

Abstract
The coronavirus responsible for the severe acute respiratory syndrome contains a small envelope protein, E, with putative involvement in host apoptosis and virus morphogenesis. To perform these functions, it has been suggested that protein E can form a membrane destabilizing transmembrane (TM) hairpin, or homooligomerize to form a TM pore. Indeed, in a recent study we reported that the α-helical putative transmembrane domain of E protein (ETM) forms several SDS-resistant TM interactions: a dimer, a trimer, and two pentameric forms. Further, these interactions were found to be evolutionarily conserved. Herein, we have studied multiple isotopically labeled ETM peptides reconstituted in model lipid bilayers, using the orientational parameters derived from infrared dichroic data. We show that the topology of ETM is consistent with a regular TM α-helix. Further, the orientational parameters obtained unequivocally correspond to a homopentameric model, by comparison with previous predictions. We have independently confirmed that the full polypeptide of E protein can also aggregate as pentamers after expression in Escherichia coli. This interaction must be stabilized, at least partially, at the TM domain. The model we report for this pentameric α-helical bundle may explain some of the permabilizing properties of protein E, and should be the basis of mutagenesis efforts in future functional studies.
INTRODUCTION
The causative agent of the severe acute respiratory syndrome (SARS) (1) is a member of the family Coronaviridae, which cause most common colds in humans and are responsible for serious diseases in other animal species. The lipid bilayer that surrounds these viruses typically embeds three proteins: spike (S), membrane (M), and envelope (E) protein. These proteins are studied for their important role in receptor binding and virion budding.
The E protein of the SARS-CoV virus is a small, 76 amino acid, integral membrane protein with one putative transmembrane α-helical (TMH) hydrophobic domain, 20–30 amino acids long, flanked by a short N-terminus (<10 amino acids) and a longer C-terminus tail, both more hydrophilic. This protein is very important for virus morphogenesis, to the point that it has been found to be critical for viral budding in mouse hepatitis virus (MHV) (2). Also, in many coronaviruses, coexpression of M and E proteins, which probably interact via their cytoplasmic domains in pre-Golgi compartments (3), is necessary for morphogenesis (4,5).
A further role suggested for protein E is to promote apoptosis (6,7). It has also been found that SARS-CoV E protein can alter membrane permeability when expressed in Escherichia coli and mammalian cells (8) and in vitro cation-selective ion channel formation has been reported using a totally synthetic polypeptide (9). This apparent ability to form channels suggests that E protein is able to form TM homooligomers, as reported in other similar systems (10,11).
To determine if, and which, TM oligomers are present in SARS-CoV E, we used recently (12) a computational method that filters nonnative interactions with the help of evolutionary conservation data (13). A search of the possible interhelical interactions in the transmembrane domain of E protein (ETM) was performed using a total of 17 homologs of SARS-CoV E, present in other viruses. We showed that only three homooligomeric models had been conserved by evolution of the viruses: a dimer, a trimer, and two similar pentamers. Due to the special nature of our simulation, totally independent from experimental measurements, and only dependent on the disturbing effect of conservative mutations on non native models, we predicted (12) that all these oligomers should be found in vivo.
This conclusion was consistent with previous data for expression of whole protein E in E. coli and mammalian cells, where dimers and trimers in SDS were observed, although no pentamers could be detected (8). We also reported in the same study (12) that these three types of oligomers of synthetic ETM were stable in SDS, which suggested a stabilizing function at the TM domain. Clearly, of these oligomeric forms, only a pentamer is consistent with the formation of a pore that could account for the ion channel behavior observed in previous reports (9), but at present the oligomeric form of protein E, and ETM, in lipid bilayers is not yet known.
Herein, we have attacked this problem examining the structure of isotopically labeled ETM when incorporated in model lipid bilayers using polarized attenuated total reflection Fourier Transform infrared spectroscopy (PATIR-FTIR). Specifically, we have synthesized ETM introducing a labeled carbonyl (13C=18O) (14) at various positions (15), for a total of nine residues along the ETM sequence. We have then determined the orientation in space of these labeled peptidic carbonyl groups using the theory of site specific infrared dichroism (SSID) (16). Like any orientational technique, SSID has certain limitations. First, it requires a certain order of the sample, and therefore is not applicable to proteins in solution. Also, it requires a known periodicity in the sample, which is found in α-helical segments. Finally, the requirement for discrimination between neighboring peptide carbonyl orientations makes it unsuitable when α-helices are either completely parallel or perpendicular to the membrane normal. SSID is therefore ideal to study TMH bundles, where all the above conditions are met (14,15,17).
We note that in several reports (14,16,18–20), we have used only two labeled residues, at consecutive positions, to give support to a particular orientation. This is appropriate when alternative models are separated by ω angles of at least 45°, because the error in ω using SSID is usually 10–20°. A more thorough validation of a particular model, however, requires a multiple labeling approach so that experimental values and predicted model can be compared at multiple sites, as reported previously for CD3ζ (15).
As in previous work (15), we have compared experimental versus predicted tilt and rotational orientation of the peptide. For the predicted values, we used those obtained from the evolutionarily conserved models during molecular dynamics simulations (12). Additionally, we have again addressed the problem of the oligomerization of E protein, i.e., the complete polypeptide, when expressed in E. coli. Our results provide an important insight into the mode of organization of E protein in lipid bilayers.
EXPERIMENTAL PROCEDURES
Expression of SARS-CoV E protein in E.coli
Plasmids of full length C-terminal Histidine tag fused SARS-CoV E protein cloned in a pET 24a vector previously described (8) were transformed into E. coli strain BL21DE3 pLysS. A single colony was grown in Luria Broth medium until the absorbance reached 0.4 at 600 nm. The culture was induced with 0.5 mM IPTG, and a part of the culture was transferred to a fresh culture vial as uninduced fraction. The induced and uninduced cultures were further incubated at 25°C for 6 hr.
Western blot analysis
Total cell lysate from the uninduced and induced fractions were prepared by sonication, and the slurry obtained was mixed with 2× SDS loading buffer (with 150 mM DTT and 2% SDS) and subjected to 15% SDS-PAGE. Proteins were transferred to nitrocellulose membrane (Pierce, Rockford, IL) and blocked overnight at 4°C in blocking solution (5% fat free milk solution in PBS pH 7.4). The membrane was incubated with a 1:1000 diluted primary antibody (anti-His tag) in blocking solution for 1 h at room temperature. After three washings with PBST, the membrane was incubated with 1:2000 diluted secondary antibody (anti rabbit HRP conjugate DAKO) in blocking solution for 1 h at room temperature. After washing for three times with PBST, the proteins were detected with a chemiluminescence detection kit (West Pico, Pierce) according to the manufacturer protocol.
Preparation of labels and peptides
Amino acids labeled with carbonyl 13C=18O were obtained as described previously (14,21). The amino acids were then derivatized with 9-fluorenyl-methyloxycarbonyl (FMOC) (22).
The peptides corresponding to ETM (Fig. 1 A) were synthesized using standard solid phase FMOC chemistry (Intavis ResPep (Gladbach, Germany) peptide synthesizer), from residue 9 to 35, adding two lysine (K) residues to both N- and C-termini (Fig. 1 B) to increase their solubility (12). The peptides were cleaved from the resin with trifluoroacetic acid (TFA) and lyophilized. The lyophilized peptides were purified by HPLC and analyzed by SDS-PAGE as described previously (12). Lyophilization was performed always in the presence of HCl (typically at an approximate molar ratio 20:1, HCl/peptide) to avoid the formation of peptide-TFA adducts; consequently, the typical TFA band at ∼1685 cm−1 was not observed in the amide I region. Peptide purity was further confirmed by mass spectrometry. During the synthesis of ETM, an amino acid bearing a 13C=18O carbonyl group was introduced at a specific position (Fig. 1 B, open circles). A mutation was introduced in two of the peptides, N15A and F23A (Fig. 1 B, residues underlined). In the first case (N15A), this was done to observe if Asn substitution had a critical destabilizing effect on the oligomers, because Asn-Asn interactions were predicted to be present in all evolutionarily conserved oligomeric models (12). The second mutation, F23A, was assumed to be critical in view of its suggested central role in the formation of a putative palindromic helical hairpin (23).
FIGURE 1.

(A) Position of the putative TMH domain (TM, see box) in the complete sequence of SARS-CoV E. The residue numbers and N- and C-termini are indicated. (B) Peptides synthesized corresponding to ETM, labeled at various positions with 13C=18O (○) and cyano-phenylalanine (•); two mutated sequences are indicated with a star (right), and the mutated residues are underlined. (C) Synthetic peptides used as a control, corresponding to the TMH domain of CD3-ζ, (top), integrin β2 (middle), and integrin αM (bottom). (D) Schematic representation of an α-helix with parameters ω (range 0–359°) and β (range (0–90°). The peptidic C=O bonds isotopically labeled are shown as spheres (C, solid; O, open). The arrows indicate the direction of the C=O bond relative to the z axis. This direction is obtained from the helix and label dichroic ratios (see Experimental Procedures) and is used to determine ω.
As a control, we used peptides that are α-helical in lipid bilayers, with their C- and N-terminus on opposite sides of the membrane, ∼25 residues buried in the membrane, and which are not known to disrupt liposome integrity. These TM peptides, of integrin β2 (β2-TM), integrin αM (αM-TM) and the component of the T-cell receptor, CD3ζ (CD3 ζ-TM), were synthesized without any labels (Fig. 1 C) and purified as described above.
Cyano-phenylalanine labeling
The nitrile-derivatized amino acid, 4-cyano-phenylalanine (Phe-CN, Sigma-Aldrich, St. Louis, MO) was introduced during the FMOC synthesis in three separate peptides at positions F20, F23, or F26 (Fig. 1 B, solid circles).
Preparation of liposomes
Unilamellar liposomes of predefined size were obtained as described previously (24). Briefly, multilamellar liposomes were obtained by mixing lipid and water and were extruded 30 times through a 200-nm pore polycarbonate filter using a mini-extruder (Avanti Polar Lipids, Alabaster, AL). DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids) liposomes containing ETM, β2-TM, αM-TM, or CD3-ζ-TM were prepared by adding the peptide to the lipid (15:1 DMPC/peptide molar ratio) prior extrusion. Typically, 1 mg of dry peptide was mixed with DMPC and 1 ml of HFIP (1,1,1,3,3,3-hexafluoro-2-propanol, Merck, Darmstadt, Germany). The solvent was evaporated with a rotary evaporator, before addition of 1 ml of H2O. Homogeneity in the diameter of the extruded liposomes was assessed using Dynamic Light Scattering (DLS), which was performed using a 90 Plus particle size analyzer (Brookhaven Instruments, Holtsville, NY).
Data collection and sample preparation for ATR-FTIR
Infrared spectra were recorded on a Nicolet Nexus spectrometer (Madison, WI) purged with N2 and equipped with a MCT/A detector, cooled with liquid nitrogen. Attenuated total reflection (ATR) spectra were measured with a 25-reflections ATR accessory from Graseby Specac (Kent, UK) and a wire grid polarizer (0.25 μm, Graseby Specac). A total of 200 interferograms collected at a resolution of 4 cm−1 were averaged for every sample and processed with 1 point zero filling and Happ-Genzel apodization. The peptide (typically ∼3 mg) was reconstituted in a 15:1 DMPC/peptide molar ratio (14) and applied onto a trapezoidal (50 mm × 2 mm × 20 mm) Ge internal reflection element (IRE). A dry, or D2O saturated, N2 stream flowing through the ATR compartment (14) was used to remove bulk water or to achieve D2O exchange, respectively.
H/D exchange
The percentage of isotopic (hydrogen/deuterium, H/D) exchange was calculated from the area ratio Amide II /Amide I, using nonpolarized spectra, before and after exchange in D2O, as described previously (25). Non polarized spectra were obtained from the parallel (‖) and perpendicular (⊥) ATR polarized spectra, using the expression 1 × (‖) + 1.44 × (⊥), as described previously (26).
Analysis of infrared data
The data was analyzed according to the theory of site-specific dichroism (SSID) presented in detail elsewhere (16). SSID uses the fact that the measured dichroic ratio of a particular transition dipole moment, e.g., peptidic C=O stretching, is a function of its spatial orientation, defined by tilt β and rotational pitch angle ω (see Fig. 1 D), which is defined arbitrarily as 0° when the transition dipole moment, the helix director, and the z axis all reside in a single plane (16). The angle α between the transition dipole moment of the vibrational transition and the helix axis is known from fiber diffraction studies (27), and was taken as 39° for the peptidic C=O bond and 29° for the N-H bond (28). The values for the electric field components, ɛx, ɛy, and ɛz, were those calculated according to a thick film approximation (29).
Dichroic ratios were calculated as the ratio between the integrated absorptions of the spectra collected with parallel and perpendicular polarized light. The area of the amide A was calculated by integration of the band centered at ∼3300 cm−1, between 3200 and 3400 cm−1 (14) when the sample was equilibrated in D2O. In these conditions, this band corresponds only to the membrane-embedded part of ETM. The absorption corresponding to the label, i.e., the 13C=18O carbonyl stretching vibration, was obtained integrating the band centered at ∼1590 cm−1 (15,16). Thus, from each measurement, two different dichroic ratios, from amide A (RHelix) and label (Rsite), were obtained (Eqs. 1 and 2, respectively) (16).
The first (RHelix, Eq. 1) is the composite dichroism that corresponds to all membrane-embedded amide N-H bonds in the helical structure for sample i. This dichroism is independent of ω and dependent solely on β and the fractional order of the preparation, fi. This fractional order is 1 if the sample is completely ordered, and 0 if randomly oriented (16). The parameters Kx,y, or z are the rotationally averaged, integrated absorption coefficients, whereas Kx,y, or z (ω) refers to the absorption coefficient at a specific ω angle.
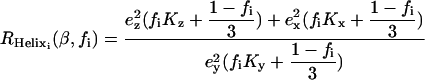 |
(1) |
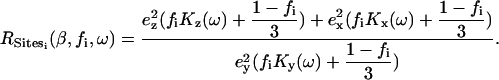 |
(2) |
The other dichroic ratio (Rsite, Eq. 2) corresponds to the isotopic label, centered at ∼1590 cm−1, which depends on three parameters: ωsite, β, and fi.
These two equations are not sufficient to obtain β, ωsite, and fi (three unknowns), therefore a second label is inserted in a different sample j, either immediately before or after the first label (16). There are 3.6 residues per turn in a canonical α-helix, therefore Δω between consecutive residues is assumed to be 100°. Thus, with the additional labeled sample, two additional equations can be obtained, RHelixj and RSitej, dependent on (β, fj) and (β, ω + 100°, fj), respectively. Solving simultaneously the four equations RHelixi, RHelixj, RSitei, and RSitej for each i and j pair, yields βij, ωij, fi, and fj, where βij and ωij are the results obtained from the combinations of sample i and sample j. These nonlinear equations were solved with Newton's method as implemented in the FindRoot function in Mathematica 5.0 (Wolfram Research, Champaign, IL). The final values of β and ω for a particular pair of labels (i, j) were obtained by averaging (at least three measurements) βij and ωij, respectively.
A total of 27 sets of dichroic ratios (Rhelix, Rsite) were obtained (for each of the 9 labeled peptides in Fig. 1 B, three independent preparations were measured). As described above, these values were combined in consecutive labels whenever possible, i.e., (i, i + 1) so that the error in the assumption Δω = 100° was a small as possible. In other cases combinations (i, i + 2) or (i, i + 3) using Δω of 200° or 300°, respectively was also tested. Specifically, the combinations used were V17/L18, L18/L19, L19/L21, L21/A22, A22/A23, A23/V24, V24/L27, and L27/L28. These combinations were used to obtain helix tilt, β, and rotational orientation ω at each i-labeled residue, as well as the contribution of disorder (f) in the samples (15), which was usually 0.7–0.9, depending on the sample.
Visualization of orientational data
To visualize the allowed values (ω, β) of a specific labeled residue for a particular measurement (Rhelix, Rsite), we used the reasonable assumption that the disorder, f, of a particular preparation, is the same for the α-helix and for its labeled site. Thus, we first plotted the allowed values of f as a function of ω and β for the measured Rsite. Second, we plotted a similar graph (f as a function of ω and β) for the measured Rhelix. As f should be the same for both helix and site, because they belong to the same sample, we obtained the intersection values of these two surfaces and projected them in the (ω,β) plane. Thus, each pair of dichroic ratios obtained in a particular preparation (Rhelix, Rsite) is represented by a curve with all the allowed values of ω and β for that particular labeled residue.
It can be shown that when Rhelix < Rsite and Rhelix > Rsite, this projection will be centered at 0° and 180°, respectively; for example, the curves for the samples with label at V17 and L18. In both cases Rhelix < Rsite (Table 1) therefore, the “smiles” for the two residues should be centered initially at 0°, but because Δω is 100°, we shift the “smiles” for L18, 100° to the left. The coordinates ω17 and β (for residues 17 and 18) can be found at the intersection of the two curves. The same strategy was followed for other combinations of labels, shifting the “smiles” of the second label to the left, according to Δω, and finding the intersection of the curves.
TABLE 1.
Dichroic ratios
| Residue | Rhelix | Rsite |
|---|---|---|
| V17 | 3.6 | 4.6 |
| V17 | 4.1 | 4.3 |
| V17 | 3.6 | 3.9 |
| L18 | 3.2 | 3.5 |
| L18 | 3.3 | 3.4 |
| L18 | 2.9 | 3.0 |
| L19 | 3.6 | 3.5 |
| L19 | 3.2 | 2.8 |
| L19 | 3.5 | 3.3 |
| L21 | 3.5 | 6.0 |
| L21 | 3.6 | 6.6 |
| L21 | 4.1 | 6.2 |
| A22 | 3.6 | 4.3 |
| A22 | 3.2 | 4.4 |
| A22 | 3.4 | 4.1 |
| A23 | 3.4 | 2.0 |
| A23 | 3.1 | 2.3 |
| A23 | 4.0 | 2.3 |
| V24 | 4.7 | 5.7 |
| V24 | 4.0 | 4.3 |
| V24 | 3.8 | 4.1 |
| L27 | 4.1 | 5.0 |
| L27 | 4.0 | 4.6 |
| L27 | 3.6 | 4.1 |
| L28 | 4.2 | 4.4 |
| L28 | 4.0 | 5.1 |
| L28 | 3.8 | 4.1 |
Dichroic ratios measured by SSID using the amide A and the isotopic label for ETM incorporated in DMPC lipid bilayers (see Experimental Procedures). The information from each labeled residue was obtained from at least three independent measurements.
The predicted values for (ω,β) were obtained from the corresponding conserved model in the molecular dynamics simulations reported previously (12), calculating the values (ω,β) for each monomer, and averaging for all the monomers in the oligomer.
Pore calculations
The dimensions of the pore for the pentameric model consistent with the experimental data were calculated using the software HOLE (30).
RESULTS
Electrophoresis
Electrophoresis performed with all the peptides shown in Fig. 1 B were identical as those obtained previously (12), showing bands consistent with dimers, trimers, and pentamers (see Fig. 2). These oligomers were present even in the case of the a priori critical mutants N15A and F23A (Fig. 1 B, underlined). These two mutants were synthesized because they could affect oligomerization via two possible mechanisms: N15 might provide stabilization for the TM oligomers via interchain hydrogen bonds (31,32), whereas F23 has been suggested to have a central role in the formation of a putative short hairpin of ETM (23). Intriguingly, we did not find a change in oligomerization pattern or electrophoretic mobility in any of these mutants, indicating that these two residues, N15 and F23, are not essential for ETM oligomerization in the presence of SDS.
FIGURE 2.

SDS-PAGE electrophoresis corresponding to the synthetic TM peptide of SARS protein E. Lane 1 shows the molecular weight markers. Lanes 2–4: peptide containing mutation N15A (see Fig. 1 B) with increasing load of peptide: 10, 20, and 40 μg, respectively. Arrows indicate the bands corresponding to the dimer, trimer, and pentameric forms of the peptide. The bands corresponding to the pentamer in lanes 2 and 3 were visible only after silver staining. Electrophoresis results for the other peptides shown in Fig. 1 B were identical, and are not shown.
Liposome morphology
Previous reports (23) have suggested that ETM inserts into lipid bilayers as a short hairpin and it may induce dramatic morphological changes in lipid bilayers.
To test this, we used dynamic light scattering (DLS) to monitor possible morphological changes of DMPC liposomes incorporating ETM, as compared to a), liposomes without peptide or b), liposomes containing a typical TMH peptide, e.g., integrin β2-TM (see Fig. 1 C). In all three cases, the calculated diameter of the liposomes was ∼170 nm (168.5 ± 1 nm, 168 ± 2 nm, and 170.5 ± 1.5 nm, respectively), which indicates no dramatic disturbing effect of ETM on liposome morphology.
ETM hydrogen/deuterium exchange
Representative infrared spectra of isotopically labeled ETM in DMPC lipid bilayers are shown in Fig. 3. As reported previously (23), the shape of the amide I absorption band, with a maximum at 1657 cm−1, is indicative of a predominant α-helical structure (33). Examination of the amide I and II bands in Fig. 3 A indicates that the percentage of hydrogen/deuterium (H/D) exchange of ETM amide groups was small (17 ± 2%), which indicates that ∼26 residues are membrane-embedded. A similar number of exchange-protected residues (25–26) was obtained with the peptides control (shown in Fig. 1 C, not shown). These results suggest that ETM and the TMH peptides shown in Fig. 1 C share the same topology, i.e., that of a regular, slightly tilted, TMH.
FIGURE 3.

Infrared spectra of ETM incorporated in DMPC lipid bilayers. (A) Nonpolarized infrared spectra of the amide I and II region in H2O, either at low hydration (solid line) or hydrated in D2O (dashed line). The small reduction in amide II intensity is due to H/D exchange. (B) Amide I region and isotopic 13C=18O label (residue A22), centered at 1590 cm−1 (see inset), obtained at 0° (solid line) or 90° (dashed line) polarization. (C) Same as in B, but corresponding to the amide A of the spectrum.
ETM backbone orientation
After confirming that ETM is inserted, and α-helical, in lipid bilayers, we investigated the tilt and rotational orientation of the peptide, comparing dichroic experimental data and predicted orientation (see Experimental Procedures) from the computational models (34). An example of the infrared dichroic data obtained at two polarizations (see Experimental Procedures) is shown for the peptide labeled at A22, for amide I and site (Fig. 3 B) and for amide A (Fig. 3 C). Spectra for other labeled peptides were similar, and are not shown.
The dichroic ratios obtained from each measurement, Rhelix and Rsite are shown in Table 1. With each pair of values (Rhelix, Rsite), we can visualize the allowed ω- and β-values for that particular site, as shown in the “smile” plots in Fig. 4, D–K (see Experimental Procedures). The “smiles” for different residues were found to intersect (i.e., converged to a solution) for the pairs 17/18 (Δω = 100°), 18/19 (Δω = 100°), 19/21 (Δω = 200°), 21/22 (Δω = 100°), 22/23 (Δω = 100°), 23/24 (Δω = 100°) and 27/28 (Δω = 100°). More importantly, Table 2 shows that using these Δω, expected for a canonical α-helix, the ω-values are consistent with the pentamer B oligomer obtained previously using global search molecular dynamics (12), except for ω27 and ω28, which are closer to pentamer A than to pentamer B. The Δω between 24 and 27 is therefore 390° (Fig. 4 J), not 300°. Indeed, when we combined 24 and 27 assuming Δω = 300° (not shown) ω24 was 40°, which is not consistent with the value ω24 = −70°, obtained with the pair 23/24. The reason for the inconsistency may be a slight bend from residues 24 to 27, and exemplifies the need to use consecutive residues in these calculations. Slices through a pentamer in agreement with these observations are represented (Fig. 4, L–Q).
FIGURE 4.

Visualization of the dichroic data obtained and proposed model for the ETM pentamer: (A) plot corresponding to the order parameter, f, of a labeled residue as a function of ω and β, assuming a Rsite of 4.2; (B) the same plot for Rhelix, when Rhelix is 3.5; (C) superposition of the two surfaces in A and B, where the intersection corresponds to (ω,β) coordinates with the same disorder f ; (D–K) coordinates (ω,β) at the intersection shown in C for pairs V17/L18 (D), L18/L19 (E), L19/L21 (F), L21/A22 (G), A22/A23 (H), A23/V24 (I), V24/L27 (J), and L27/L28 (K); (L–Q), slices for the model in agreement with our dichroic data: 11–13 (L), 14–18 (M), 19–22 (N), 23–26 (O), 27–30 (P) and 31–34 (Q). The residue numbers are indicated. Color code: L, green; V, cyan; I, salmon; A, marine; F, blue; N, orange; and S and T, red.
TABLE 2.
Experimental versus predicted orientational values for ETM incorporated in model lipid bilayers
| Residue | Dimer | Trimer | Pentamer A | Pentamer B | ω (exp) | β (exp) |
|---|---|---|---|---|---|---|
| V17 | 61 (12) | −60 (36) | −31 (25) | −80 (21) | −47 ± 5 | 20 ± 2 |
| L18 | −176 (6) | 52 (37) | 57 (24) | 8 (26) | 15 ± 8 | 14 ± 2 |
| L19 | −54 (12) | −178 (30) | 164 (17) | 110 (22) | 105 ± 3 | 27 ± 1 |
| L21 | 99 (9) | 9 (39) | 6 (27) | −30 (26) | −44 ± 4 | 29 ± 2 |
| A22 | −127 (6) | 137 (32) | 106 (23) | 60 (25) | 50 ± 7 | 19 ± 3 |
| A23 | −3 (13) | −113 (32) | −128 (34) | −183 (18) | −170 ± 7 | 16 ± 1 |
| V24 | 40 (14) | −21 (39) | −32 (27) | −83 (23) | −70 ± 4 | 19 ± 1 |
| L27 | −2 (14) | −52 (36) | −64 (22) | −113 (20) | −48 ± 4 | 20 ± 2 |
| L28 | 66 (11) | 66 (36) | 22 (26) | −15 (26) | 52* |
Comparison of the rotational orientation (ω) and helix tilt (β, within parentheses) obtained computationally (14) for the dimeric, trimeric, and pentameric (pentamer A and pentamer B) models with the values for ω and β obtained herein by SSID (exp, shown in last two columns).
Obtained adding 100° to w27.
We point out that a solution was found consistent with the expected helix periodicity and rotational orientation of pentamer B (see Table 2) when using the pairs 18/19 or 19/21, even with the mutated peptide N15A (isotopically labeled at L19). This suggests that this mutation not only does not perturb the oligomeric forms present in SDS (see Fig. 2) but also does not alter significantly the structure of the oligomer in lipid bilayers. Similarly, our results show that the substitution F23A did not affect the orientation of the α-helices: when the dichroic data from the label introduced at position A23 (Fig. 1 B, asterisks) was combined with V24 or A22, a solution was found which was consistent with the ω-values expected for the pentameric B model. Together with the electrophoresis results, which show that none of the oligomeric forms are affected by mutation F23A (Fig. 2), this suggests that F23 does not play any essential role in either maintaining the oligomeric structure or in defining a particular topology.
Phe-CN labeling
If the topology of ETM is a regular TMH, the side chains of the 3 Phe residues (F20, F23, and F26) must be at or near the center of the lipid bilayer. To confirm this topology, we labeled ETM with Phe-CN, a nitrile derivatized phenylalanine, at these three positions (see Fig. 1 B, solid dots). The rationale for this experiment is provided by the sensitivity of the C≡N stretching vibration of this nitrile derivatized side chain to the hydration and polarity of the environment (35,36). If the side chain is fully hydrated, the band is centered at 2235 cm−1 in contrast, when inserted deep in phospholipid bilayers, this band shifts to lower wavenumbers, at 2229 cm−1, and becomes narrower (35). Fig. 5 shows the C≡N stretching vibration of the three nitrile derivatized amino acids when ETM is reconstituted in DMPC lipid bilayers. Clearly, for all three Phe residues, the band is centered at ∼2230 cm−1. This indicates a generally hydrophobic and dehydrated environment, consistent with these Phe residues being deeply buried in the lipid bilayer.
FIGURE 5.

Spectral features of the C≡N stretching vibration for F20 (dotted line), F23 (dashed line), and F26 (solid line) cyano-phenylalanine when ETM was reconstituted in DMPC lipid bilayers.
Homopentamers detected after E expression in E. coli cells
To add support to a pentameric form of SARS-CoV E in biological membranes, we expressed the full length of protein E (shown in Fig. 1 A) in E. coli cells (see Experimental Procedures). After electrophoresis of the extract in reducing conditions, where disulfide covalent bonds are not possible, mainly monomers (∼9 kDa) and pentamers (∼46 kDa) were detected (Fig. 6, lane 2), and also some dimers (∼17 kDa). This suggests that not only protein E pentameric oligomerization may be present in vivo, but also that is mediated, at least partly, by TM interactions.
FIGURE 6.

Expression and oligomerization of SARS-CoV E protein. The His-tagged E protein expressed in E. Coli BL21DE3pLysS cells was analyzed for expression as uninduced and induced total cell lysate fractions (lanes 1 and 2), respectively, on a 15% SDS polyacrylamide gels in the presence of 150 mM DTT and 2% SDS. The proteins were transferred to a nitrocellulose membrane and probed with polyclonal anti His tag antibody in 1:1000 dilution. This was by followed incubating with horse radish-peroxidase conjugated anti rabbit IgG and the E protein was detected by a chemiluminescence detection kit. Numbers on the right indicate molecular masses (kDa).
DISCUSSION
We have shown that SDS-resistant oligomers of ETM, consistent with dimers, trimers, and pentamers, can be observed when ETM is synthesized with two flanking lysine residues. This modification, which we also used in a previous report (12) was made to improve solubility and purification (37). It has been demonstrated (38,39) for various TM oligomers, e.g., monomeric epidermal growth factor receptor (EGFR), dimeric glycophorin A (GpA), and tetrameric M2 from Influenza A, that even with up to four flanking lysines at each C- and N-termini, oligomerization is not affected during SDS PAGE electrophoresis. Also, electrophoretic mobility has been reported to be reduced ∼30% when three lysines are added at each terminus (38). In this work, with only two lysines on each side, the error in molecular weight estimation should not be higher than 20%. In support that a pentameric form of whole E protein is also present in SDS, we show bands consistent with pentamers (∼46 kDa), along with monomers (∼9 kDa) and some dimers (∼17 kDa) of E protein when expressed in E. coli in reducing conditions. In a previous work (8) only monomers (∼9 kDa), dimers (∼18 kDa), and trimers (∼27 kDa) were observed, but only under nonreducing conditions. We are unable to explain these variations in the oligomeric species observed, but they may be related to the complex functionality of SARS- CoV E. In any case, the band at (∼46 kDa) cannot be interpreted as being trimers, because these were observed previously at ∼27 kDa (8). Incidentally, a pentameric homooligomer has also been observed in other similarly sized viral membrane proteins, e.g., the small (63 residues) hydrophobic protein in the respiratory syncytial virus (40,41). It is possible therefore that these two proteins perform similar functions. In this respect, E protein has been found to induce in vivo permeabilization of biological membranes to bulky molecules such as o-nitrophenyl-β-D-galactopyranoside and hygromycin B (8). Although a pentameric porelike structure cannot explain permeabilization to these big molecules, it can explain the permeability to cations observed in an in vitro system (9).
If model B were correct, the narrowest constriction site along the lumen occurs at F26 (see Fig. 7), with a diameter of only 1.66 Å (the effective diameter of a water molecule is ∼2 Å). Clearly, the model pentamer B is not consistent with an open channel (the diameter of naked Na+ or Ca2+ is ∼2 Å, and that of K+ is even larger, 2.66 Å). We note that similar constriction diameters have been found for other channels in a closed state, e.g., 1 Å in KirBac (42), a bacterial homolog of mammalian inward rectifier K channels. Considering the model A orientation from residues 24–27 however, the channel appears more open. Our experimental data can only be explained by a bend of the α-helices around residues 25–27, with residues 17–24 consistent with pentamer B and 24 onward consistent with pentamer A. The only partial consistency with the computational models may be explained by the fact that during the simulations of ETM (12) we restrained the helix tilt to a particular value, and effects such as bends or kinks, if existed in the native structure, were not accounted for. The result of this simplistic assumption may have been that, instead of a single pentameric model, two different models were obtained with only a partially correct orientation in different parts of the sequence. This conclusion is supported by the fact that none of these two models contained all the lowest energy structures for the 17 sequences analyzed, although most of them (16) were in agreement with model A and only 1 (PHEV) with model B.
FIGURE 7.

Plot showing the pore radius for the model pentamer B (solid line) and A (dashed line) as a function of the z coordinate (normal to the bilayer). As the orientation of residues 27 and 28 are more similar to pentamer A; we also show the plot as a hybrid between pentamer A and B, as a thicker line.
We have estimated the ion conductance of this putative channel, using the pore radius profile (30). The result, 74 pS, is remarkably similar to that reported experimentally (80 pS) using a synthetic polypeptide of SARS-CoV E (9). This value is also similar to the 50–70 pS predicted for phospholamban (43), also suggested to be a cation channel, for which conductance was experimentally reported to be 100 pS (44).
We note that our results are in general inconsistent with previous reports (23,45) that suggest that ETM forms a membrane-destabilizing, totally embedded, α-helical transmembrane hairpin. Indeed, we show by DLS that ETM has no disturbing effect on lipid bilayers, and does not induce tubular morphologies.
Further, we show by H/D exchange that only 25–26 residues are inserted in the membrane, and this is inconsistent with a totally embedded ETM, expected for a hairpin model (23,45).
Also, the hairpin model predicts that F23 side chain will be at the tip of the hairpin, and located at, or near, the polar headgroups of the lipids (23,45). Our result after labeling the F residues of ETM with cyanophenylalanine favor a TM α-helical topology of ETM, because the frequency of vibration in all F residues is similar and centered around 2230 cm−1, and not closer to 2235 cm−1, which would be expected for a more polar environment.
Our dichroic data is also consistent with a regular TM α-helix, i.e., the solution to the system of equations (see Experimental Procedures) (16) for the pairs around F23, i.e., 22/23, or 23/24, converges to a sensible result when assuming α-helical canonical periodicity between consecutive residues (Δω = 100°). Equally for the pair 22/24, if Δω ∼ 200°. We also note that if the helix tilt determined previously without any isotopic label (23) was ≤6°, we should be unable to calculate ω-angles for the different residues, because the transition dipole moment of any peptidic C=O bond would have a similar orientation relative to the z axis. In other words, SSID can only provide orientational information if the tilt is at least 10°. In this work, the helix tilt found is in the range 20–30°. We conclude therefore that the topology of ETM in our preparation is not the same as the preparation used to report a TM hairpin (23,45).
One possible explanation for the discrepancy may be found in the fact that our peptide contains 2 lysines flanking ETM at both N- and C-termini, whereas these lysines were not present in the latter reports. It is possible that the presence of these positively charged lysines favors a transmembrane topology, and not a hairpin. In any case, the pentameric form we report is unlikely to be an artifact, because these TM interactions and oligomeric size are consistent with evolutionary conservation data and expression of the complete SARS-CoV E polypeptide, and also consistent with the reported ion channel activity. Clearly, more than one topology is possible for ETM, especially considering the range of attributed roles for this small protein.
Finally, although channel-like activity appears to be more compatible with a regular α-helix than with a short transmembrane hairpin, the latter may explain better the role of E protein in viral morphogenesis. However, we note that a palindromic sequence similar to the one described for E protein, and which has been suggested to be essential, is only present in the SARS sequence, but not in the rest of the homologs we have considered (12), which should presumably have a similar function.
CONCLUSION
We have obtained experimental data that shows that ETM, flanked with two lysine residues, incorporated in model lipid bilayers adopts an α-helical conformation, with a transmembrane topology, i.e., C- and N-termini in opposite sides of the lipid bilayer. By comparison with previous computational studies, our orientational data of synthetic ETM labeled isotopically is only consistent with a homopentameric model. This pentameric form could be responsible for the ion channel effects observed in vitro in the full length polypeptide of protein E.
Acknowledgments
J.T. thanks the financial support of the Biomedical Research Council of Singapore.
Jaume Torres and Krupakar Parthasarathy contributed equally to this work.
References
- 1.Rota, P. A., M. S. Oberste, S. S. Monroe, W. A. Nix, R. Campagnoli, J. P. Icenogle, S. Penaranda, B. Bankamp, K. Maher, M. H. Chen, S. Tong, A. Tamin, L. Lowe, M. Frace, J. L. DeRisi, Q. Chen, D. Wang, D. D. Erdman, T. C. Peret, C. Burns, T. G. Ksiazek, P. E. Rollin, A. Sanchez, S. Liffick, B. Holloway, J. Limor, K. McCaustland, M. Olsen-Rasmussen, R. Fouchier, S. Gunther, A. D. Osterhaus, C. Drosten, M. A. Pallansch, L. J. Anderson, and W. J. Bellini. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 300:1394–1399. [DOI] [PubMed] [Google Scholar]
- 2.Fischer, F., C. F. Stegen, P. S. Masters, and W. A. Samsonoff. 1998. Analysis of constructed E gene mutants of mouse hepatitis virus confirms a pivotal role for E protein in coronavirus assembly. J. Virol. 72:7885–7894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim, K. P., and D. X. Liu. 2001. The missing link in coronavirus assembly. Retention of the avian coronavirus infectious bronchitis virus envelope protein in the pre-Golgi compartments and physical interaction between the envelope and membrane proteins. J. Biol. Chem. 276:17515–17523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bos, E. C., W. Luytjes, H. V. van der Meulen, H. K. Koerten, and W. J. Spaan. 1996. The production of recombinant infectious DI-particles of a murine coronavirus in the absence of helper virus. Virology. 218:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Corse, E., and C. E. Machamer. 2000. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J. Virol. 74:4319–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.An, S., C. J. Chen, X. Yu, J. L. Leibowitz, and S. Makino. 1999. Induction of apoptosis in murine coronavirus-infected cultured cells and demonstration of E protein as an apoptosis inducer. J. Virol. 73:7853–7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, C. J., S. An, and S. Makino. 2001. Induction of apoptosis in murine coronavirus-infected 17Cl-1 cells. Adv. Exp. Med. Biol. 494:615–620. [DOI] [PubMed] [Google Scholar]
- 8.Liao, Y., J. Lescar, J. P. Tam, and D. X. Liu. 2004. Expression of SARS-coronavirus envelope protein in Escherichia coli cells alters membrane permeability. Biochem. Biophys. Res. Commun. 325:374–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilson, L., C. McKinlay, P. Gage, and G. Ewart. 2004. SARS coronavirus E protein forms cation-selective ion channels. Virology. 330:322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang, J., S. Kim, F. Kovacs, and T. A. Cross. 2001. Structure of the transmembrane region of the M2 protein H+ channel. Protein Sci. 10:2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adams, P. D., I. T. Arkin, D. M. Engelman, and A. T. Brunger. 1995. Computational searching and mutagenesis suggest a structure for the pentameric transmembrane domain of phospholamban. Nat. Struct. Biol. 2:154–162. [DOI] [PubMed] [Google Scholar]
- 12.Torres, J., J. Wang, K. Parthasarathy, and D. X. Liu. 2005. The transmembrane oligomers of coronavirus protein E. Biophys. J. 88:1283–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Briggs, J. A., J. Torres, and I. T. Arkin. 2001. A new method to model membrane protein structure based on silent amino acid substitutions. Proteins. 44:370–375. [DOI] [PubMed] [Google Scholar]
- 14.Torres, J., P. D. Adams, and I. T. Arkin. 2000. Use of a new label, 13C=18O, in the determination of a structural model of phospholamban in a lipid bilayer. Spatial restraints resolve the ambiguity arising from interpretations of mutagenesis data. J. Mol. Biol. 300:677–685. [DOI] [PubMed] [Google Scholar]
- 15.Torres, J., J. A. Briggs, and I. T. Arkin. 2002. Multiple site-specific infrared dichroism of CD3-zeta, a transmembrane helix bundle. J. Mol. Biol. 316:365–374. [DOI] [PubMed] [Google Scholar]
- 16.Arkin, I. T., K. R. MacKenzie, and A. T. Brunger. 1997. Site-directed dichroism as a method for obtaining rotational and orientational constraints for oriented polymers. J. Am. Chem. Soc. 119:8973–8980. [Google Scholar]
- 17.Kukol, A., P. D. Adams, L. M. Rice, A. T. Brunger, and T. I. Arkin. 1999. Experimentally based orientational refinement of membrane protein models: A structure for the Influenza A M2 H+ channel. J. Mol. Biol. 286:951–962. [DOI] [PubMed] [Google Scholar]
- 18.Forrest, L. R., A. Kukol, I. T. Arkin, D. P. Tieleman, and M. S. Sansom. 2000. Exploring models of the influenza A M2 channel: MD simulations in a phospholipid bilayer. Biophys. J. 78:55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kukol, A., and I. T. Arkin. 1999. VPU transmembrane peptide structure obtained by site-specific Fourier transform infrared dichroism and global molecular dynamics searching. Biophys. J. 77:1594–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kukol, A., and I. T. Arkin. 2000. Structure of the influenza C virus CM2 protein transmembrane domain obtained by site-specific infrared dichroism and global molecular dynamics searching. J. Biol. Chem. 275:4225–4229. [DOI] [PubMed] [Google Scholar]
- 21.Torres, J., A. Kukol, J. M. Goodman, and I. T. Arkin. 2001. Site-specific examination of secondary structure and orientation determination in membrane proteins: the peptidic 13C=18O group as a novel infrared probe. Biopolymers. 59:396–401. [DOI] [PubMed] [Google Scholar]
- 22.Wellings, D. A., and E. Atherton. 1997. Standard FMOC protocols. Methods Enzymol. 289:44–67. [DOI] [PubMed] [Google Scholar]
- 23.Arbely, E., Z. Khattari, G. Brotons, M. Akkawi, T. Salditt, and I. T. Arkin. 2004. A highly unusual palindromic transmembrane helical hairpin formed by SARS coronavirus E protein. J. Mol. Biol. 341:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayer, L. D., M. J. Hope, and P. R. Cullis. 1986. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta. 858:161–168. [DOI] [PubMed] [Google Scholar]
- 25.McGhie, E. J., P. J. Hume, R. D. Hayward, J. Torres, and V. Koronakis. 2002. Topology of the Salmonella invasion protein SipB in a model bilayer. Mol. Microbiol. 44:1309–1321. [DOI] [PubMed] [Google Scholar]
- 26.Marsh, D. 1999. Quantitation of secondary structure in ATR infrared spectroscopy. Biophys. J. 77:2630–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsuboi, M. 1962. Infrared dichroism and molecular conformation of α-form poly-γ-benzyl-L-glutamate. J. Polym. Sci. [B]. 59:139–153. [Google Scholar]
- 28.Marsh, D., M. Muller, and F. J. Schmitt. 2000. Orientation of the infrared transition moments for an alpha-helix. Biophys. J. 78:2499–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harrick, N. J. 2003. Principles of internal reflection spectroscopy. In Internal reflection spectroscopy. Harrick Scientific, Ossining, NY. 13–65.
- 30.Smart, O.S., J.G. Neduvelil, X. Wang, B.A. Wallace, and M.S. Sansom. 1996. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J Mol. Graph. 14:354–376. [DOI] [PubMed] [Google Scholar]
- 31.Zhou, F. X., H. J. Merianos, A. T. Brunger, and D. M. Engelman. 2001. Polar residues drive association of polyleucine transmembrane helices. Proc. Natl. Acad. Sci. USA. 98:2250–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choma, C., H. Gratkowski, J. D. Lear, and W. F. DeGrado. 2000. Asparagine-mediated self-association of a model transmembrane helix. Nat. Struct. Biol. 7:161–166. [DOI] [PubMed] [Google Scholar]
- 33.Byler, D. M., and H. Susi. 1986. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers. 25:469–487. [DOI] [PubMed] [Google Scholar]
- 34.Torres, J., J. A. Briggs, and I. T. Arkin. 2002. Convergence of experimental, computational and evolutionary approaches predicts the presence of a tetrameric form for CD3-zeta. J. Mol. Biol. 316:375–384. [DOI] [PubMed] [Google Scholar]
- 35.Getahun, Z., C. Y. Huang, T. Wang, B. De Leon, W. F. DeGrado, and F. Gai. 2003. Using nitrile-derivatized amino acids as infrared probes of local environment. J. Am. Chem. Soc. 125:405–411. [DOI] [PubMed] [Google Scholar]
- 36.Tucker, M. J., Z. Getahun, V. Nanda, W. F. DeGrado, and F. Gai. 2004. A new method for determining the local environment and orientation of individual side chains of membrane-binding peptides. J. Am. Chem. Soc. 126:5078–5079. [DOI] [PubMed] [Google Scholar]
- 37.Therien, A. G., and C. M. Deber. 2002. Oligomerization of a peptide derived from the transmembrane region of the sodium pump gamma subunit: effect of the pathological mutation G41R. J. Mol. Biol. 322:583–590. [DOI] [PubMed] [Google Scholar]
- 38.Melnyk, R. A., A. W. Partridge, and C. M. Deber. 2001. Retention of native-like oligomerization states in transmembrane segment peptides: application to the Escherichia coli aspartate receptor. Biochemistry. 40:11106–11113. [DOI] [PubMed] [Google Scholar]
- 39.Melnyk, R. A., A. W. Partridge, J. Yip, Y. Wu, N. K. Goto, and C. M. Deber. 2003. Polar residue tagging of transmembrane peptides. Biopolymers. 71:675–685. [DOI] [PubMed] [Google Scholar]
- 40.Collins, P. L., and G. Mottet. 1993. Membrane orientation and oligomerization of the small hydrophobic protein of human respiratory syncytial virus. J. Gen. Virol. 74:1445–1450. [DOI] [PubMed] [Google Scholar]
- 41.Kochva, U., H. Leonov, and I. T. Arkin. 2003. Modeling the structure of the respiratory syncytial virus small hydrophobic protein by silent-mutation analysis of global searching molecular dynamics. Protein Sci. 12:2668–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuo, A., J. M. Gulbis, J. F. Antcliff, T. Rahman, E. D. Lowe, J. Zimmer, J. Cuthbertson, F. M. Ashcroft, T. Ezaki, and D. A. Doyle. 2003. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 300:1922–1926. [DOI] [PubMed] [Google Scholar]
- 43.Sansom, M. S., G. R. Smith, O. S. Smart, and S. O. Smith. 1997. Channels formed by the transmembrane helix of phospholamban: a simulation study. Biophys. Chem. 69:269–281. [DOI] [PubMed] [Google Scholar]
- 44.Kovacs, R. J., M. T. Nelson, H. K. Simmerman, and L. R. Jones. 1988. Phospholamban forms Ca2+-selective channels in lipid bilayers. J. Biol. Chem. 263:18364–18368. [PubMed] [Google Scholar]
- 45.Khattari, Z., G. Brotons, M. Akkawi, E. Arbely, I. T. Arkin, and T. Salditt. 2006. SARS coronavirus E protein in phospholipid bilayers: an x-ray study. Biophys. J. 90:2038–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]