

Abstract
The physical forces that underlie the exclusion of solutes from macromolecular surfaces can be probed in a similar way as the measurement of forces between macromolecules in condensed arrays using the osmotic stress technique and x-ray scattering. We report here the dependence of alcohol exclusion or, equivalently, the preferential hydration of DNA on the spacing between helices in condensed arrays. The actual forces describing exclusion are quite different from the commonly assumed steric crowding coupled with weak binding. For a set of 12 nonpolar alcohols, exclusion is due to repulsive hydration interactions with the charged DNA surface. Exclusion amplitudes do not depend simply on size, but rather on the balance between alkyl carbons and hydroxyl oxygens. Polyols are included at very close spacings. The distance dependence of polyol inclusion, however, is quite different from nonpolar alcohol exclusion, suggesting the underlying mechanism of interaction is different.
INTRODUCTION
The exclusion of small solutes from macromolecular surfaces can strongly affect protein stability, ligand binding, recognition reactions, and conformational changes (1–6). Energies of exclusion can be quite substantial. Despite the many measurements, the physical basis of exclusion is still unclear. Many theories assume a steric exclusion perhaps balanced by weak specific binding, see, e.g., Schurr et al. (7) and references therein for an excellent discussion. We have previously probed the distance dependence characterizing the exclusion of 2-propanol and 2-methyl-2,4-pentanediol (MPD) from spermidine (Spd3+) condensed DNA arrays (8) and of several salts and polar solutes from the hydrophobically modified polymer hydroxypropyl cellulose, HPC, (9). We concluded that water structuring forces dominate the interaction between these solutes and macromolecules. These hydration forces are manifested in an exponentially varying solute concentration gradient with an approximate 3–4 Å decay length that reflects a water-water correlation length. This functional form results in a “preferential hydration” where the number of included waters or, equivalently, the solute partition coefficient is insensitive to the bulk concentration of solute but varies substantially with the chemical nature and size of the solute and of the macromolecular surface probed. The energy associated with exclusion can be calculated as a Π-V work, the solute osmotic pressure acting on the included water. We also showed that the number of included water molecules extracted using this method is consistent with the dependence of the critical Spd3+ concentration necessary for DNA precipitation from dilute solution on 2-propanol or MPD concentration and with the dependence of the cloud point temperature of HPC on salt concentration.
We infer changes in solute exclusion from the changes in spacing between DNA helices in condensed arrays as the solute concentration and the osmotic pressure of a polymer that is excluded from the DNA phase are varied, using a Maxwell relationship of the Gibbs-Duhem equation. The osmotic stress technique has been used to measure forces between several biomacromolecules in condensed arrays, including DNA, collagen, lipid bilayers, and several types of polysaccharides (10–17). It was the commonality of observed forces at high osmotic pressures and small spacings (the last 10–15 Å of surface separation) for systems that are charged, zwitterionic, and uncharged polar or nonpolar, which led to the conclusion that the energetics of structuring water in confined spaces dominates the interactions between macromolecules.
We extend our previous observations to compare the exclusion of MPD from cobaltic hexammine ( )-, Spd3+-, and Na+-DNA arrays. Even though the forces between DNA helices are very different, helices are attractive in
)-, Spd3+-, and Na+-DNA arrays. Even though the forces between DNA helices are very different, helices are attractive in  and Spd3+, but repulsive in Na+, the exclusion of MPD is strikingly similar for all of these DNA arrays. MPD interacts with the DNA surface directly rather than, for example, simply changing DNA-DNA forces through the dielectric constant.
and Spd3+, but repulsive in Na+, the exclusion of MPD is strikingly similar for all of these DNA arrays. MPD interacts with the DNA surface directly rather than, for example, simply changing DNA-DNA forces through the dielectric constant.
We also measure the exclusion magnitudes of 15 alcohols from Spd3+-DNA assemblies to parse the relative contributions of solute size and chemical nature. Our previous measurements showed that MPD was about twice as excluded as 2-propanol and is also twice the molecular weight (8). However, size did not seem to be the critical parameter since glycerol, which is intermediate in size, interacts only weakly with DNA. With this extended set of alcohols we confirm that size alone plays a negligible role in exclusion within the range of interhelical spacings examined. Rather, it is the relative numbers of nonpolar alkyl carbons and polar hydroxyl groups that determines the magnitude of the interaction. The exclusion of all the nonpolar alcohols examined show a similar 3.5–4 Å decay length for the exponential concentration gradient.
The polyols, glycerol, threitol, and sorbitol, show a much different behavior. There is a slight inclusion of glycerol at small interhelical spacings, whereas the inclusion magnitude grows substantially larger for threitol and sorbitol. Since inclusion still varies linearly with polyol concentration, it is not classical binding. If analyzed as an exponential, the distance dependence of sorbitol and threitol inclusion would have ∼1 Å decay length. We postulate that inclusion of these polyols minimizes the unfavorable energies associated with the distorted hydrogen-bonded network of water confined in small spaces.
METHODS AND MATERIALS
Materials
High molecular weight chicken blood DNA was prepared as described previously (18). Polyethylene glycol (molecular weight (MW) 8000), spermidine·3HCl, t-butanol, 1,4 butanediol, sorbitol, and 2-methyl-2,4-pentanediol were purchased from Fluka Chemical (Buchs, Switzerland) (micro select grade). Ethanol, ethylene glycol, glycerol, and 2-propanol were purchased from J T Baker (Phillipsburg, NJ) (analytic grade). Methanol, 2-butanol, 2,3-butanediol, 1,2,4-butanetriol, 1,3-propanediol, 1,2 propanediol, and threitol were purchased from the Sigma-Aldrich Chemical (St. Louis, MO). Cobaltic hexammine trichloride was purchased from the Eastman Kodak (Rochester, NY). All chemicals were used without further purification.
Osmotic stress
The method for direct force measurement by osmotic stress has been described in detail by (19). In brief, condensed DNA arrays are equilibrated against a bathing polymer solution, typically polyethylene glycol (PEG), of known osmotic pressure. PEG is excluded from the condensed DNA phase and applies a force on it. Water, salt, and small solutes are free to exchange between the PEG and condensed DNA phases. After equilibrium is achieved, the osmotic pressures in both phases are the same, as necessarily are the chemical potentials of all permeating species. The interhelical spacing, Dint, can be determined as a function of the applied PEG osmotic stress by Bragg scattering of x-rays.
 and Spd3+ precipitated DNA were prepared by slowly adding the trivalent ion in 0.2 mM steps with mixing to a 1 mg/ml (∼3 mM DNA-phosphate) DNA solution to a final nominal concentration of ∼2 mM. Condensed DNA samples for NaBr/PEG were prepared by ethanol precipitating DNA from sodium acetate solutions. DNA pellets (∼0.2–0.3 mg) were equilibrated against ∼1 ml PEG solutions containing varying concentrations of alcohols and 2 mM Spd3+, 2 mM
and Spd3+ precipitated DNA were prepared by slowly adding the trivalent ion in 0.2 mM steps with mixing to a 1 mg/ml (∼3 mM DNA-phosphate) DNA solution to a final nominal concentration of ∼2 mM. Condensed DNA samples for NaBr/PEG were prepared by ethanol precipitating DNA from sodium acetate solutions. DNA pellets (∼0.2–0.3 mg) were equilibrated against ∼1 ml PEG solutions containing varying concentrations of alcohols and 2 mM Spd3+, 2 mM  , 1.2 M NaBr, or 20 mM NaBr, all in 10 mM TrisCl (pH 7.5) at room temperature for ∼2–3 weeks with two changes of PEG solution and occasional mixing. Osmotic pressures of the PEG/alcohol solutions were measured directly using a Vapro Vapor Pressure Osmometer (model 5520, Wescor, Logan, UT).
, 1.2 M NaBr, or 20 mM NaBr, all in 10 mM TrisCl (pH 7.5) at room temperature for ∼2–3 weeks with two changes of PEG solution and occasional mixing. Osmotic pressures of the PEG/alcohol solutions were measured directly using a Vapro Vapor Pressure Osmometer (model 5520, Wescor, Logan, UT).
X-ray scattering
An Enraf-Nonius Service (Bohemia, NY) fixed copper anode Diffractis 601 x-ray generator (National Institutes of Health, Bethesda, MD) equipped with double focusing mirrors (Charles Supper, Natick, MA) was used for x-ray scattering. DNA samples were sealed with a small amount of equilibrating solution in the sample cell, and then mounted into a temperature-controlled holder at 20°C. A helium filled Plexiglas cylinder with Mylar windows was between the sample cell and image plate, a distance of ∼16 cm. Diffraction patterns were recorded by direct exposure of Fujifilm BAS image plates and digitized with a Fujifilm BAS 2500 scanner. The images were analyzed using the FIT2D (copyright A. P. Hammersley, European Synchrotron Radiation Facility) and SigmaPlot 9.01 (SPSS, Chicago, IL) software programs. The sample to image plate distance was calibrated using powdered p-bromobenzoic acid. Mean pixel intensities between scattering radii r − 0.05 mm and r + 0.05 mm averaged over all angles of the powder pattern diffraction, 〈I(r)〉, were used to calculate integrated radial intensity profiles, 2πr〈I(r)〉. The sharp, intense ring corresponds to interaxial Bragg diffraction from DNA helices packed in a hexagonal array. X-ray scattering patterns were reproducible over at least several months of storage. No sample degradation was apparent.
Critical Spd3+ concentrations for DNA precipitation
The critical concentration of Spd3+ necessary for the precipitation of DNA from dilute solution was determined as described basically in (20). A series of DNA samples were prepared with varying Spd3+ concentration but fixed solute concentration in 0.1 M NaCl, 10 mM TrisCl (pH 7.5). The DNA concentration was ∼15 μM basepairs in 1 ml total volume. After incubating at room temperature for ∼2 h, the solution was centrifuged at ∼16,000 × g for 10 min and the absorbance at 260 nm of the supernatant measured. The Spd3+ concentration was varied around the critical concentration, taken at half loss of absorbance, in steps of 0.25 mM.
Thermodynamics
The thermodynamic analysis of the effects of solutes on the forces between DNA helices has been developed in more detail elsewhere (8,9). We only briefly outline the results here. A macroscopic phase of ordered DNA helices is in equilibrium with a solution of salt, osmolyte, and a polymer as PEG that is excluded from the DNA phase. The salt and osmolyte are free to equilibrate between the DNA and bulk solution phases. We consider that PEG simply applies an osmotic pressure, ΠPEG, on the DNA phase. A difference in solute concentration between the bulk solution and DNA phase can be equivalently analyzed as the solute contribution to the osmotic pressure, Πsolute, acting on an excess or deficit number of water molecules in the DNA phase, Γw, or the solute chemical potential acting on an excess or deficit number of solute molecules in the DNA phase. Since we observe that Γw is constant with changing solute concentration, we focus on the contribution of solute to osmotic pressure. The Gibbs-Duhem equation becomes
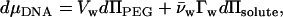 |
(1) |
where μDNA is the DNA chemical potential per basepair, Vw is the volume of water per basepair, and 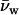 is the molecular volume of water (assumed 30 Å3). Since forces between DNA helices are insensitive to salt concentration under the experimental conditions examined, we have neglected the contribution of salt to the Gibbs-Duhem equation. The number of excess water molecules is given by the difference in concentration between the bulk solution and DNA phase. If the DNA phase contains Ns and Nw solute and water molecules per basepair, respectively, and the bulk solution contains a ratio ns/nw of solute/water molecules, then Γw per basepair is
is the molecular volume of water (assumed 30 Å3). Since forces between DNA helices are insensitive to salt concentration under the experimental conditions examined, we have neglected the contribution of salt to the Gibbs-Duhem equation. The number of excess water molecules is given by the difference in concentration between the bulk solution and DNA phase. If the DNA phase contains Ns and Nw solute and water molecules per basepair, respectively, and the bulk solution contains a ratio ns/nw of solute/water molecules, then Γw per basepair is
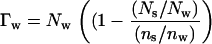 |
(2) |
The ratio (Ns/Nw)/(ns/nw) is the solute partition coefficient defined by (21).
Rearrangement of the Maxwell relation of Eq. 1 gives the change in the number of excess waters as helices move closer as a function of the change in PEG osmotic pressure needed to maintain constant Vw as the solute osmotic pressure is varied:
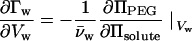 |
(3) |
for hexagonal packing of helices with spacing Dint, dVw = √3 L Dint dDint, where L is rise per basepair (= 3.4 Å for B-form DNA). We have previously observed that ΠPEG and Πsolute are linearly interdependent at constant Vw for the exclusion of MPD and 2-propanol from Spd3+-DNA arrays. For a linear interdependence, the slope ∂ΠPEG/∂Πsolute is simply given by the solute osmotic pressure, Π0, and the difference in PEG osmotic pressures at a constant interhelical spacing with and without added solute, the apparent Πexcess. For a concentration m of solute
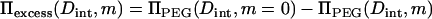 |
(4) |
and
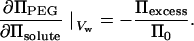 |
(5) |
A linear interdependence of ΠPEG and Πsolute at constant Dint means Πexcess/Π0 will be insensitive to osmolyte concentration. The number of excess waters, Γw, can be calculated by integrating Eq. 3.
By vapor pressure osmometry measurements, we found the osmotic pressures of mixtures of PEG and ethylene glycol, glycerol, butanediol, propanediol, butanetriol, or MPD are additive to within ∼25% or less at 40% PEG, the highest concentration examined. We assume that osmotic pressures of PEG and the more volatile solutes that we cannot measure by vapor pressure osmometry are also additive. Osmotic pressures with threitol and sorbitol are more nonideal reaching ∼50–60% excess pressure in 40% PEG and 1 molal sorbitol. We assume that to within 10%, this excess pressure results entirely from an increase in solute activity, due to an exclusion from PEG, as we observed for salt-PEG mixtures (9). For  -DNA and Spd3+-DNA arrays, the data for these solutes in the absence of PEG overlaps the data with added PEG suggesting we have correctly accounted for PEG-solute interactions. We neglect any effect of 1.2 M NaBr on MPD activity. We found previously that PEG and salt osmotic pressures are additive at high concentrations to within ∼10% (see supporting information available in Chik et al. (9)).
-DNA and Spd3+-DNA arrays, the data for these solutes in the absence of PEG overlaps the data with added PEG suggesting we have correctly accounted for PEG-solute interactions. We neglect any effect of 1.2 M NaBr on MPD activity. We found previously that PEG and salt osmotic pressures are additive at high concentrations to within ∼10% (see supporting information available in Chik et al. (9)).
RESULTS
Differences in the interactions of DNA helices with water and solutes will necessarily result in a different concentration of solute in the vicinity of DNA relative to the bulk solution. Changes in the excess or deficit of solutes, or water, near DNA must then also consequently affect forces between helices as they approach. Thermodynamic forces between DNA helices in condensed arrays can be inferred from the dependence of interhelical spacing, measured with x-ray scattering, on the osmotic pressure of a polymer as PEG that is excluded from the DNA phase. As the spacing between DNA helices in condensed arrays changes, there will be a change in the excess number of solute and water molecules associated with the DNA phase. The Gibbs-Duhem equation provides a framework to relate changes in the number of excess water or solute associated with the condensed DNA array to changes in spacing between helices at constant PEG osmotic pressure as the solute concentration is varied (Eq. 3).
Fig. 1 A shows thermodynamic force curves for DNA equilibrated against 1.2 M NaBr and 2 mM Co(NH3)6Cl3, with and without added 1 molal MPD. DNA spontaneously assembles in  , and helices only swell to an interaxial spacing of 27.75 Å in the absence of PEG osmotic pressure. In 1.2 M NaBr, two force regimes are observed. At high osmotic pressures, interhelical forces appear to increase exponentially with decreasing spacing with a characteristic 3–4 Å decay length. Force amplitudes in this region are relatively insensitive to salt concentration. At lower osmotic pressures, DNA-DNA interactions are dominated by the entropic fluctuations of helices confined by intermolecular forces. For salt concentrations above ∼1 M, force amplitudes are insensitive to ionic strength with an exponential decay length ∼7 Å. Typical x-ray scattering profiles for DNA condensed by 2 mM Co(NH3)6Cl3, with and without added 1 molal MPD, are illustrated in Fig. 1 B.
, and helices only swell to an interaxial spacing of 27.75 Å in the absence of PEG osmotic pressure. In 1.2 M NaBr, two force regimes are observed. At high osmotic pressures, interhelical forces appear to increase exponentially with decreasing spacing with a characteristic 3–4 Å decay length. Force amplitudes in this region are relatively insensitive to salt concentration. At lower osmotic pressures, DNA-DNA interactions are dominated by the entropic fluctuations of helices confined by intermolecular forces. For salt concentrations above ∼1 M, force amplitudes are insensitive to ionic strength with an exponential decay length ∼7 Å. Typical x-ray scattering profiles for DNA condensed by 2 mM Co(NH3)6Cl3, with and without added 1 molal MPD, are illustrated in Fig. 1 B.
FIGURE 1.

Effect of MPD on DNA force curves. (A) The spacing between DNA helices, Dint, measured by x-ray scattering is shown as a function of the osmotic pressure of PEG, ΠPEG, acting on the condensed DNA array. The salt concentration was 1.2 M NaBr for shaded squares and solid squares and 2 mM Co(NH3)6Cl3 for shaded circles and solid circles, both with 10 mM TrisCl, pH 7.5, at 20°C. The MPD concentration was 0 in solid squares and solid circles and 1 molal in shaded squares and shaded circles. The arrows indicate the equilibrium interhelical spacing in the absence of PEG for Co3+-DNA with and without added MPD. Changes in the number of water molecules in the DNA phase that exclude MPD can be determined from the change in interhelical spacing as the MPD concentration is varied at constant ΠPEG. An excess MPD osmotic pressure can be calculated from the difference in PEG osmotic pressures with and without added MPD at a constant spacing. (B) The x-ray scattering profiles of DNA condensed with 2 mM Co(NH3)6Cl3 in the absence of PEG and with an MPD concentration of 0 (solid line) and 1 molal (dashed line), both with 10 mM TrisCl, pH 7.5, at 20°C. MPD exclusion creates an osmotic pressure on the Co3+-DNA array that results in a 0.9 Å decrease in Dint, as observed by the shift in the Bragg scattering peak maximum to higher scattering vector, q.
With added MPD, interaxial spacings decrease at constant ΠPEG for both  -DNA and Na+-DNA. We previously have observed a linear interplay between ΠPEG and ΠMPD with Spd3+-DNA to maintain constant spacing (8). This linearity means that the change in the number of excess waters included in the DNA phase is independent of solute concentration. An apparent excess osmotic pressure, Πexcess, due to the difference in solute concentrations in the bulk solution and the DNA phase can be calculated from the difference in PEG osmotic pressures with and without added solute at constant spacing. This excess pressure is directly related to changes in excess water molecules associated with DNA using Eqs. 3 and 5.
-DNA and Na+-DNA. We previously have observed a linear interplay between ΠPEG and ΠMPD with Spd3+-DNA to maintain constant spacing (8). This linearity means that the change in the number of excess waters included in the DNA phase is independent of solute concentration. An apparent excess osmotic pressure, Πexcess, due to the difference in solute concentrations in the bulk solution and the DNA phase can be calculated from the difference in PEG osmotic pressures with and without added solute at constant spacing. This excess pressure is directly related to changes in excess water molecules associated with DNA using Eqs. 3 and 5.
Exclusion of MPD from Co(NH3)63+-DNA, Spd3+-DNA, and Na+-DNA arrays
Fig. 2 shows the distance dependence of the excess osmotic pressure normalized by the total solute osmotic pressure, Πexcess/Π0, for the exclusion of MPD from DNA arrays equilibrated against 2 mM Co(NH3)6Cl3, 2 mM SpdCl3, and two concentrations of NaBr: 20 mM and 1.2 M. We have chosen these salt conditions since changes in Dint are not due to changes in salt activities caused by the addition of MPD. Force curves in  and Spd3+ are independent of trivalent ion concentration between 0.25 and 10 mM. Force curves are only weakly dependent on salt concentration between 1 and 40 mM NaBr at high osmotic pressure and for concentrations >∼1 M at all osmotic pressures examined. At high ΠPEG, the force magnitude is greater at 20 mM NaBr, compared to 1.2 M NaBr, reflecting an additional contribution of electrostatics to repulsive hydration interactions at close spacings. PEG (8000 MW) remains phase separated from DNA arrays in low NaBr only at high pressures (log(Π, erg/cm3) ∼ 7.55 for 20 mM).
and Spd3+ are independent of trivalent ion concentration between 0.25 and 10 mM. Force curves are only weakly dependent on salt concentration between 1 and 40 mM NaBr at high osmotic pressure and for concentrations >∼1 M at all osmotic pressures examined. At high ΠPEG, the force magnitude is greater at 20 mM NaBr, compared to 1.2 M NaBr, reflecting an additional contribution of electrostatics to repulsive hydration interactions at close spacings. PEG (8000 MW) remains phase separated from DNA arrays in low NaBr only at high pressures (log(Π, erg/cm3) ∼ 7.55 for 20 mM).
FIGURE 2.

Distance dependence of the change in excess water that excludes MPD. The apparent excess pressure applied by MPD (Eq. 4) normalized by the total MPD osmotic pressure in the bathing solution is shown as dependent on the interhelical spacing. From Eqs. 3 and 5, Πexcess/Π0 = −dΓw/dV, where Γw is the excess water associated with the DNA phase. MPD exclusion is shown for DNA arrays equilibrated against 1.2 M NaBr (squares), 20 mM NaBr (triangles), 2 mM SpdCl3 (circles), and 2 mM Co(NH3)6Cl3 (diamonds). The MPD concentration for the solid symbols was 1 molal, 0.5 molal for the symbols with inner crosses, and 2 molal for symbols with inner dots. The different MPD concentrations overlap for each salt condition, within experimental error, indicating that ΔΓw is independent of solute concentration at any fixed spacing. To a good first order approximation, excess water varies exponentially with the distance between helices. This is illustrated by the linear fits on the logarithmic scale (solid lines) to the NaBr and trivalent ion data.
All four curves in Fig. 2 show a very similar distance dependence of MPD exclusion. The apparent exponential decay length varies between ∼3.5 and 4 Å. This exponential exclusion even extends to the low pressure regime (Dint > 30 Å) with 1.2 M NaBr that shows fluctuation enhanced repulsion between double helices. The data for 0.5 and 1 molal MPD overlap within experimental error for the four salt conditions. This indicates the number of waters that preferentially hydrate DNA in the presence of MPD is constant, as was reported previously for Spd3+-DNA arrays (8). Equivalently, it indicates that the solute partition coefficient (see Eq. 2) at fixed Dint is independent of alcohol concentration. Little difference is observed in the exclusion of MPD from  -DNA and Spd3+-DNA arrays. Also, little difference is seen in the exclusion of MPD from DNA arrays equilibrated against 20 mM and 1.2 M NaBr. The exclusion amplitude for the interaction of MPD with Na+-DNA, however, is ∼30% greater than for
-DNA and Spd3+-DNA arrays. Also, little difference is seen in the exclusion of MPD from DNA arrays equilibrated against 20 mM and 1.2 M NaBr. The exclusion amplitude for the interaction of MPD with Na+-DNA, however, is ∼30% greater than for  -DNA and Spd3+-DNA. This difference could be due either to the effect of the dielectric constant of this solute on electrostatic interactions between DNA helices or to differences in the preferential interaction of MPD with bound
-DNA and Spd3+-DNA. This difference could be due either to the effect of the dielectric constant of this solute on electrostatic interactions between DNA helices or to differences in the preferential interaction of MPD with bound  , Spd3+, and Na+.
, Spd3+, and Na+.
The total number of included waters, ΔNw, can be obtained by integrating Πexcess/Π0 from infinite separation to the dry spacing of 20.9 Å, assuming Πexcess/Π0 can attain a maximal value of 1. We also have assumed the bulk solution value for the volume per water molecule, 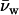 = 30 Å3. Exponential decay lengths (λ), amplitudes at Dint = 27 Å (Πexcess(27 Å)/Π0), and ΔNw per basepair are given in Table 1.
= 30 Å3. Exponential decay lengths (λ), amplitudes at Dint = 27 Å (Πexcess(27 Å)/Π0), and ΔNw per basepair are given in Table 1.
TABLE 1.
Exclusion of alcohols from condensed DNA arrays
| DNA | Alcohol | λ, Å | Πexcess(27Å)/Π0 | ΔNw, per basepair |
|---|---|---|---|---|
| Na+-DNA | MPD | 4.0 ± 0.2 | 0.65 ± 0.02 | 42 ± 3 |
| Co3+-DNA | MPD | 3.65 ± 0.35 | 0.46 ± 0.02 | 34 ± 5 |
| Spd3+-DNA | MPD | 3.55 ± 0.25 | 0.49 ± 0.02 | 35 ± 4 |
| Δ(C-O) = 3* | 3.6 ± 0.2 | 0.41 ± 0.02 | 32 ± 3 | |
| Δ(C-O) = 2† | 3.7 ± 0.2 | 0.28 ± 0.02 | 25 ± 3 | |
| Δ(C-O) = 1‡ | 3.65 ± 0.25 | 0.22 ± 0.02 | 20 ± 3 | |
| Methanol | 4.45 ± 0.5 | 0.13 ± 0.03 | 11 ± 4 | |
| Ethylene glycol | – | 0.05 ± 0.01 | ||
| Glycerol | – | 0.00 ± 0.02 | ||
| Threitol | 0.95 ± 0.25 | −0.10 ± 0.03 | ||
| Sorbitol | 1.25 ± 0.15 | −0.37 ± 0.04 |
The data shown in Figs. 2–4 were fit to an exponential function to determine an apparent decay length and best fit amplitude at 27 Å. The total number of excess waters was determined by integrating the exponential fit for Πexcess/Π0 from Dint = ∞ to 20.9 Å (the spacing measured for dried Spd3+-DNA arrays) using Eqs. 3 and 5 and dV = √3 l Dint dDint, where V is the water volume/basepair in the DNA phase and l is the rise/basepair assumed 3.4 Å.
An average of the data for 1-butanol, 2-butanol, and t-butanol.
An average of the data for 1-propanol, 2-propanol, 1,4-butanediol, and 2,3-butanediol.
An average of the data for ethanol, 1,3-propanediol, and 1,2-propanediol.
Exclusion of alcohols from Spd3+-DNA arrays
In Fig. 3, the distance profile for exclusion from Spd3+-DNA of 12 alcohols with differing numbers of alkyl carbons and hydroxyl oxygens is given. Two concentrations differing by a factor of two are shown for each alcohol. Once again, exclusion scales linearly with concentration within experimental error, i.e., Πexcess/Π0 is insensitive to alcohol concentration. Exclusion amplitudes vary by a factor of ∼5 between methanol and MPD. The apparent exponential decay lengths vary between 3.4 and 4.4 Å. The average decay length is λ = 3.7 ± 0.3 Å.
FIGURE 3.

Dependence of the change in excess water for Spd3+-DNA, Πexcess/Π0, on interhelical spacing is shown for a set of 12 nonpolar alcohols. Condensed DNA arrays were equilibrated against 2 mM SpdCl3 and 10 mM TrisCl (pH 7.5), PEG, and varied concentrations of alcohols, at 20°C. The alcohols are coded such that solutes with the same excess number of alkyl carbons over hydroxyl groups have the same color. The alcohols are methanol (1.5 and 3 molal), red circles; ethanol (1 and 2 molal), green circles; 1,3-propanediol (1 and 2 molal), green squares; 1,2-propanediol (1 and 2 molal), green diamonds; 1-propanol (2 molal), yellow circles; 2-propanol (1 and 2 molal); yellow squares; 1,4-butanediol (1 and 2 molal), yellow diamond; 2,3-butanediol (1 and 2 molal), yellow hexagon; 1-butanol (0.5 and 1 molal), aqua circles; 2-butanol (0.5 and 1 molal), aqua squares; t-butanol (0.5 and 1 molal)-(0.5 and 1 molal), aqua diamonds; and MPD (0.5 and 1 molal), pink square. Data for the lower alcohol concentration are given by the symbols with inner dots. The decay length of the apparent exponential, calculated from linear fits on the logarithmic scale (solid lines), varies between ∼3.5 and 4 Å. The amplitude or preexponential factor varies significantly with the chemical nature of the osmolyte, but not its size.
To a good first order approximation, the exclusion amplitude for this set of alcohols depends simply on the number of alkyl carbons in excess of hydroxyl groups. Overall size is secondary. The data points in Fig. 3 have been colored appropriately to show this grouping. For example, 1-propanol, 2-propanol, and 1,4- or 2,3-butanediol (yellow, two excess carbons over hydroxyls: Δ(C-O) = 2) are similarly excluded. These four alcohols are more highly excluded than ethanol and 1,2- or 1,3-propanediol (green, Δ(C-O) = 1), but less excluded than 1-butanol, 2-butanol or t-butanol (blue, Δ(C-O) = 3) and MPD (pink, Δ(C-O) = 4). Exponential decay lengths, exclusion amplitudes, and integrated numbers of waters are summarized in Table 1.
The exclusion amplitudes seen in these osmotic stress experiments for 2-butanol and 1,4-butanediol correlate well with their effect on the critical SpdCl3 concentration needed for precipitation of DNA from dilute solution in 0.1 M NaCl. We previously reported that in the absence of added osmolyte, DNA precipitates at ∼10 mM SpdCl3 and that this is reduced to ∼5.4 mM SpdCl3 in 1 molal 2-propanol and ∼3.5 mM SpdCl3 in 1 molal MPD (8). We further showed that this decrease in critical Spd3+ concentration is quantitatively consistent with the measured exclusion of alcohol. The critical concentration is ∼5.6 mM SpdCl3 in 1 molal 1,4-butanediol (Δ(C-O) = 2), very close to that for 2-propanol (Δ(C-O) = 2), and ∼4.2 mM SpdCl3 in 1 m 2-butanol (Δ(C-O) = 3), intermediate between MPD (Δ(C-O) = 4) and 2-propanol.
Interaction of oligo- and polyols with Spd3+-DNA arrays
Fig. 4 shows the distance dependence of Πexcess/Π0 for methanol, ethylene glycol, glycerol, threitol, sorbitol, and 1,2,4-butanetriol. Within experimental error, methanol resembles the other alcohols, where exclusion increases as the spacing between DNA helices decreases. If analyzed as an exponential, the apparent 4.4 Å decay length for methanol is similar to the other alcohols shown in Fig. 3. Ethylene glycol shows a small, constant exclusion of ∼5% over the entire 25–29 Å range of spacings. Glycerol shows no preferential interaction until high PEG pressures, and then there is an apparent 15–20% inclusion at 26 Å. The spacing between helices actually increases at high ΠPEG with added glycerol. At a constant Dint, the extent of inclusion increases with polyol size for threitol and sorbitol. The spacing between helices in Spd3+-DNA arrays increases with added sorbitol even in the absence of applied PEG pressure. At the highest PEG pressures used, Πexcess/Π0 would suggest there is almost twice as much sorbitol in the Spd3+-DNA array as in the bulk solution. This apparent inclusion of sorbitol in the Spd3+-DNA array is consistent with an increased concentration of SpdCl3 necessary to precipitate DNA from dilute solution in 0.1 M NaCl. In 1 molal sorbitol, the critical concentration is ∼13.3 mM SpdCl3. If analyzed as an exponential (the solid line in Fig. 4), the apparent decay length is 1.25 Å, which is a much different length than found for the exclusion of alcohols from DNA.
FIGURE 4.

Interaction of more polar alcohols and polyols with Spd3+-DNA is different from the nonpolar alcohols. Negative values of Πexcess/Π0 mean that helices move further apart as osmolyte is added. The alcohols and polyols are methanol (1.5 and 3 molal), circles; ethylene glycol (2 molal), upward triangle; glycerol (1 and 2 molal), triangle; threitol (1 and 2 molal), squares; sorbitol (0.4 and 0.8 molal), diamonds; and 1,2,4-butanetriol (1 and 2 molal), hexagon. Data for the lower concentrations are shown as the symbols with inner circles. The solid line is the best exponential fit to the sorbitol data with a 1.25 Å decay length. Solutes with multiple hydroxyl groups tend to be included at close interhelical spacings.
Exclusion of 1,2,4-butanetriol also shows a different behavior from the other alcohols seen in Fig. 3. The exclusion amplitude for this triol is distinctly smaller than for the other Δ(C-O) = 1 solutes of its class. Additionally, there is a marked decrease in the exclusion at the highest PEG pressure used analogous to the abrupt inclusion of glycerol, threitol, and sorbitol at close spacings.
Effect of alcohol size and chemical nature on exclusion amplitude
Fig. 5 shows the average exclusion at 27 Å as a function of the excess number of carbons for two cases. The exclusion for each Δ(C-O) calculated by averaging the data for the alcohols shown in Fig. 3 is given by the circles. Methanol is the sole representative for Δ(C-O) = 0. The exclusion for Δ(C-O) = 2, 3, and 4 varies linearly with the number of excess alkyl carbons. The total exclusion can be represented as a simple sum of the exclusion amplitudes for the individual chemical moieties comprising the solute.
FIGURE 5.

Exclusion of alcohols scales with the number of alkyl carbons in excess of hydroxyl groups, Δ(C-O). Values of Πexcess/Π0 at 27 Å versus Δ(C-O) are shown for two cases: circles, the data for all 12 nonpolar alcohols shown in Fig. 3 are averaged, methanol is the only representative for Δ(C-O) = 0; squares, only alcohols with four carbons are included, 1- 2-, and t-butanol, 1,4- and 2,3-butanediol, 1,2,4-butanetriol, and threitol. The data for Δ(C-O) = 2 − 4 varies approximately linearly, suggesting that exclusion amplitudes can be calculated by simply summing over the constituent chemical groups. The diverging behavior at Δ(C-O) = 0 and 1 reflects the attractive interactions of alcohols with multiple hydroxyl groups with Spd3+-DNA.
The squares show the exclusion considering only solutes with four carbons. Threitol and 1,2,4-butanetriol are the representatives for Δ(C-O) = 0 and 1, respectively. The difference between the two data sets illustrates that the interaction of solutes having multiple hydroxyl groups and DNA is qualitatively different from the more nonpolar alcohols.
DISCUSSION
The study of the interactions of small solutes with biological macromolecules has grown increasingly sophisticated. Several experiments measuring total exclusion, the integral of the water or solute distribution functions, have probed the chemical features of solutes and macromolecules that dominate interactions. Bolen and co-workers have parsed the interactions of several osmolytes into peptide backbone and side-chain contributions (6,22). Record and co-workers have suggested that betaine glycine is strongly excluded from anionic groups on macromolecules (21,23,24) and have used the differing interactions of urea and glycine betaine to dissect the lac repressor-operator binding reaction into protein folding and phosphate binding contributions (25).
There has been additionally much recent theoretical work connecting pairwise potentials with solute distribution functions and finally to the thermodynamic consequences of osmolyte inclusion or exclusion, particularly using the Kirkwood-Buff formalism (7,26,27). Unfortunately, in the absence of a measured distribution, these treatments attempt to connect with experiments using ad hoc assumptions concerning excluded water, assuming the pairwise potential simply contains excluded volume and site binding contributions, or relying on molecular dynamics simulations.
The data of Harries et al. (28) shows that solute-surface interactions are more complicated than simple steric exclusion would predict. The effect of several salts and solutes on the cyclodextrin - adamantine binding reaction was investigated using isothermal titration calorimetry. Several salts and solutes acted through a difference in the preferential hydration of products and reactants, i.e., the energetic contribution of solutes could be expressed as a 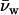 Π ΔNw work. The free energy changes associated with binding in the presence of osmolytes, however, were almost entirely due to changes in enthalpy, not entropy as would be expected for simple sterics. Even those solutes that affected the reaction minimally, i.e., ΔNw was small, typically showed compensating changes in enthalpy and entropy rather than no changes. Physical forces other than steric exclusion dominate these solute-surface interactions.
Π ΔNw work. The free energy changes associated with binding in the presence of osmolytes, however, were almost entirely due to changes in enthalpy, not entropy as would be expected for simple sterics. Even those solutes that affected the reaction minimally, i.e., ΔNw was small, typically showed compensating changes in enthalpy and entropy rather than no changes. Physical forces other than steric exclusion dominate these solute-surface interactions.
Our approach has been to measure the sensitivity of forces between macromolecules in condensed arrays to the solute concentration in the bulk solution. We use the osmotic pressure of a polymer, as PEG, that is excluded from the macromolecular phase to apply a force on the ordered array. A Maxwell relationship of the fundamental Gibbs-Duhem equation relates the change in the number of excess solute or water molecules in the macromolecular phase to the change in spacing between macromolecules as the applied PEG pressure is kept constant. Excess water or solute can be defined through the ratio of the numbers of solutes and waters in the condensed phase and the bulk solution as given in Eq. 2. It does not matter if we focus on the number of excess waters or the deficit in solutes since the two are straightforwardly connected.
We emphasize that only changes in the number of excess water or solute molecules as DNA helices are pushed together, dΓw/dVw, are measured with this method. Direct binding of solutes to DNA or the exclusion of solutes from only water sequestered in grooves, for example, would not likely be seen until the spacing between helices starts probing these very close distances. We have confirmed previously, however, that the measurements for 2-propanol and MPD are consistent with a simple exclusion of these osmolytes from DNA and that glycerol is neither appreciably included nor excluded (8).
MPD is seen similarly excluded from condensed arrays of Spd3+-DNA,  -DNA, and Na+-DNA (Fig. 2). Spd3+ and
-DNA, and Na+-DNA (Fig. 2). Spd3+ and  mediate spontaneous assembly of DNA, but helices are repulsive in NaBr. In the absence of added solute, the interhelical force for Spd3+-DNA and
mediate spontaneous assembly of DNA, but helices are repulsive in NaBr. In the absence of added solute, the interhelical force for Spd3+-DNA and  -DNA arrays shows an approximate 2 Å exponential decay length at the highest PEG pressures. Force amplitudes are insensitive to changes in trivalent ion concentration from ∼0.5 to 20 mM. In 1.2 M NaBr and high osmotic pressures (log(ΠPEG, erg/cm3) > 7.0), the apparent exponential decay length is ∼3–4 Å. Force magnitudes in this pressure region are relatively insensitive to salt concentration. At lower pressures, the observed interaction is due to fluctuation enhanced forces (15,29,30), where interactions are dominated by motions of the helices in a confining force field. The apparent exponential decay length is ∼7–8 Å, about double the high pressure, bare force decay length. Force magnitudes in this regime are insensitive to NaBr ionic strengths for concentrations >1 M. At lower ionic strengths, fluctuation enhanced, salt concentration sensitive, electrostatic double layer repulsion is observed. In 20 mM NaBr and high pressures, force amplitudes are ∼25% greater than in 1.2 M NaBr, reflecting the added contribution from electrostatic repulsion to hydration forces at low salt concentrations. The Na+-DNA array does not remain phase separated from PEG at low osmotic pressures.
-DNA arrays shows an approximate 2 Å exponential decay length at the highest PEG pressures. Force amplitudes are insensitive to changes in trivalent ion concentration from ∼0.5 to 20 mM. In 1.2 M NaBr and high osmotic pressures (log(ΠPEG, erg/cm3) > 7.0), the apparent exponential decay length is ∼3–4 Å. Force magnitudes in this pressure region are relatively insensitive to salt concentration. At lower pressures, the observed interaction is due to fluctuation enhanced forces (15,29,30), where interactions are dominated by motions of the helices in a confining force field. The apparent exponential decay length is ∼7–8 Å, about double the high pressure, bare force decay length. Force magnitudes in this regime are insensitive to NaBr ionic strengths for concentrations >1 M. At lower ionic strengths, fluctuation enhanced, salt concentration sensitive, electrostatic double layer repulsion is observed. In 20 mM NaBr and high pressures, force amplitudes are ∼25% greater than in 1.2 M NaBr, reflecting the added contribution from electrostatic repulsion to hydration forces at low salt concentrations. The Na+-DNA array does not remain phase separated from PEG at low osmotic pressures.
Despite the large differences in interhelical force characteristics, the distance dependence of MPD exclusion is remarkably similar for Spd3+-DNA,  -DNA, and Na+-DNA arrays. The apparent exponential decay length is ∼3.5–4.0 Å even in the fluctuation enhanced force regime in 1.2 m NaBr. There is surprisingly little difference in force amplitude between Spd3+ and
-DNA, and Na+-DNA arrays. The apparent exponential decay length is ∼3.5–4.0 Å even in the fluctuation enhanced force regime in 1.2 m NaBr. There is surprisingly little difference in force amplitude between Spd3+ and  . The exclusion amplitude is somewhat greater with Na+, but there is little difference between 20 mM and 1.2 M NaBr. The extra repulsion in 20 mM NaBr due to electrostatics does not translate into significantly increased exclusion of MPD. The overlap of data for two MPD concentrations indicates that the excess number of included water molecules is insensitive to MPD concentration and, therefore, DNA is preferentially hydrated.
. The exclusion amplitude is somewhat greater with Na+, but there is little difference between 20 mM and 1.2 M NaBr. The extra repulsion in 20 mM NaBr due to electrostatics does not translate into significantly increased exclusion of MPD. The overlap of data for two MPD concentrations indicates that the excess number of included water molecules is insensitive to MPD concentration and, therefore, DNA is preferentially hydrated.
The distance dependencies of exclusion for the 12 alcohols from Spd3+-DNA condensed arrays shown in Fig. 3 are consistent with a 3.5–4 Å decay length exponential distribution function. Only the preexponential amplitude factor is dependent on the nature of the alcohol. We have observed that the 3–4 Å decay length exponential force dominates the repulsion for the last 10–15 Å surface separations between many kinds of macromolecules, including lipid bilayers, polysaccharides, and DNA (10,11,14–17,31). Since this common force is observed for biopolymers that are charged, zwitterionic, or wholly uncharged, we have postulated that it is due to the energetics of restructuring of water in confined spaces. The hydrogen-bonded network of water between surfaces with incompatible hydration structures is strained. Within the hydration force formalism, the ∼4 Å decay length is a water-water correlation length. The force amplitude is dependent on the hydration energy and orientation of water structured on the apposing surfaces. The close correspondence of the distance dependence of alcohol exclusion and hydration forces between macromolecular surfaces indicates that these alcohols interact directly with DNA also through water structuring forces.
We also have reported a similar distance dependence for solute-macromolecule interactions in a very different system: the exclusion of salts and polar solutes from hydrophobically modified and neutral hydroxypropyl cellulose, HPC (9). The dependence of the amplitude of exclusion on the identity of the salt follows the well known Hofmeister series that has long been thought connected to water structuring (32). The common exclusion characteristics of these two, very different systems reinforces the conclusion that hydration water structuring forces likely underlie the repulsive interactions of most small solutes with macromolecular surfaces. Additionally, there have been several recent molecular dynamics simulations that suggest water structuring may mediate the interactions between various molecules in aqueous solution, particularly between charged and nonpolar or uncharged groups (33–35).
The dependence of exclusion amplitude on the size and chemical nature of the alcohol is quite instructive. To a first order approximation, the exclusion amplitude of alcohols simply scales with the excess number of alkyl carbons over hydroxyl oxygens, regardless of overall size. For Δ(C-O) = 2, 3, and 4, Fig. 5 indicates that exclusion varies linearly with Δ(C-O). In this case, preferential hydration is not simply determined by solute size and steric exclusion. It is not clear to what extent soft, flexible molecules can be replaced by the hard sphere approximations that have been considered. Nor can combining steric exclusion with specific site binding account for the very similar distance dependencies of exclusion for all the alcohols. The solute distribution function observed indicates that the interaction potential between these alcohols and DNA is quite different from what has been generally assumed for repulsive solute-macromolecule interactions. For the larger alcohols, the exclusion amplitude is a simple, additive function of the individual alkyl carbon and hydroxyl oxygen contributions. This additivity easily could be mistaken for a steric exclusion if only homologous solutes are examined (3,36).
We would presume that the exclusion of alcohols from DNA is dominated by the interaction of the alkyl carbons in excess over hydroxyl groups with the phosphate and counterion charge on the DNA backbone. The exclusion of alcohol alkyl groups from the charges on DNA should then be comparable with our previous results for the exclusion of kosmotropic salts from methyl groups on HPC (9). Assuming additive interactions, the 42 waters/basepair calculated for exclusion of MPD (Δ(C-O) = 4) from Na+-DNA (Table 1) translates into ∼5.25 waters/NaPhosphate/excess alkyl group. We reported a preferential hydration of 55 waters/disaccharide (10 Å) for the exclusion of KF from HPC. The phosphate and F− anions are generally ranked similarly in the Hofmeister series (32). Each sugar unit comprising HPC has ∼3 hydroxypropyl groups (2 excess alkyl groups/hydroxylpropyl) corresponding then to ∼4.6 waters/KF/ excess alkyl group. The somewhat increased exclusion seen for DNA may indicate that phosphate groups are only partially responsible for exclusion.
A different behavior is seen for those solutes with Δ(C-O) = 0. Methanol shows an exclusion that is characteristic of the other alcohols, albeit the exclusion is only ∼15% at the closest spacings. Ethylene glycol (HO-CH2-CH2-OH) shows a very slight exclusion, ∼5%, with very little apparent distance variation. Glycerol (HO-CH2-CHOH-CH2-OH) shows essentially no interaction with DNA until close spacings. At 26 Å separation between DNA helices, there is an apparent 15% inclusion of glycerol. The trend of increasing inclusion continues for threitol (HO-CH2-(CH(OH))2-CH2-OH) and sorbitol (HO-CH2-(CHOH)4-CH2-OH) with apparent inclusions of ∼30% and 80%, respectively, at ∼26 Å. The apparent inclusion of sorbitol is consistent with the slightly increased concentration of Spd3+ needed to precipitate DNA in 0.1 M NaCl. A propensity for inclusion of polyols at close spacings is also evident for 1,2,4-butanetriol shown in Fig. 4. The exclusion amplitude for this alcohol is significantly smaller at low ΠPEG than for the other Δ(C-O) = 1 alcohols, more closely resembling methanol. The exclusion amplitude then decreases at the higher ΠPEG, or smaller spacings, suggesting an increasing contribution from inclusion at close distances.
The interaction of these polyols with DNA shows very different force characteristics from the nonpolar alcohols. The distance dependence is far steeper for the polyols, λ ∼ 1.25 Å for sorbitol. Secondly, unlike the alcohols, polyol inclusion has a very steep dependence on solute size. Indeed, the excess inclusion pressure acting at ∼26 Å for the series glycerol, threitol, and sorbitol varies almost as (MW)2. We suspect that this inclusion relieves a strain in water structuring associated with tight packing. The inclusion of polyols can replace a number of water molecules that may not have a normal complement of hydrogen bonds. In this view, polyols interact only indirectly with DNA through the frustrated water structuring that is a consequence of DNA-DNA interactions at close spacings, not through direct DNA-solute forces.
CONCLUSIONS
The preferential hydration of DNA in the presence of nonpolar alcohols is due to a hydration force driven exclusion of these osmolytes. Reactions or conformational transitions that decrease the extent of exclusion will be favored with increasing alcohol concentration. The effect of these alcohols on solution dielectric constant and DNA electrostatic forces is secondary. Indeed, the strong exclusion of alcohol from DNA would mean a distance dependent dielectric constant that would greatly complicate electrostatic calculations. The observation that water structuring forces underlie preferential hydration and osmolyte exclusion offers a new perspective for understanding the interactions between molecules in aqueous solution.
Acknowledgments
The authors gratefully acknowledge Drs. V. A. Parsegian, B. A. Todd, and D. Harries for valuable discussions and suggestions.
This research was supported by the Intramural Research Program of the National Institute of Child Health and Human Development and the National Institutes of Health.
References
- 1.Timasheff, S. N. 1993. The control of protein stability and association by weak interactions with water: How do solvents affect these processes? Annu. Rev. Biophys. Biomol. Struct. 22:67–97. [DOI] [PubMed] [Google Scholar]
- 2.Parsegian, V. A., R. P. Rand, and D. C. Rau. 1995. Macromolecules and water: Probing with osmotic stress. In Methods in Enzymology. M. L. Johnson and G. K. Ackers, editors. Academic Press, New York. 259:43–94. [DOI] [PubMed]
- 3.Davis-Searles, P. R., A. J. Saunders, D. A. Erie, D. J. Winzor, and G. J. Pielak. 2001. Interpreting the effects of small uncharged solutes on protein-folding equilibria. Annu. Rev. Biophys. Biomol. Struct. 30:271–306. [DOI] [PubMed] [Google Scholar]
- 4.Record, M. T., Jr., W. Zhang, and C. F. Anderson. 1998. Analysis of effects of salts and uncharged solutes on protein and nucleic acid equilibria and processes: A practical guide to recognizing and interpreting polyelectrolyte effects, Hofmeister effects, and osmotic effects of salts. Adv. Protein Chem. 51:281–353. [DOI] [PubMed] [Google Scholar]
- 5.Parsegian, V. A., R. P. Rand, and D. C. Rau. 2000. Osmotic stress, crowding, preferential hydration, and binding: A comparison of perspectives. Proc. Natl. Acad. Sci. USA. 97:3987–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bolen, D. W., and I. V. Baskakov. 2001. The osmophobic effect: Natural selection of a thermodynamic force in protein folding. J. Mol. Biol. 310:955–963. [DOI] [PubMed] [Google Scholar]
- 7.Schurr, J. M., D. P. Rangel, and S. R. Aragon. 2005. A contribution to the theory of preferential interaction coefficients. Biophys. J. 89:2258–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hultgren, A., and D. C. Rau. 2004. Exclusion of alcohols from spermidine-DNA assemblies: Probing the physical basis for preferential hydration. Biochemistry. 43:8272–8280. [DOI] [PubMed] [Google Scholar]
- 9.Chik, J., S. Mizrahi, S. Chi, V. A. Parsegian, and D. C. Rau. 2005. Hydration forces underlie the exclusion of salts and of neutral polar solutes from hydroxypropylcellulose. J. Phys. Chem. B. 109:9111–9118. [DOI] [PubMed] [Google Scholar]
- 10.Bonnet-Gonnet, C., S. Leikin, S. Chi, D. C. Rau, and V. A. Parsegian. 2001. Measurement of forces between hydroxypropylcellulose polymers: Temperature favored assembly and salt exclusion. J. Phys. Chem. B. 105:1877–1886. [Google Scholar]
- 11.Leikin, S., V. A. Parsegian, D. C. Rau, and R. P. Rand. 1993. Hydration forces. Annu. Rev. Phys. Chem. 44:369–395. [DOI] [PubMed] [Google Scholar]
- 12.Leikin, S., D. C. Rau, and V. A. Parsegian. 1994. Direct measurement of forces between self-assembled proteins: Temperature-dependent exponential forces between collagen triple helices. Proc. Natl. Acad. Sci. USA. 91:276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leikin, S., D. C. Rau, and V. A. Parsegian. 1995. Temperature-favoured assembly of collagen is driven by hydrophilic not hydrophobic interactions. Nat. Struct. Biol. 2:205–210. [DOI] [PubMed] [Google Scholar]
- 14.Petrache, H. I., N. Gouliaev, S. Tristam-Nagle, R. Zhang, R. M. Suter, and J. F. Nagle. 1998. Interbilayer interactions: High resolution X-ray study. Phys. Rev. E. 57:7014–7024. [Google Scholar]
- 15.Podgornik, R., H. H. Strey, K. Gawrisch, D. C. Rau, A. Rupprecht, and V. A. Parsegian. 1996. Bond orientational order, molecular motion, and free energy of high-density DNA mesophases. Proc. Natl. Acad. Sci. USA. 93:4261–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rau, D. C., and V. A. Parsegian. 1990. Direct measurement of forces between linear polysaccharides xanthan and schizophyllan. Science. 249:1278–1281. [DOI] [PubMed] [Google Scholar]
- 17.Strey, H. H., R. Podgornik, D. C. Rau, and V. A. Parsegian. 1998. DNA-DNA interactions. Curr. Opin. Struct. Biol. 8:309–313. [DOI] [PubMed] [Google Scholar]
- 18.Rau, D. C., and V. A. Parsegian. 1992. Direct measurement of the intermolecular forces between counterion-condensed DNA double helices: Evidence for long range attractive hydration forces. Biophys. J. 61:246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parsegian, V. A., R. P. Rand, N. L. Fuller, and D. C. Rau. 1986. Osmotic stress for the direct measurement of intermolecular forces. Methods Enzymol. 127:400–416. [DOI] [PubMed] [Google Scholar]
- 20.Pelta, J., F. Livolant, and J.-L. Sikorav. 1996. DNA aggregation induced by polyamines and cobalthexamine. J. Biol. Chem. 271:5656–5662. [DOI] [PubMed] [Google Scholar]
- 21.Felitsky, D. J., and M. T. Record Jr. 2004. Application of the local-bulk partitioning and competitive binding models to interpret preferential interactions of glycine betaine and urea with protein surface. Biochemistry. 43:9276–9288. [DOI] [PubMed] [Google Scholar]
- 22.Auton, M., and D. W. Bolen. 2005. Predicting the energetics of osmolyte-induced protein folding/unfolding. Proc. Natl. Acad. Sci. USA. 102:15065–15068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Felitsky, D. J., J. G. Cannon, M. W. Capp, J. Hong, A. W. Van Wynsberghe, C. F. Anderson, and M. T. Record Jr. 2004. The exclusion of glycine betaine from anionic biopolymer surface: Why glycine betaine is an effective osmoprotectant but also a compatible solute. Biochemistry. 43:14732–14743. [DOI] [PubMed] [Google Scholar]
- 24.Hong, J., M. W. Capp, C. F. Anderson, R. M. Saecker, D. J. Felitsky, M. W. Anderson, and M. T. Record Jr. 2004. Preferential interactions of glycine betaine and of urea with DNA: Implications for DNA hydration and for effects of these solutes on DNA stability. Biochemistry. 43:14744–14758. [DOI] [PubMed] [Google Scholar]
- 25.Hong, J., M. W. Capp, R. M. Saecker, and M. T. Record Jr. 2005. Use of urea and glycine betaine to quantify coupled folding and probe the burial of DNA phosphates in lac repressor-lac operator binding. Biochemistry. 44:16896–16911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chitra, R., and P. E. Smith. 2001. Preferential interactions of cosolvents with hydrophobic solutes. J. Phys. Chem. B. 105:11513–11522. [Google Scholar]
- 27.Smith, P. E. 2004. Cosolvent interactions with biomolecules: Relating computer simulation data to experimental thermodynamic data. J. Phys. Chem. B. 108:18716–18724. [Google Scholar]
- 28.Harries, D., D. C. Rau, and V. A. Parsegian. 2005. Solutes probe hydration in specific association of cyclodextrin and adamantane. J. Am. Chem. Soc. 127:2184–2190. [DOI] [PubMed] [Google Scholar]
- 29.Podgornik, R., D. C. Rau, and V. A. Parsegian. 1989. The action of interhelical forces on the organization and DNA double helices: Fluctuation-enhanced decay of electrostatic double-layer and hydration forces. Macromolecules. 22:1780–1786. [Google Scholar]
- 30.Podgornik, R., D. C. Rau, and V. A. Parsegian. 1994. Parametrization of direct and soft steric-undulatory forces between DNA double helical polyelectrolytes in solutions of several different anions and cations. Biophys. J. 66:962–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McIntosh, T. J. 2000. Short-range interactions between lipid bilayers measured by X-ray diffraction. Curr. Opin. Struct. Biol. 10:481–485. [DOI] [PubMed] [Google Scholar]
- 32.Collins, K. D., and M. W. Washabaugh. 1985. The Hofmeister effect and the behaviour of water at interfaces. Q. Rev. Biophys. 18:323–422. [DOI] [PubMed] [Google Scholar]
- 33.Bennion, B. J., and V. Daggett. 2004. Counteraction of urea-induced protein denaturation by trimethylamine N-oxide: A chemical chaperone at atomic resolution. Proc. Natl. Acad. Sci. USA. 101:6433–6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh, T., A. Kalra, and S. Garde. 2005. On the salt-induced stabilization of pair and many-body hydrophobic interactions. J. Phys. Chem. B. 109:642–651. [DOI] [PubMed] [Google Scholar]
- 35.Hassan, S. A. 2005. Amino acid side chain interactions in the presence of salts. J. Phys. Chem. B. 109:21989–21996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davis-Searles, P. R., A. S. Morar, A. J. Saunders, D. A. Erie, and G. J. Pielak. 1998. Sugar-induced molten-globule model. Biochemistry. 37:17048–17053. [DOI] [PubMed] [Google Scholar]