

In a recent article, Colquhoun et al. (Colquhoun, D., K. A. Dowsland, M. Beato, and A. J. Plested. 2004. Biophys. J. 86:3510–3518) present a method to impose detailed balance on a complex reaction mechanism, which relies on identifying reaction cycles. We propose a method to impose detailed balance without explicitly considering reaction cycles. In our method, the detailed balance constraint is imposed by parameterizing the model in terms of the concentrations and balanced fluxes at equilibrium. A general rate matrix satisfying detailed balance is derived from these parameters. We illustrate this technique for voltage- and ligand-dependent single molecule kinetics. In the single molecule case, when ligand binding obeys the law of mass action, we point out that our parameterization correctly gives the ligand dependence of the equilibrium probabilities without solving any equations. We also show how to impose detailed balance for general nonlinear mass-action kinetics. The techniques for obtaining a minimal reaction network subject to the detailed balance constraint are also presented and illustrated on a large network.
INTRODUCTION
The principle of microscopic reversibility implies detailed balance—the statement that, at thermodynamic equilibrium, each individual reaction is balanced. That is, at equilibrium each individual reaction occurs with equal forward and backward fluxes. A reaction system that satisfies detailed balance does not consume or dissipate free energy at thermodynamic equilibrium (1). Although reaction systems with no closed loops (or acyclic systems) always satisfy detailed balance, most complex reaction schemes involve reaction cycles, and satisfying detailed balance requires that the product of equilibrium constants around a reaction cycle equals one. Colquhoun et al. (2) recently presented methods to impose detailed balance on complex reaction mechanisms, such as ion channel kinetic schemes modeled with finite Markov chains. The methods rely on finding a fundamental cycle basis with respect to a spanning tree of a graph representing the reaction topology. In this comment, we discuss an alternative method that balances the forward and backward fluxes of each reversible reaction and does not require direct consideration of reaction cycles. The method will be discussed for single molecule dynamics and mass-action kinetics. We will also discuss techniques for identifying a minimal reaction network for imposing detailed balance.
SINGLE MOLECULE DYNAMICS
Idealized single molecules, such as an ion channel with a finite number of states, are often modeled by homogeneous Markov processes, which can in general be described by the so-called forward equation,
 |
where element pij(t) in matrix P(t) is the probability that the system is in state j at time t, provided it was in state i at time 0, and Q is called the generator matrix, whose element qij denotes the rate constant for the transition from state i to j, i ≠ j. Each diagonal entry  so that Qu = 0, where u is a column vector containing all ones. Detailed balance holds at equilibrium implying that all fluxes balance: wiqij = wjqji (3,4), where w is the equilibrium distribution of states such that wTQ = 0. Defining a diagonal matrix W = diag(w), the detailed balance conditions are written as
so that Qu = 0, where u is a column vector containing all ones. Detailed balance holds at equilibrium implying that all fluxes balance: wiqij = wjqji (3,4), where w is the equilibrium distribution of states such that wTQ = 0. Defining a diagonal matrix W = diag(w), the detailed balance conditions are written as
 |
(1) |
Imposing detailed balance on a reaction network requires that Q satisfies Eq. 1. It is evident that the matrix Qs = WQ is a symmetric generator matrix and entries in Qs are equilibrium fluxes. Q can be written in terms of Qs and W as follows:
 |
(2) |
Note that W need not be normalized for Q to obey detailed balance. Normalization can be incorporated in Qs. However, if W is normalized then it gives the equilibrium distribution and Qs gives the equilibrium fluxes. By specifying the equilibrium distribution (entries in W) and the equilibrium fluxes (entries in Qs), one can impose detailed balance on Q using the above equation, which in fact provides an alternative parameterization scheme to that of directly specifying a system using rate constants in Q. This way of specifying a system is especially useful in situations where equilibrium distribution and fluxes can be obtained from measured data. Constraints in addition to detailed balance, such as measured rate constants or dependence between rate constants, which can be expressed by equations that are linear with respect to the logarithms of rate constants, are also linear with respect to the logarithms of the components of Qs and W. Therefore, the technique to calculate constrained parameters in terms of free parameters using linear equations proposed by Colquhoun et al. (2) and Qin et al. (5) also applies.
In practice, a model reaction scheme is given a priori. The topology of this scheme can be encoded within Qs (i.e., a nonzero entry indicates a reaction and a zero entry indicates no reaction), and topology is preserved in Eq. 2. In the case of finding a Q matrix that best fits data, via a maximum likelihood method (6), Eq. 2 can be substituted directly into the likelihood formula. The following outlines a procedure for using Eq. 2 in a maximum likelihood method (assuming no other constraints):
Specify a symmetric generator Qs with arbitrary positive entries consistent with a given reaction scheme. Specify an arbitrary equilibrium distribution vector w.
Calculate Q = W−1Qs, where W = diag(w).
For fitting data using a maximum likelihood method, maximize the likelihood function by adjusting w and fluxes in Qs iteratively, until satisfactory parameters are found.
The values Qs and w can be enforced to be positive during the course of model fitting by using constrained optimization or following Qin et al. (5).
Parameter dependence: ligand concentration, voltage
The above program can be carried out in the case that there is dependence on other physical parameters such as ligand concentration, pressure, temperature, voltage, etc. In principle, one can choose Qs and W to be any desired functions of the parameters.
Note that if detailed balance is to be satisfied for all physical parameters, the ratio of the product of rate constants one way around any loop to the product the other way around the loop must necessarily be independent of parameter values. Here we give a physically motivated prescription for enforcing detailed balance, without considering loops, for reactions with exponential voltage dependence and ligand dependence governed by the law of mass action. Mass action reactions have monomial ligand dependence, so in the case of a single ligand with concentration [L],
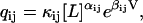 |
where αij are integers, and κij and βij are constants. This formulation can be made to enforce positivity of qij (which is sometimes not desirable (7)) by replacing κij with  (5). From Eq. 2, we know that under detailed balance, each rate constant qij is the ratio of the equilibrium flux
(5). From Eq. 2, we know that under detailed balance, each rate constant qij is the ratio of the equilibrium flux  to the equilibrium probability wi. Parameterizing
to the equilibrium probability wi. Parameterizing 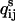 as
as  (
( ) and wi as
) and wi as 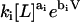 results in the factorization
results in the factorization
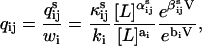 |
(3) |
where 
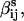 and
and  are symmetric in i and j. This equation provides a formula for imposing detailed balance in a model with ligand- and voltage-dependence. If positivity is desired,
are symmetric in i and j. This equation provides a formula for imposing detailed balance in a model with ligand- and voltage-dependence. If positivity is desired,  and ki can be written as exponentials (5):
and ki can be written as exponentials (5): 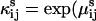 ) and
) and 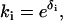 so that ln(qij) =
so that ln(qij) = 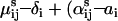 ) ln(L) + (
) ln(L) + (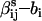 )V. For mass-action kinetics, ai is the number of ligands bound in the ith state and
)V. For mass-action kinetics, ai is the number of ligands bound in the ith state and 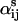 is the total number of ligands present in the reversible reaction from state i to state j. (To be specific, if the state i has p ligands bound and state j has p + r ligands bound and the reaction is
is the total number of ligands present in the reversible reaction from state i to state j. (To be specific, if the state i has p ligands bound and state j has p + r ligands bound and the reaction is 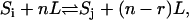 then
then 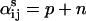 for mass action.) The bi and βij are the net charge displacements along the electric field in state i and the transition state between states i and j, respectively. With this formulation, the wi values are specified in terms of explicit parameters of the model, thus giving the equilibrium probabilities, up to a normalization scalar that is introduced below. The case of more than one species of ligand is handled similarly and treated in the following example.
for mass action.) The bi and βij are the net charge displacements along the electric field in state i and the transition state between states i and j, respectively. With this formulation, the wi values are specified in terms of explicit parameters of the model, thus giving the equilibrium probabilities, up to a normalization scalar that is introduced below. The case of more than one species of ligand is handled similarly and treated in the following example.
We illustrate how to impose detailed balance on the de Young-Keizer model (8) of the IP3 receptor, which is a calcium channel that binds two ligands, Ca2+ and IP3. The model, which has six fundamental cycles, is shown in Fig. 1.
FIGURE 1.

State transition model of an IP3 receptor in the endoplasmic reticulum with calcium dependence. The ion channel is gated by two ligands: calcium ([Ca2+]) and inositol 1,4,5-trisphosphate ([IP3]).
The equilibrium vector with proper ligand dependence is simply written down according to the prescription,
 |
where the ligand concentrations [Ca2+] and [IP3] are denoted as C and I, respectively, and where aiC and aiI are the number of calcium ions and IP3 molecules, respectively, bound in state i(i = 0, …, 7). For the de Young-Keizer model,
 |
where η is the normalization scalar defined by 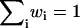 :
:
 |
Note that we have arrived at the formula for wi, including w6 = k6IC, which exhibits the celebrated bell-shaped calcium-dependence of the open probability in the de Young-Keizer model (8), without actually solving any equations.
For mass-action kinetics involving noncatalytic ligands, the exponents  which give the ligand dependence of Qs, obey
which give the ligand dependence of Qs, obey
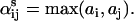 |
(4) |
Symmetry of αs follows from the symmetry of the max function. For models with multiple ligands, Eq. 4 holds for each ligand separately. For the de Young-Keizer model,
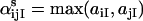 |
 |
The fluxes are then given by
 |
where the diagonal entries xi are minus the sum of the other elements in each row, as is required for a generator.
DEGREES OF FREEDOM
To calculate the number of free parameters in Eq. 2, let Ns and Nr denote the numbers of states and reversible reactions (i.e., the number of links in the state-transition diagram), respectively. Without any constraint, the transition matrix Q contains 2Nr free parameters. If detailed balance is the only physical constraint that the system must obey, the number of free parameters is Nr + Ns – 1 because there are Nr free parameters in the symmetric matrix Qs and Ns – 1 in w (with a normalization constraint wTu = 1). Colquhoun et al. (2) obtained the same number of free parameters by counting the number of connections constrained by detailed balance around a set of fundamental cycles ((2), their Eq. 9). Detailed balance still holds for Q even if w is not normalized because a multiplication constant that normalizes w can always be factored out of Qs. Here we consider the example shown in Colquhoun et al. ((2), their Fig. 3). For that scheme, Q can be written as
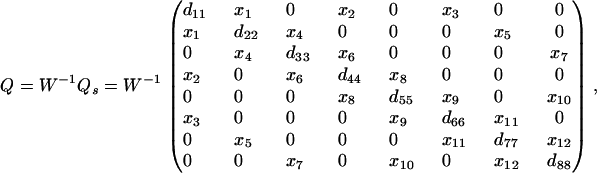 |
where each xi denotes a nonzero entry. The diagonal entries dii assure that every row sums to zero. The parameters are the xi and the wi. In agreement with Colquhoun et al. (2), W and Qs have 19 free parameters: the 12 xi and 7 independent wi.
MASS-ACTION KINETICS
Here we consider a general biochemical network with mass-action rate laws. An idealized reaction model is implied, i.e., all reactions proceed in an isothermal and well-mixed container of a constant volume. Whereas before we treated ligand concentrations as parameters, now the concentration of each participant is a dynamic variable. A network has a set of m biochemical species (e.g., molecules, proteins and protein complexes, etc.) x = {xi|i = 1, .., m}. A reaction-group G is defined as a set of species participating in a reaction, either as reactants or products. Any reaction within the network is a process that converts reactants into products, and a unidirectional reaction is denoted as a group transition: Gr → Gp. Note that reactants in one reaction could be products in another. Assuming the network has n distinct reaction groups, we can compile information about stoichiometry into the m × n stoichiometric matrix S. The components sij are the stoichiometric coefficients for species i in reaction group j (note that sij ≥ 0). All the reaction groups can be listed using an n by 1 column vector 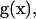 where each entry
where each entry 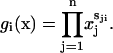 A biochemical network is characterized by kinetic rate constants for the reactions. This kinetic information can be compiled into an n × n kinetics matrix K, where kij(i ≠ j) is the kinetic rate constant for the reaction that converts reaction group i into group j. For a mass-action rate law, gi(x)kij is the corresponding reaction rate. The diagonal entry kii of K consolidates rate constants for all the outgoing fluxes from reaction group gi(x), i.e.,
A biochemical network is characterized by kinetic rate constants for the reactions. This kinetic information can be compiled into an n × n kinetics matrix K, where kij(i ≠ j) is the kinetic rate constant for the reaction that converts reaction group i into group j. For a mass-action rate law, gi(x)kij is the corresponding reaction rate. The diagonal entry kii of K consolidates rate constants for all the outgoing fluxes from reaction group gi(x), i.e.,  The flux rates for all the groups can be expressed as a vector f = KTg(x) and the whole system can be described by the following dynamic equation,
The flux rates for all the groups can be expressed as a vector f = KTg(x) and the whole system can be described by the following dynamic equation,
 |
(5) |
This description of general mass-action kinetics was developed by Horn and Jackson (9). Notations adopted here are similar to those of Chaves et al. (10). The detailed balance constraint can be described as gi(xeq)kij = gi(xeq)kji, where xeq denotes the equilibrium concentrations. The constraint can also be written in matrix form as
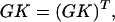 |
where G = diag(g(xeq)). Thus,
 |
(6) |
where Ks = GK is a symmetric matrix. To impose detailed balance on this system using the method described above, one can first specify the equilibrium concentration vector xeq, subsequently construct vector g(xeq), and then use Eq. 6 to calculate K. Analogous to the analysis for single molecule dynamics one can use steady-state data to estimate free parameters in G and K. A simple example of a chemical reaction model with a single reaction cycle is shown in Fig. 2. If we only consider the reaction scheme in the dashed box, we have the chemical species vector x = [A B C D]T. The stoichiometric matrix is
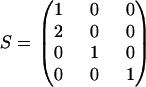 |
and the rate matrix is
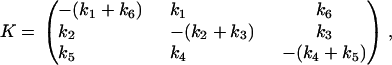 |
and g(x) = [AB2 C D]T. For a specified equilibrium vector of concentrations xeq = [Aeq Beq Ceq Deq], we have
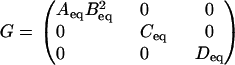 |
If Ks is constructed as
 |
then by Eq. 6 the rate constant matrix that satisfies detailed balance is
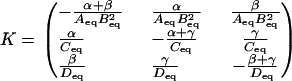 |
As can be easily confirmed, this matrix satisfies the detailed balance constraint derived from the loop-based approach, k1k3k5 = k2k4k6.
FIGURE 2.

A hypothetical chemical reaction model with one cycle. The network in the dashed box is the minimal network upon which the detailed balance must be imposed.
MINIMAL REACTION SCHEME
All chemical reactions obey detailed balance because chemistry obeys the principle of microscopic reversibility. However, for systems that are out of equilibrium, detailed balance is violated because of nonequilibrium constraints. Many biochemical reaction systems are open systems that contain irreversible and energy-driven reactions (e.g., protein synthesis, degradation, and phosphorylation reactions, transport across a system boundary, etc.). Still, as a physical law, detailed balance must be obeyed by all reversible and non-energy-driven reactions at equilibrium. The consequence is that, in a model, detailed balance must be imposed for any non-energy-driven reaction cycle to satisfy the physical constraint even if a system may always function away from equilibrium due to constant energy dissipation or externally imposed fluxes. To impose detailed balance on the relevant part of a reaction network, it is necessary to distinguish irreversible energy-driven reactions from non-energy-driven reversible reactions. We take this information as given in the model specification.
We note that reversible reactions outside any reaction loop trivially satisfy detailed balance. Rate constants for these noncyclic reactions need not be part of an imposed detailed balance constraint. Therefore, the reaction scheme on which we need to impose detailed balance may be much smaller than the full scheme. As an example, let us consider the overall reaction scheme in Fig. 2. We can construct a binary matrix T encoding only the reaction scheme as
 |
The vector g(x) = [AB2 C D E F]T. To obtain the minimal reaction scheme that must satisfy detailed balance, one can remove the irreversible reaction E → A + 2B by setting T41 = 0 and remove the reversible acyclic reactions by setting T52 = 0 and then T25 = 0. The minimal scheme is enclosed in the dashed box in Fig. 2.
Now we discuss how to automate the process of finding the minimal reaction scheme for large networks. Given a binary matrix T encoding the overall reaction scheme (i.e., tij = 1 if there is a transition from state i to j or from reaction group i to j in mass-action networks, otherwise tij = 0) and a binary matrix Te encoding all energy-driven reactions, we can obtain the minimal reaction scheme for imposing detailed balance by removing three types of reactions:
Remove all energy-driven reactions, by subtracting Te from T, i.e., T′ = T – Te.
Remove all the irreversible reactions, by setting T′ij = 0 if T′ij ≠ 0 and T′ji = 0. We denote the resulting matrix as T″.
Remove all the reversible acyclic reactions. We note that a state (or a reaction group in a mass action network) that is connected to only one other state is a state in an acyclic reaction and it can be removed from the full scheme. For any row i in T″, we set t″ij = 0 and t″ji = 0 if t″ij is the only nonzero entry in the row. Remove all such states iteratively until states in the resulting matrix
 are connected to at least two other states.
are connected to at least two other states.
The above procedure, however, cannot remove acyclic reactions that bridge disjoint cycles. Even though the number of such reactions is probably small in most biochemical reaction networks (e.g., it appears that there is no such acyclic reaction in the example that follows), these reactions can be removed by the following numerical method.
It is clear that acyclic reactions are always balanced at steady-state. Letting Q be an arbitrary generator consistent with the reduced scheme  we can solve wTQ = 0 for the steady-state distribution w, and any reaction that satisfies wiqij = wjqji is an acyclic reaction and can be removed. With extremely low probability, reactions in cycles might be balanced by chance and erroneously removed because Q is arbitrary and w is solved numerically. To eliminate such cases, one can run the procedure multiple times to increase the confidence and can always verify whether detailed balance is satisfied by the full scheme. We note that
we can solve wTQ = 0 for the steady-state distribution w, and any reaction that satisfies wiqij = wjqji is an acyclic reaction and can be removed. With extremely low probability, reactions in cycles might be balanced by chance and erroneously removed because Q is arbitrary and w is solved numerically. To eliminate such cases, one can run the procedure multiple times to increase the confidence and can always verify whether detailed balance is satisfied by the full scheme. We note that 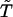 may contain disjoint schemes. Thus, the null space of Q may have more than one dimension. In such cases, one can construct w as a linear combination of the basis of the null space and ensure it contains no zero entries.
may contain disjoint schemes. Thus, the null space of Q may have more than one dimension. In such cases, one can construct w as a linear combination of the basis of the null space and ensure it contains no zero entries.
We have applied the above methods to find the minimal reaction network in a recently developed model for signaling by epidermal growth factor receptor (EGFR) (11). The data file and the MatLab code for this example are available online as Supplementary Material. The model contains 356 biochemical species, 1667 reaction groups, and 3749 unidirectional reactions. This model is generated by a rule-based and automated reaction network generator program called BioNetGen (12). The model has 701 energy-driven protein phosphorylations and dephosphorylations and 2460 acyclic reactions. The minimal reaction network contains 588 unidirectional reactions in cycles and 258 reaction groups, a much reduced reaction scheme. It is worth noting that the minimal network of the EGFR model contains 34 disjoint schemes (the number of the null space dimension of the minimal K matrix). In contrast to our method, a cycle-based approach would need to identify all the disjoint schemes and a fundamental cycle basis would need to be found for each one.
CONCLUSION
Imposing detailed balance in a complex reaction mechanism using the previously proposed method (2) involves techniques to identify a cycle basis in the reaction scheme. It has been shown that finding a minimum cycle basis (which gives rise to simpler equations than does an arbitrary fundamental cycle basis) has computational complexity O((m + n)3.376) (13), where m and n denote the number of undirected edges (reversible transitions or reactions) and the number of vertices (states or reaction groups), respectively. For some applications with small networks, this cost may not be significant or rate-limiting. The computational cost will be high for finding reaction cycles in a large network such as that of the EGFR model considered above. In this article, we present an alternative method of imposing detailed balance for both single molecule kinetics and nonlinear mass-action kinetics. The method provides a parameterization procedure for reaction rate constants without explicitly considering reaction cycles. We give a method to identify the minimal reaction scheme for imposing detailed balance whenever a system contains energy-driven, irreversible, and acyclic reversible reactions. For single molecule kinetics with exponential voltage dependence and mass-action ligand binding, we have shown how to fully parameterize the generator to satisfy detailed balance. This parameterization explicitly gives the correct ligand- and voltage-dependence for the equilibrium states.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank James R. Faeder for providing the reaction scheme data for the EGFR model.
This work was supported by National Institutes of Health grants No. GM65830 and No. RR18754 and Department of Energy contract No. W-7405-ENG-36.
References
- 1.Alberty, R. A. 2004. Principle of detailed balance in kinetics. J. Chem. Edu. 81:1206–1209. [Google Scholar]
- 2.Colquhoun, D., K. A. Dowsland, M. Beato, and A. J. Plested. 2004. How to impose microscopic reversibility in complex reaction mechanisms. Biophys. J. 86:3510–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kolmogorov, A. 1936. Theory of Markov chains. Annal. Mathematics. 112:155–160. [Google Scholar]
- 4.Fredkin, D. R., M. Montal, and J. Rice. 1985. Identification of aggregated Markovian models: application to the nicotinic acetylcholine receptor. Proc. Berkeley Conf. in Honor of Jerzy Neyman and Jack Kiefer. 1:269–289. [Google Scholar]
- 5.Qin, F., A. Auerbach, and F. Sachs. 1996. Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys. J. 70:264–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qin, F., A. Auerbach, and F. Sachs. 1997. Maximum likelihood estimation of aggregated Markov processes. Proc. R. Soc. Lond. B Biol. Sci. 264:375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruno, W. J., J. Yang, and J. E. Pearson. 2005. Using independent open-to-closed transitions to simplify aggregated Markov models of ion channel gating kinetics. Proc. Natl. Acad. Sci. USA. 102:6326–6331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Young, G., and J. Keizer. 1992. A single-pool inositol 1,4,5-trisphosphate-receptor-based model for agonist-stimulated oscillations in Ca2+ concentration. Proc. Natl. Acad. Sci. USA. 89:9895–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horn, F., and R. Jackson. 1972. General mass action kinetics. Arch. Rational Mech. Anal. 47:81–116. [Google Scholar]
- 10.Chaves, M., E. D. Sontag, and R. J. Dinerstein. 2004. Steady-states of receptor-ligand dynamics: a theoretical framework. J. Theor. Biol. 227:413–428. [DOI] [PubMed] [Google Scholar]
- 11.Blinov, M. L., J. R. Faeder, B. Goldstein, and W. S. Hlavacek. 2006. A network model of early events in epidermal growth factor receptor signaling that accounts for combinatorial complexity. Biosystems. 83:136–151. [DOI] [PubMed] [Google Scholar]
- 12.Blinov, M. L., J. R. Faeder, B. Goldstein, and W. S. Hlavacek. 2004. BioNetGen: software for rule-based modeling of signal transduction based on the interactions of molecular domains. Bioinformatics. 20:3289–3291. [DOI] [PubMed] [Google Scholar]
- 13.Golynski, A., and J. D. Horton. 2002. A polynomial time algorithm to find the minimum cycle basis of a regular matroid. Lect. Notes Comput. Sci. 2368:200–209. [Google Scholar]