

Abstract
The bacterial protein tyrosine phosphatase YopH is an essential virulence determinant in Yersinia spp., causing gastrointestinal diseases and the plague. Like eukaryotic PTPases, YopH catalyzes the hydrolysis of the phosphate moiety of phosphotyrosine within a highly conserved binding pocket, which is also characterized by the closure of the so-called “WPD loop” upon ligand binding. In this study, we investigate the conformational changes and dynamics of the WPD loop by molecular dynamics simulations. Consistent with experimental observations, our simulations show that the WPD loop of YopH is intrinsically flexible and fluctuates between the open and closed conformation with a frequency of ∼4 ns for the apo, native protein. The region of helix α4 spanning loop 384-392, which has been revealed experimentally as a second substrate-binding site in YopH, is found to be highly associated with the WPD loop, stabilizing it in the closed, active conformation, and providing a structural basis for the cooperation of the second-substrate binding site in substrate recognition. Loop L4 (residues 323–327) is shown to be involved in a parallel, correlated motion mode with the WPD loop that contributes the stabilization of a more extended open conformation. In addition, we have simulated the loop reopening in the ligand-bound protein complex by applying the locally enhanced sampling method. Finally, the dynamic behavior of the WPD loop for the C403S mutant differs from the wild-type YopH remarkably. These results shed light on the role of the WPD loop in PTPase-mediated catalysis, and are useful in structure-based design for novel, selective YopH inhibitors as antibacterial drugs.
INTRODUCTION
Yersinia spp. are causative agents of human diseases ranging from gastrointestinal syndromes to the plague (1). Yersinia pestis has recently gained increased attention due to the emergence of antibiotic resistance in this pathogen and concerns with its potential use as a biological weapon (2). The Yersinia genus utilizes a contact-dependent type III secretion machinery to inject cytotoxic effector proteins, referred to as Yops (Yersinia outer proteins), directly into the cytosol of mammalian cells. One of these virulence factors, YopH, is a highly active bacterial protein tyrosine phosphatase (PTPase) (3,4). Upon translocation into host cell, YopH disrupts the host cytoskeleton by dephosphorylating a variety of pTyr-containing protein targets such as the focal adhesion kinase, paxilin, p130Cas, and Fyn-binding protein (5). Because its PTPase activity is essential for Yersinia pathogenicity, YopH has emerged as a potential therapeutic protein target for antibacterial agents in structure-based drug design.
Like eukaryotic protein tyrosine phosphatases, YopH catalyzes the hydrolysis of the phosphate moiety of phosphotyrosine with a highly conserved signature motif, or “P-loop”, that includes the nucleophilic Cys-403 within the binding pocket (Fig. 1) (6). The active site is also defined with a flexible “WPD loop” that harbors the general acid/base Asp-356 essential for catalysis (7). PTPases utilize a two-step mechanism for the catalysis reaction (8,9). In the first step, the phosphoryl group from the pTyr-containing substrate is transferred to the active site cysteine residue within the signature motif leading to the formation of a cysteinyl-phosphate intermediate. The WPD loop is required to adopt a closed conformation that brings the catalyst Asp-356 close to the scissile oxygen of pTyr to transfer a proton to the substrate during the catalysis. In the second step, the phospho-enzyme intermediate is subsequently hydrolyzed by an activated water molecule. Asp-356 acts as a base to abstract a proton from water in the breakdown of the intermediate, presumably after the opening up of the WPD loop for the release of product and binding of new substrate.
FIGURE 1.

Schematics of the YopH structure. The WPD loop is shown in the open (green) and closed (magenta) conformation. The P-loop is colored in yellow. Other loops are named according to the observation with increased fluctuations in molecular dynamics simulations (see Fig. 2 B). The figure was prepared using the PyMol software.
The movement of the WPD loop plays a central role in the PTPase-mediated catalytic process. Crystallographic structures have revealed that the WPD loop adopts either an open, enzymatically inactive, or a closed, active conformation under different conditions, and the closed conformation is generally associated with ligand binding (10–12). Juszczak and co-workers have examined the loop dynamics of YopH using time-resolved fluorescence anisotropy and steady-state ultraviolet resonance Raman (UVRR) spectroscopies (13). The results have shown that under physiological condition, the WPD loop in the ligand-free native enzyme alternates between the open and closed form with a rate constant of ∼2.6 × 108 s−1 (∼3.8 ns of frequency). The rate of loop closure is independent of the presence of ligand, whereas in the presence of ligand the rate of opening is dramatically reduced, resulting in a closed conformation. Wang and co-workers have further monitored the dynamics of the WPD loop of YopH using H/D exchange combined with electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry analysis (14). The results have revealed conformational changes of the WPD loop upon ligand binding, as well as several significant changes in segment exposure and flexibility that are not evident from x-ray structures.
A better understanding of the dynamics of the WPD loop and conformational changes upon ligand binding will not only provide insight into the mechanism of catalysis, but also will facilitate the structure-based design of novel, selective YopH inhibitors as antibacterial agents. As the dynamic properties of the flexible WPD loop have generally been obtained from the crystallographic and biochemical studies, a detailed computational study with molecular dynamics simulations should offer new insight on the WPD loop dynamics and interactions at the atomic level that cannot be elucidated by experiments. Previous molecular dynamics (MD) simulations for the human protein tyrosine phosphatase PTP1B and its complex with substrate have shown that the flexibility of the protein and the WPD loop were reduced by ∼10% upon ligand binding, and interactions with substrate are mainly dominated by electrostatic forces (15,16). Since these MD simulations were only conducted within a very short time course of 1 ns, the dynamics of the WPD loop and conformational transition were not fully investigated.
In the work presented here, we carried out large-scale MD simulations for both the wild-type YopH and the C403S mutant protein to probe the dynamic properties of the WPD loop. To our knowledge, this is the first detailed computational study of the dynamics of the WPD loop using extended MD simulations. Several simulation protocols were employed for the apo and ligand-bound structures starting with either an open or closed conformation to fully explore the loop transition. These simulations allowed us to detect the opening-closure of the WPD loop in solution, and in agreement with experimental data, with a frequency of ∼4 ns for the apo, native protein. Applying the locally enhanced sampling (LES) method (17), we have successfully simulated the loop reopening in the ligand-bound protein complex. The dynamic behavior of the WPD loop for the C403S mutant exhibited in the MD simulations were significantly different from the wild-type, providing a structural mechanism for the different thermodynamic and functional properties of the mutant protein.
METHODS
Protocol setup
The initial coordinates of YopH structures were taken from the Protein Data Bank (PDB): the apo, wild-type YopH in the open conformation (PDB id 1ypt), the ligand-free, wild-type YopH in the closed conformation (PDB id 1ytn, removal of the ligand), and the ligand-bound, wild-type YopH in complex with a phosphate anion (PDB id 1lyv). The C403S mutant was constructed from the apo, wild-type YopH with the open conformation (PDB id 1ypt). According to the charges and protonation states of active residues at physiological conditions (18), residue Cys403 in the wild-type structure adopts a negatively charged form, and Asp-356 in the wild-type and mutant structures adopts a protonated neutral form. All systems were solvated by a cubic box of TIP3P water molecules extended 12 Å from protein atoms using the AMBER 8.0 package (19). The solvated protein systems were neutralized by counterions and subjected to a thorough minimization before MD simulations. The minimization was performed on two steps: first the water molecules were minimized while holding solute frozen (1000 steps using the steepest descent algorithm), then the whole system was minimized with 5000 steps of conjugate gradient to remove close contacts and to relax the system.
Molecular dynamics simulations
MD simulations were carried out using the SANDER module of the AMBER 8.0 package and the Parm99 force field (20). Bond lengths involving bonds to hydrogen were constrained with SHAKE (21) and the time step for all MD simulations was 2 fs. A nonbonded cutoff of 10 Å was used, and the nonbonded pair list was updated every 25 time steps. Periodic boundary conditions were applied to simulate a continuous system. The particle mesh Ewald method (22) was employed to calculate the long-range electrostatic interactions. The simulated system was first subjected to a gradual temperature increase from 0 K to 300 K over 20 ps, and then equilibrated for 500 ps at 300 K, followed by the production run of 10 ns length in total. Constant temperature and pressure (300 K/1 atm) were maintained using the Berendsen coupling algorithm (23) with a time constant for heat bath coupling of 0.2 ps.
The resulting trajectories were analyzed using the PTRAJ module of AMBER 8.0 package (19). All counterions and water molecules were stripped from the MD structures. The root mean-square deviations (RMSD) of the backbone atoms of YopH were calculated from the trajectories at 1 ps interval, with the initial structure as the reference. The mean-square positional fluctuations about the average structure,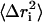 , were calculated based on the superposition of all Cα atoms. The root mean-square fluctuation is related to the B-factors, which can be obtained experimentally from x-ray crystallographic according to (24)
, were calculated based on the superposition of all Cα atoms. The root mean-square fluctuation is related to the B-factors, which can be obtained experimentally from x-ray crystallographic according to (24)
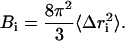 |
Essential dynamics analysis
The essential dynamics (ED) analysis is a technique that reduces the complexity of the data and extracts the concerted motion in simulations that are essentially correlated and presumably meaningful for biological function (25). In the ED analysis, a variance/covariance matrix was constructed from the trajectories after removal of the rotational and translational movements. A set of eigenvectors and eigenvalues was identified by diagonalizing the matrix. The eigenvalues represented the amplitude of the eigenvectors along the multidimensional space, and the displacements of atoms along each eigenvector showed the concerted motions of protein along each direction. An assumption of ED analysis is that the correlated motions for the function of the protein are described by eigenvectors with large eigenvalues. The movements of protein in the essential subspace were identified by projecting the Cartesian trajectory coordinates along the most important eigenvectors from the analysis. The ED analysis was performed using the module in the AMBER 8.0 package (19).
Locally enhanced sampling
LES is a mean-field approach to improve sampling efficiency by replacing a given portion of protein with multiple copies, thus providing a smoothing effect of the energy landscape (17). The LES MD simulations were conducted using the final structure with an open conformation obtained from previous simulations as starting coordinates. The WPD loop was divided into three LES segments (352-354, 355-358, 359-361) and each residue in the LES regions was assigned five copies. The topology file and restart file were generated using the ADDLES module in the AMBER 8.0 package (19).
RESULTS
Dynamics of the open conformation
The MD simulations for the apo, native YopH protein were performed in explicit solvent starting with an open conformation. To ensure the stability of the trajectory, we inspected the potential energy of the system and the RMSD throughout the simulations. The potential energies reached a steady state during equilibration phase and the average energies remained constant (data not shown). Fig. 2 A depicts the RMSD calculated for the backbone atoms of protein compared to the initial structure over the 10 ns of MD simulations. The trajectory reached a plateau after 2 ns with an average RMSD value <2.0 Å, indicating that the system evolved to a stable state and has reasonably converged. To detect the regional motion in the protein structure, the atomic positional fluctuations for residues (Cα) were calculated from the MD simulations in comparison to the experimental B-factors obtained from the crystal structure. As shown in Fig. 2 B, the calculated B-factors are generally in agreement with the experimentally measured data. Increased atomic fluctuations are mainly observed at several loop regions including the WPD loop (352-361) and loop L2 (286-297) surrounding the catalytic binding pocket, and the putative focal complex targeting loop L1 (223-226). The WPD loop exhibits a high level of flexibility throughout the simulations. Surprisingly, the largest motions are found in a broad region at the end of helix α4 and the following loop L6 (384-392). In contrast, experimental B-factors within this region in the crystal structure are quite low, due perhaps to the environment in the crystalline state.
FIGURE 2.

(A) RMSD of the backbone atoms for the apo, wild-type YopH protein with respect to the initial structure over 10 ns of MD simulations.(B) Calculated B-factors of Cα atoms of YopH from MD simulations (solid) in comparison to the experimental data obtained from crystal structure (shaded).
Comparison of crystal structures of YopH with the open and closed conformations revealed that the characterized movements of the WPD loop occur with residues 355–358, which form a type I β-turn at one of the loop hinges in the open conformation (11). When the loop is closed, this β-turn converts to a type II configuration that orients residues Asp-356 and Gln-357 over the active site to participate in interactions with ligand. Gln-357 is on the third position of the β-turn and crucial to the displacement of the WPD loop for complete closure. The extent of the loop movements is up to 6 Å between the ligand-free and ligand-bound states. We therefore monitored the distances between the backbone atoms (Cα) of residues Gln-357 and Val-407, the latter of which is located on the bottom of the binding pocket. The distances serve as a direct measurement of the “gate” of the WPD loop opening/closure during the MD simulations.
Fig. 3 shows the measured distances of the “gate” of the binding pocket over 10 ns of simulations. The distances in the crystal structures for the open and closed conformations are 17 Å and 11 Å on average. It can be seen that, starting with an open conformation, the WPD loop of the apo protein begins to bend over the active site at ∼3 ns and reaches the most extended closed conformations at ∼4 ns with an average distance of 13 Å. The closed conformation is not as complete as compared to that in the crystal structure. As discussed later, the completely closed form is probably induced by ligand binding and may not exist in the apo structure. The WPD loop remains in the closed state for a short time period (∼400 ps), and opens up slowly to extend to an even more open state with an average distance up to 20 Å after ∼6 ns. There is a tendency to close over again at ∼9.0 ns, but even more incompletely, after which it opens up rapidly.
FIGURE 3.

Measured distances between Cα atom of residues Gln-357 and Val-407 throughout the time course of 10 ns MD simulations. The experimental values for the open and closed conformations are 17 Å and 11 Å on average, and denoted by solid lines on the graph.
Conformational changes of the WPD loop in the MD simulations were analyzed by measuring the dihedral angle χ2,1 of Trp-354 on the loop hinge and the β-turn changes on the other hinge of the WPD loop. The dihedral angle χ2,1 of the invariant Trp-354 varies between −60° and −90° associated with the open conformation during the simulations, consistent with the experimental observation of −70° in the crystal structure. It is up to −100° in the extended open conformation. The large variation reflects the intensive fluctuation of the open form during the simulations. For the closed conformation, the dihedral angle χ2,1 of Trp-354 fluctuates between −20° and −40° in the MD simulations. The β-turn of residues 355–358 is in the type I and type II configurations for the closed and open conformations in general. Fig. S1 (Supplementary Material) shows some snapshots of the WPD loop conformations observed in the simulations in comparison with the open and closed forms in the crystal structures. The most extensive movements, which are coupled to the loop transition, are the conformational changes of the side chain of Gln-357, which points out of the protein surface in the open form and flips over the binding pocket in the closed conformation. In the extended open conformation, it points out of the protein surface with significantly increased fluctuation.
To characterize the WPD loop motion, we applied ED analysis to reduce the complexity of the data and extract the concerted motion in simulations (25). The ED analysis was performed on Cα atoms of the protein, and the first few modes of essential motions were analyzed. Fig. 4 shows the motion modes of the WPD loop by projecting the MD trajectory onto the eigenvector and superimposing the minimum and maximum structures. The first essential mode is the opening-closure of the WPD loop (Fig. 4 A). The maximum distance of the displacement between the open and closed form is up to 10 Å, suggesting that the loop motion in the MD simulations is more extensive. In contrast to the crystal structure, the open conformation of the essential mode is in a more extended state, whereas the closed from is typically half-closed. Coupled with the WPD loop conformational changes, two other regions also exhibit significant movements. One lies in loop L2 (286-297) in the active site, which shows correlated closure with the WPD loop. The other flexible region is located at loop L6 (380-392) extending to helix α4. In the open state of the WPD loop, loop L6 (384-392) typically protrudes out of the protein surface and the last two turns of helix α4 (380-383) are in a loose coil form, whereas in the closed conformation, helix α4 becomes more ordered (Fig. 4 C). Further analysis of the dynamical behavior of loop L6 throughout the MD simulations reveals that L6 fluctuates intensely in the open state. With the loop closure, the motions of L6 are significantly reduced.
FIGURE 4.

Essential modes of YopH visualized by projecting the MD trajectory onto the eigenvector and superimposing the minimum and maximum structures from MD simulations. (A) The first essential mode shows the opening-closure of the WPD loop and correlated internal motions with loop L2 and loop L6. (B) The second essential mode shows the dominant open conformation with correlated motions between the WPD loop and loop L4. (C) Cartoon representations of helix α4 and the following loop L6 associated with the open (gray and yellow) and closed (blue and green) conformations in the first essential mode.
The second essential mode is a novel loop motion involving loop L4 (323-327) that is adjacent to the WPD loop. As shown in Fig. 4 B of the superimposed minimum and maximum structures from ED analysis, both conformations are in the open form. Therefore, this is a loop-opening mode, reflecting the major motion of the open conformation during the simulations. The essential mode shows that loop L4 is highly correlated with the WPD loop. In the maximum structure of the more extended open conformation, the WPD loop moves in a parallel manner with loop L4, as seen in the open conformation of the first essential mode. Such a parallel correlated motion forms a kind of stacking interaction that may contribute to the stability of the open form. In comparison, the WPD loop in the less extended open conformation of the second essential mode is situated on a position perpendicularly to loop L4. In fact, the experimentally observed open conformation in the crystal structure is an intermediate between these two conformations. Notably, helix α7 in the second essential mode appears to play a role in keeping the WPD loop in the open form. To change from the open conformation to the closed conformation, the WPD loop needs to cross an energy barrier generated by helix α7. This probably involves a conformational rearrangement of helix α7, as well as helix α4 shown in the first essential mode.
Dynamics of the closed conformation
All crystal structures of YopH in the closed conformation are complexed with a ligand. MD simulations of YopH/ligand complex demonstrate that both the protein and the WPD loop are quite stable in the closed state. Fig. 5 shows the RMSD of the WPD loop in the ligand-bound protein compared to the ligand-free, open conformation. The movements of the WPD loop in the ligand-bound complex are significantly reduced, suggesting that the ligand plays a stabilizing role for the WPD loop. Previous MD simulation studies of the PTP1B/substrate complex revealed the same results of reduced flexibility of the WPD loop upon complex formation (15,16). These MD results are consistent with experimental data that the average B-value of residues in the WPD loop in the crystal structures for the apo protein is ∼30 Å2, whereas it is ∼10 Å2 in the ligand-bound complex with the closed conformation.
FIGURE 5.

RMSD of the backbone atoms of the WPD loop for the ligand-bound YopH complex in comparison to the RMSD of the WPD loop for the ligand-free protein during MD simulations.
An intriguing question is whether one can simulate the loop reopening within the ligand-bound complex. The ability to reopen the WPD loop is critical to the PTP-mediated hydrolysis to allow the release of substrate after dephosphorylation. Since the bound ligand would trap the closed conformation in a local minimum, it is much more challenging to sample the structural transition with standard MD simulations. The occurrence of loop reopening in the ligand-bound complex is expected to be more energetically demanding and slower than the event of loop closure in the apo protein. To fully explore the dynamic behavior of the WPD loop, we applied the LES method to augment the MD simulations to a more extended sampling space. The enhanced sampling is achieved by replacing the WPD loop with five copies in the simulations, thus providing a reduced barrier to conformational transition compared to the original system. The LES approach has been successfully applied in many studies to improve the sampling efficiency of simulations (26–28).
Applying LES MD at 360 K, we detected the loop reopening starting with the closed conformation in complex with a phosphate anion. Fig. 6 shows the measured distances of the loop “gate” using both standard MD simulations and LES under different conditions. Because the five copies of the WPD loop in LES showed similar dynamical changes with respect to the opening and closure of the WPD loop in the simulations, only the trajectory of the first copy was analyzed in comparison with the standard MD simulations. As shown in Fig. 6, the WPD loop opened up completely at ∼500 ps with the LES MD at 360 K. In contrast, the loop remained in a very stable, closed state using either standard MD at 400 K or LES MD at room temperature. The electrostatic and hydrogen bonding interactions between the WPD loop, the ligand, and residues within the binding pocket play a major role in stabilizing the WPD loop in the closed conformation. Such a high energy barrier prevented the loop from opening until the enhanced sampling method was used with a high temperature in the simulations. After opening up, the WPD loop fluctuated mostly in the open conformation as seen in the apo protein, but no loop closure was observed again even when we extended the LES simulations to 10 ns.
FIGURE 6.

Measured distances between Cα atom of residues Gln-357 and Val-407 for the ligand-bound YopH complex under different conditions of MD simulations. For the LES MD simulations, the trajectory of the first copy is used for the analysis.
We also examined the dynamics of the phosphate anion in the binding pocket during the LES MD simulations. Whereas the WPD loop underwent significant conformational changes in opening, the phosphate ligand remained stably within the active site. The hydrogen bonds between the phosphate and residue Arg-409 were well maintained throughout the simulations. We speculate that the ligand facilitates the loop reopening under physiological conditions and diffuses out of the binding site. Because the loop closure and opening in the catalytic process involve the making/breaking of the cysteinyl phosphate intermediate and activated water molecules, the simple model used here cannot reflect the dynamical process of loop reopening in the protein/ligand complex. Considering the proton transfer and cleavage of monophosphate-ester bond, the H-bonding network between ligand and protein, as well as the charge balance between the deprotonated, negatively charged Asp-356, the negatively charged Cys-403, and the positively charged Arg-409 are expected to be disrupted, both should facilitate the reopening of the WPD loop and the release of substrate.
Dynamics of the C403S mutant
One major goal of our MD studies is to examine the different dynamic properties of the C403S mutant with respect to the wild-type YopH protein. The invariant Cys residue in the PTP signature motif is absolutely required for activity. Biochemical and site-directed mutagenesis experiments show that, although the mutant is capable of binding substrates and inhibitors, mutation of Cys to Ser results in a catalytically inactive enzyme (29). Further studies indicate that the lack of activity may be caused by structural perturbations in the active site (29). The crystal structure of the C403S mutant YopH bound with sulfate is similar to that of the ligand-bound native protein (11). UVRR spectroscopic studies have shown that, in contrast to the wild-type YopH, both the ligand-bound and ligand-free C403S mutants remain in the closed conformation (13). Interestingly, the experimentally determined x-ray crystal structure of apo PTP1B C215S mutant reveals that the WPD loop is in an open conformation, and the P-loop undergoes dramatic conformational changes as compared to the wild-type PTP1B (30).
Because the structure of apo YopH C403S mutant is not available, the mutation was constructed from the wild-type YopH with an open conformation and subjected to full-energy minimization and equilibration before standard MD simulations. Fig. 7 shows the measured distances of the “gate” between Gln-357 and Val-407 as function of 10 ns of simulations for the mutant protein. Unlike the dynamical behavior in the apo, wild-type YopH, the WPD loop of mutant began to close at ∼1.5 ns and quickly reached a rather stable state. The closed conformation was remarkably stable over the time course from ∼2 ns to ∼4 ns. With an abrupt opening up to a more open state, the WPD loop flipped back to the closed form and fluctuated mostly between half-closed conformations, and finally evolved to the open conformation during the rest time of MD simulations.
FIGURE 7.

Measured distances between Cα atom of residues Gln-357 and Val-407 for the C403S YopH mutant as a function of simulation time.
Compared to the MD simulations of the wild-type YopH (Fig. 3), there are several intriguing differences associated with the dynamics of the WPD loop for the C403S mutant. First, the mutant protein remains in the closed conformation much longer than the wild-type throughout the simulations. The ratio of time occupancy between the closed (and half-closed) and open conformation of the mutant is ∼70:30, indicating that the closed conformation is more favorable to the mutant protein. Second, unlike the typical half-closed conformation observed in the wild-type, the closed conformation of the mutant in the MD simulations flipped more deeply into the binding pocket, similarly to those observed in the crystal structures of ligand-bound complexes. Because the replacement of the charged thiolate with a neutral hydroxyl group is believed to disrupt the subtle charge balance within the binding pocket and cause conformational arrangement of the active site in the mutant, the energy barrier for the loop closure is expected to be lower than that in the wild-type. Finally, we note that the open conformation becomes dominant for the mutant protein after ∼8 ns of simulation time.
We further investigated the dynamical changes within the active site of the C403S mutant as compared to the wild-type protein. Fig. 8 shows the RMSD of the P-loop for both the wild-type and mutant protein during the 10 ns of MD simulations. The P-loop in the wild-type YopH is quite stable across the entire trajectories with an average RMSD of 0.3 Å. For the mutant, the P-loop shows a similar dynamical behavior to the wild-type initially. However, noticeable differences are observed when the simulation time is extended to ∼8 ns. The RMSD is increased up to 0.6 Å in comparison to the initial RMSD of 0.3 Å, indicating that the P-loop undergoes more significant conformational changes. Further inspection reveals that the P-loop of the open conformation moves upward and protrudes out of the active site after 8 ns of simulations as compared to the closed conformation within the time course from ∼2 ns to ∼4 ns (Fig. 9). In fact, this result is similar to the observation in the structure of PTP1B C215S mutant that the extended P-loop prevents the WPD loop closure (30). Consistent with experimental data, the YopH mutant in the more extended open conformation shows increased exposure of solvent surface of the WPD loop (29). In addition, a number of active site residues such as Arg-409 and Gln-446, which are critical to the closure of WPD loop and catalytic activity, also exhibit increased fluctuation in the mutant protein.
FIGURE 8.

RMSD of the backbone atoms of the P-loop for the C403S YopH mutant over 10 ns of MD simulations in comparison to the RMSD of the P-loop in the apo, wild-type protein.
FIGURE 9.

Average structure of C403S YopH mutant after 8 ns of MD simulations (green) in comparison to the average structure of the closed conformation during the 2 ∼ 4 ns of simulations (magenta).
DISCUSSION
The mobility of the WPD loop plays an important role in the catalytic process of PTPases. Many studies have been applied to investigate the dynamics of the loop motion in solution. Our MD simulations are generally consistent with those experimental results. The fluctuation of the WPD loop between the open and closed conformations in the apo, native state is an intrinsic property of the enzyme that is closely associated with its functional role. Ligand binding significantly reduces the protein flexibility and constrains the WPD loop predominately in the closed form. Experiments have revealed that the WPD loop in the apo enzyme alternates between the open and closed conformations with a frequency of ∼3.8 ns, whereas in the ligand-bound state it remains in the closed conformation without significant dynamics on the nanosecond timescale (13). We have detected the opening and closure of the WPD loop in the apo protein during the MD simulations. Starting with an open conformation, the closure of the WPD loop occurs at ∼4.0 ns and ∼9.0 ns, separated mainly by a period of open form fluctuation. Indeed, when we further extended the simulation time to ∼25 ns, the sign of loop closure was observed again at ∼13 ns, ∼17 ns, and ∼22 ns (Fig. S2, Supplementary Material). This frequency of loop transition observed in the MD simulations is in good agreement with the experimental results.
Unlike the transition of the WPD loop between the open and closed conformations in the wild-type protein, the apo C403S mutant remains in the closed conformation with remarkable stability. This is consistent with the experimental observation from the UVRR spectroscopic studies (13,14). Notably, the P-loop in the active site of the mutant is found to become more dynamic and undergo conformational changes that protrude out of the active site after ∼8 ns of simulations, resulting in an open conformation dominated at later time points. Denaturation experiments also demonstrate that the structure of C403S mutant is less stable than the wild-type (29). Therefore, substitution of sulfur with oxygen atom in the C403S mutant not only results in an inefficient nucleophilic attack, but also leads to substantial conformational rearrangements in the active site, as revealed by the increased mobility in the active site and the extended P-loop causing the WPD loop in an open conformation during the simulations.
The closed conformation observed in the MD simulations for the apo, native protein is typically in a half-closed state. It is likely that the completely closed conformation observed in the crystal structure is induced by ligand binding, because the H-bond network between the WPD loop, the ligand, and active site residues plays a major driving force for the closure of the WPD loop in the ligand-bound complex. In the apo state, our MD simulation results show that the WPD loop alternates between open and closed conformations, but the open conformation appears to dominate for a long time course throughout the simulations. The closure of the WPD loop becomes more rapid and remains only for a short time period within the extended simulations, and the open conformation is much longer lived than the closed form. Further dynamic analysis shows that loop L4 is highly correlated with the WPD loop forming a stacking interaction, which might contribute to the stabilization of the more open form observed in the simulations. These results suggest that the open conformation is likely more favorable and stable than the closed conformation for the apo, native enzyme in solution. A hypothesis is that the flexible WPD loop in the apo state generally favors an open, inactive conformation. Substrate binding in the active site facilitates the closure of the WPD loop, and the closed, active form is stabilized by the substrate binding in the catalytic binding pocket, as well as the binding at the second substrate binding site, as discussed below.
It is worthy to note that the most extensive conformational changes observed in the MD simulations occur at the loop region L6 extending to helix α4. An ordered α4 helix is found to correlate with the closure of the WPD loop, and the flexibility of loop L6 is highly associated with the WPD loop movements. These findings are significant because such conformational changes are not apparent and cannot be inferred from the crystal structures. Recent crystallographic studies of YopH with its substrate reveal that this region constitutes a second substrate binding site promoting the recognition of substrate at the catalytic active site (31). Since the second substrate binding site is far from the catalytic binding site, the basis of this cooperation has not been well understood. Our simulations reveal that substrate binding at the second substrate site may induce a more ordered form in helix α4, stabilizing the motion of the WPD loop in the closed, active conformation.
Notably, crystallographic studies of PTP1B with small molecular inhibitors have also revealed that the region of helices α3 and α6 (corresponding to helices α4 and α7 in YopH) constitutes an allosteric binding site (32). The allosteric inhibition of PTP1B activity is achieved by perturbation along helices α3 and α6, although no significant conformational changes are observed with α3-α6 helices in the crystal structures upon inhibitor binding (32). Our simulation results reveal that the corresponding helices α4 and α7 in YopH is highly correlated with the motion of the WPD loop, providing structural insights on the role of these helices in allosteric inhibition. Moreover, experiments have shown that an ordered helix α7 at the C-terminal end of PTP1B is associated with the WPD loop, serving as a regulatory region to stabilize the closure of the WPD loop for full enzyme activity (32). This is quite similar to the result of loop L4 in YopH that plays a stabilizing role for the open conformation. A comparison of YopH and PTP1B structures shows that the C-terminal end of YopH does not position at the α4-α7 region. The end of PTP1B α7 helix is partly overlapped with loop L4 in YopH, and the corresponding loop in PTP1B is relatively short and negligible. Therefore, the regulatory role of loop L4 in YopH activity may be worth further experimental investigation. The finding that the second substrate binding site is correlated with the dynamics of the WPD loop via helices α4 and α7, as well as loop L4, suggests that they are potential allosteric binding sites for the design of novel, selective YopH inhibitors as antibacterial agents.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank R. Bennett, C. Pepper, A. Gazes, and G. Latter of The Rockefeller University Information Technology Resource Center for computational facilities.
This work was funded in part by program grant 1U19AI056510 from the National Institute of Allergy and Infectious Disease and research funds from The Rockefeller University.
References
- 1.Brubaker, R. R. 1991. Factors promoting acute and chronic diseases caused by Yersiniae. Clin. Microbiol. Rev. 4:309–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hawley, R. J., and E. M. Eitzen Jr. 2001. Biological weapons—a primer for microbiologists. Annu. Rev. Microbiol. 55:235–253. [DOI] [PubMed] [Google Scholar]
- 3.Cornelis, G. R. 2002. The Yersinia Ysc-Yop ‘type III’ weaponry. Nat. Rev. Mol. Cell Biol. 3:742–752. [DOI] [PubMed] [Google Scholar]
- 4.Guan, K. L., and J. E. Dixon. 1990. Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science. 249:553–556. [DOI] [PubMed] [Google Scholar]
- 5.Black, D. S., A. Marie-Cardine, B. Schraven, and J. B. Bliska. 2000. The Yersinia tyrosine phosphatase YopH targets a novel adhesion-regulated signalling complex in macrophages. Cell. Microbiol. 2:401–414. [DOI] [PubMed] [Google Scholar]
- 6.Zhang, Z. Y., Y. Wang, L. Wu, E. B. Fauman, J. A. Stuckey, H. L. Schubert, M. A. Saper, and J. E. Dixon. 1994. The Cys(X)5Arg catalytic motif in phosphoester hydrolysis. Biochemistry. 33:15266–15270. [DOI] [PubMed] [Google Scholar]
- 7.Zhang, Z. Y., Y. Wang, and J. E. Dixon. 1994. Dissecting the catalytic mechanism of protein-tyrosine phosphatases. Proc. Natl. Acad. Sci. USA. 91:1624–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guan, K. L., and J. E. Dixon. 1991. Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate. J. Biol. Chem. 266:17026–17030. [PubMed] [Google Scholar]
- 9.Zhang, Z. Y., W. P. Malachowski, R. L. Van Etten, and J. E. Dixon. 1994. Nature of the rate-determining steps of the reaction catalyzed by the Yersinia protein-tyrosine phosphatase. J. Biol. Chem. 269:8140–8145. [PubMed] [Google Scholar]
- 10.Stuckey, J. A., H. L. Schubert, E. B. Fauman, Z. Y. Zhang, J. E. Dixon, and M. A. Saper. 1994. Crystal structure of Yersinia protein tyrosine phosphatase at 2.5 A and the complex with tungstate. Nature. 370:571–575. [DOI] [PubMed] [Google Scholar]
- 11.Schubert, H. L., E. B. Fauman, J. A. Stuckey, J. E. Dixon, and M. A. Saper. 1995. A ligand-induced conformational change in the Yersinia protein tyrosine phosphatase. Protein Sci. 4:1904–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fauman, E. B., C. Yuvaniyama, H. L. Schubert, J. A. Stuckey, and M. A. Saper. 1996. The X-ray crystal structures of Yersinia tyrosine phosphatase with bound tungstate and nitrate. Mechanistic implications. J. Biol. Chem. 271:18780–18788. [DOI] [PubMed] [Google Scholar]
- 13.Juszczak, L. J., Z. Y. Zhang, L. Wu, D. S. Gottfried, and D. D. Eads. 1997. Rapid loop dynamics of Yersinia protein tyrosine phosphatases. Biochemistry. 36:2227–2236. [DOI] [PubMed] [Google Scholar]
- 14.Wang, F., W. Li, M. R. Emmett, C. L. Hendrickson, A. G. Marshall, Y. L. Zhang, L. Wu, and Z. Y. Zhang. 1998. Conformational and dynamic changes of Yersinia protein tyrosine phosphatase induced by ligand binding and active site mutation and revealed by H/D exchange and electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Biochemistry. 37:15289–15299. [DOI] [PubMed] [Google Scholar]
- 15.Peters, G. H., T. M. Frimurer, J. N. Andersen, and O. H. Olsen. 1999. Molecular dynamics simulations of protein-tyrosine phosphatase 1B. I. ligand-induced changes in the protein motions. Biophys. J. 77:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peters, G. H., T. M. Frimurer, J. N. Andersen, and O. H. Olsen. 2000. Molecular dynamics simulations of protein-tyrosine phosphatase 1B. II. substrate-enzyme interactions and dynamics. Biophys. J. 78:2191–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elber, R., and M. Karplus. 1990. Enhanced sampling in molecular-dynamics: use of the time-dependent Hartree approximation for a simulation of carbon-monoxide diffusion through myoglobin. J. Am. Chem. Soc. 112:9161–9175. [Google Scholar]
- 18.Dillet, V., R. L. Van Etten, and D. Bashford. 2000. Stabilization of charges and protonation states in the active site of the protein tyrosine phosphatases: a computational study. J. Phys. Chem. B. 104:11321–11333. [Google Scholar]
- 19.Case, D. A., T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, K. M. Merz, B. Wang, D. A. Pearlman, M. Crowley, S. Brozell, V. Tsui, H. Gohlke, J. Mongan, V. Hornak, G. Cui, P. Beroza, C. Schafmeister, J. W. Caldwell, W. S. Ross, and P. A. Kollman. 2004, AMBER 8, University of California, San Francisco.
- 20.Wang, J., P. Cieplak, and P. A. Kollman. 2000. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21:1049–1074. [Google Scholar]
- 21.Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1997. Numerical integration of the Cartesian equation of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23:327–341. [Google Scholar]
- 22.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 23.Ichiye, T., and M. Karplus. 1991. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular-dynamics and normal mode simulations. Proteins. 11:205–217. [DOI] [PubMed] [Google Scholar]
- 24.McCammon, J. A., and S. C. Harvey. 1987. Dynamics of Proteins and Nucleic Acids. Cambridge University Press, Cambridge, UK.
- 25.Amadei, A., A. B. M. Linssen, and H. J. C. Berendsen. 1993. Essential dynamics of proteins. Proteins. 17:412–425. [DOI] [PubMed] [Google Scholar]
- 26.Roitberg, A., and R. Elber. 1991. Modeling side-chains in peptides and proteins: application of the locally enhanced sampling and the simulated annealing methods to find minimum energy conformations. J. Chem. Phys. 95:9277–9287. [Google Scholar]
- 27.Simmerling, C., J. L. Miller, and P. A. Kollman. 1998. Combined locally enhanced sampling and particle mesh Ewald as a strategy to locate the experimental structure of a nonhelical nucleic acid. J. Am. Chem. Soc. 120:7149–7155. [Google Scholar]
- 28.Simmerling, C., M. R. Lee, A. R. Ortiz, A. Kolinski, J. Skolnick, and P. A. Kollman. 2000. Combining MONSSTER and LES/PME to predict protein structure from amino acid sequence: application to the small protein CMTI-1. J. Am. Chem. Soc. 122:8392–8402. [Google Scholar]
- 29.Zhang, Z. Y., and L. Wu. 1997. The single sulfur to oxygen substitution in the active site nucleophile of the Yersinia protein-tyrosine phosphatase leads to substantial structural and functional perturbations. Biochemistry. 36:1362–1369. [DOI] [PubMed] [Google Scholar]
- 30.Scapin, G., S. Patel, V. Patel, B. Kennedy, and E. Asante-Appiah. 2001. The structure of apo protein-tyrosine phosphatase 1B C215S mutant: More than just an S → O change. Protein Sci. 10:1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ivanov, M. I., J. A. Stuckey, H. L. Schubert, M. A. Saper, and J. B. Bliska. 2005. Two substrate-targeting sites in the Yersinia protein tyrosine phosphatase co-operate to promote bacterial virulence. Mol. Microbiol. 55:1346–1356. [DOI] [PubMed] [Google Scholar]
- 32.Wiesmann, C., K. J. Barr, J. Kung, J. Zhu, D. A. Erlanson, W. Shen, B. J. Fahr, M. Zhong, L. Taylor, M. Randal, R. S. McDowell, and S. K. Hansen. 2004. Allosteric inhibition of protein tyrosine phosphatase 1B. Nat. Struct. Mol. Biol. 11:730–737. [DOI] [PubMed] [Google Scholar]