layden, and A. S. Perelson. 1998. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282:103-107. [DOI] [PubMed] [Google Scholar]
- 31.Newhouse, M., J. Sanchis, and J. Bienenstock. 1976. Lung defense mechanisms (first of two parts). N. Engl. J. Med. 295:990-998. [DOI] [PubMed] [Google Scholar]
- 32.Nowak, M. A., S. Bonhoeffer, A. M. Hill, R. Boehme, H. C. Thomas, and H. McDade. 1996. Viral dynamics in hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 93:4398-4402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32a.Office of Technology Assessment, Congress of the United States. 1981. Cost-effectiveness of influenza vaccination, vol. 052-003-00855-6. Government Printing Office, Washington, D.C.
- 33.Pavia, D., J. R. Bateman, and S. W. Clarke. 1980. Deposition and clearance of inhaled particles. Bull. Eur. Physiopathol. Respir. 16:335-366. [PubMed] [Google Scholar]
- 34.Perelson, A. S., P. Essunger, Y. Cao, M. Vesanen, A. Hurley, K. Saksela, M. Markowitz, and D. D. Ho. 1997. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387:188-191. [DOI] [PubMed] [Google Scholar]
- 35.Perelson, A. S., D. E. Kirschner, and R. J. De Boer. 1993. Dynamics of HIV infection of CD4+ T cells. Math. Biosci. 114:81-125. [DOI] [PubMed] [Google Scholar]
- 36.Perelson, A. S., A. U. Neumann, M. Markowitz, J. M. Leonard, and D. D. Ho. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582-1586. [DOI] [PubMed] [Google Scholar]
- 37.Ramratnam, B., S. Bonhoeffer, J. Binley, A. Hurley, L. Zhang, J. E. Mittler, M. Markowitz, J. P. Moore, A. S. Perelson, and D. D. Ho. 1999. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet 354:1782-1785. [DOI] [PubMed] [Google Scholar]
- 38.Richman, D. D., B. R. Murphy, S. Baron, and C. Uhlendorf. 1976. Three strains of influenza A virus (H3N2): interferon sensitivity in vitro and interferon production in volunteers. J. Clin. Microbiol. 3:223-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts, N. J., Jr., R. Douglas, Jr., R. M. Simons, and M. E. Diamond. 1979. Virus-induced interferon production by human macrophages. J. Immunol. 123:365-369. [PubMed] [Google Scholar]
- 40.Samuel, C. E. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14:778-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sedmak, J. J., and S. E. Grossberg. 1973. Interferon bioassay: reduction in yield of myxovirus neuraminidases. J. Gen. Virol. 21:1-7. [DOI] [PubMed] [Google Scholar]
- 42.Seo, S. H., E. Hoffmann, and R. G. Webster. 2002. Lethal H5N1 influenza viruses escape host anti-viral cytokine responses. Nat. Med. 8:950-954. [DOI] [PubMed] [Google Scholar]
- 43.Simonsen, L., K. Fukuda, L. B. Schonberger, and N. J. Cox. 2000. The impact of influenza epidemics on hospitalizations. J. Infect. Dis. 181:831-837. [DOI] [PubMed] [Google Scholar]
- 44.Smith, W., C. H. Andrewes, and P. P. Laidlaw. 1933. A virus obtained from influenza patients. Lancet 222:66-68. [Google Scholar]
- 45.Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227-264. [DOI] [PubMed] [Google Scholar]
- 46.Thompson, W. W., D. K. Shay, E. Weintraub, L. Brammer, N. Cox, L. J. Anderson, and K. Fukuda. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179-186. [DOI] [PubMed] [Google Scholar]
- 47.Tsiang, M., J. F. Rooney, J. J. Toole, and C. S. Gibbs. 1999. Biphasic clearance kinetics of hepatitis B virus from patients during adefovir dipivoxil therapy. Hepatology 29:1863-1869. [DOI] [PubMed] [Google Scholar]
- 48.Van Reeth, K. 2000. Cytokines in the pathogenesis of influenza. Vet. Microbiol. 74:109-116. [DOI] [PubMed] [Google Scholar]
- 49.Vilcek, J., and G. C. Sen. 1996. Orthomyxoviruses, p. 375-399. In B. N. Fields, D. M. Knipe, P. M. Howley, R. M. Chanock, J. L. Melnick, T.P.Monath, B. Roizman, and S. E. Straus (ed.), Fields virology. Lippincott-Raven Publishers, Philadelphia, Pa.
- 50.Wang, X., M. Li, H. Zheng, T. Muster, P. Palese, A. A. Beg, and A. Garcia-Sastre. 2000. Influenza A virus NS1 protein prevents activation of NF-B and induction of alpha/beta interferon. J. Virol. 74:11566-11573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wright, P., and R. G. Webster. 2001. Orthomyxoviruses, p. 1533-1579. In D. M. Knipe, P. M. Howley, D. E. Griffin, M. A. Martin, R. A. Lamb, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed. Lippincott Williams and Wilkins, Philadelphia, Pa.
- 52.Zdanov, V. M., and A. G. Bukrinskaja. 1969. Myxoviruses reproduction. Medicina, Moscow, Russia.
- 53.Zhang, L., P. J. Dailey, T. He, A. Gettie, S. Bonhoeffer, A. S. Perelson, and D. D. Ho. 1999. Rapid clearance of simian immunodeficiency virus particles from plasma of rhesus macaques. J. Virol. 73:855-860. [DOI] [PMC free article] [PubMed] [Google Scholar]