

Abstract
Adeno-associated virus (AAV) is a nonpathogenic parvovirus that requires adenovirus (Ad) or another helper virus for a fully permissive infection. AAV-mediated inhibition of Ad is well documented, yet many details of this interaction remain unclear. In this study, we observed a maximum 50-fold decrease in infectious virus production and a 10- to 40-fold reduction in Ad DNA synthesis during coinfections with AAV. With the exception of the E3 gene, AAV decreased all steady-state Ad mRNA levels at 24 h postinfection (hpi) in a dose-dependent manner. However, not all transcription units were affected equally. E4 and late transcription were the most strongly inhibited, and E1A and E2A were the least affected. The temporal effects of AAV on Ad mRNA transcript levels also varied among the Ad genes. Ad protein expression paralleled mRNA levels at 24 hpi, suggesting that coinfecting AAV does not exert substantial effects on translation. In plasmid transfection assays, Rep78 protein most effectively limited Ad amplification, while Rep40 had no effect. Since E2a and E4 proteins are essential for efficient Ad DNA amplification, we examined the relationship between reduced E2A and E4 expression and decreased DNA amplification. Transfected Rep78 did not reduce E2A and E4 transcript levels prior to DNA replication. Also, AAV-induced inhibition of E2A and E4 mRNA production did not occur in the presence of hydroxyurea. It is therefore unlikely that decreased early gene expression is solely responsible for AAV's suppression of Ad DNA replication. Our results suggest that AAV amplification and/or Rep gene expression inhibits Ad DNA synthesis.
The adenovirus serotype 5 (Ad5) genome is a 35,938-bp double-stranded linear DNA molecule that is organized into five early (E1A, E1B, E2, E3, and E4) transcription units and one late unit based on their temporal expression (reviewed in reference 29). Expression of the early proteins begins shortly after cellular entry and is required for entry into the late phase. Late gene expression begins after the onset of DNA replication, between 6 and 10 h postinfection (hpi). In general, each Ad transcription unit is dedicated to a set of related functions. Transcriptional activation induced by the E1A gene products sets the stage for infection by altering cellular gene expression and inducing entry into S phase. In addition, the E1A proteins are essential for the efficient transcription of all other Ad genes. The E1B region functions primarily to counter the apoptotic effects of E1A proteins. The proteins encoded by the E3 transcription unit modulate the cellular environment to prevent detection by the host immune system and are not required for Ad replication in cell culture. Two transcription units, E2 and E4, have significant effects on Ad DNA replication. The E2 region is dedicated to DNA replication and encodes three proteins that are central to the process: single-stranded DNA-binding protein, Ad polymerase, and terminal binding protein precursor. In the presence of accessory cellular proteins and an Ad terminal binding protein-DNA template, the E2 proteins are sufficient to reproduce DNA replication in vitro (11). The functions of the E4-encoded proteins are more diverse. Although E4 proteins are not directly required for DNA replication, they are essential for efficient template amplification. As a result, Ad mutants lacking the E4 region have severe defects in DNA replication, late viral mRNA accumulation, and late protein synthesis (16, 30).
In 1965, a contaminating virus was identified in human and simian adenoviral preparations. Due to its inability to replicate in the absence of adenovirus, this defective parvovirus was named adeno-associated virus (AAV). Subsequent studies identified additional helper viruses, including herpes simplex virus (1, 4, 5), cytomegalovirus (22), and human papillomavirus (35). The AAV serotype 2 (AAV2) single-stranded DNA genome encodes four nonstructural replication proteins (Rep78, Rep68, Rep52, and Rep40) and three structural capsid (Cap) proteins (reviewed in reference 24). As their names suggest, Rep proteins are essential for the replication of AAV DNA. In addition to the helicase activity common to all four Rep proteins, Rep78 and Rep68 exhibit endonuclease activity and modulate AAV transcription.
Even though AAV requires the assistance of a helper virus, it inhibits Ad replication during coinfection. Previous studies document up to a 100-fold decrease in Ad production and up to a 10-fold decrease in Ad DNA replication in the presence of AAV (6, 8, 21). Although the effects of AAV on Ad transcription during coinfection have not been thoroughly studied, there is a growing body of evidence suggesting that Ad gene expression may be transcriptionally regulated by AAV Rep proteins (14, 19, 25). For example, many of the proteins known to interact with Rep proteins are involved in RNA transcription or its regulation (e.g., HMG1, PC4, SP1, and TBP) (13, 17, 26, 27, 32, 36). Additionally, previous reports indicate that the interactions with the carboxyl terminus of Rep78 and Rep52 inhibit cyclic AMP-dependent PKA and its homolog, PrKX (10, 14). By inhibiting PKA and PrKX, Rep78/52 may decrease expression of downstream cyclic AMP-inducible genes, including Ad E1a, E3, and E4. Furthermore, our laboratory has previously reported that Rep proteins decrease E2a protein and steady-state mRNA levels but not mRNA stability (19, 25). The effects of AAV and its Rep proteins during coinfection on the expression of other Ad genes are unknown.
Although it has been known for almost 40 years that AAV inhibits Ad propagation, the details of this interaction remain unclear. Before the mechanisms of AAV-mediated inhibition can be fully understood, it is essential that we obtain fundamental knowledge about Ad gene expression in the presence of AAV. In this study, we examined AAV's effects on Ad production, DNA replication, and the expression of mRNA and proteins from individual transcription units. We also conducted a temporal analysis of Ad mRNA levels during coinfection and explored the relationship between decreased Ad E2A and E4 gene expression and the inhibition of Ad DNA replication. These studies provide the groundwork for mechanistic studies and offer new insight into the interactions between AAV and Ad.
MATERIALS AND METHODS
Cells, plasmids, and viruses.
HeLa cells (human cervical epithelium; ATCC CCL-2) were grown as monolayers in Eagle minimum essential medium supplemented with 10% fetal bovine serum, penicillin (50 mg/ml), streptomycin (50 mg/ml), gentamicin (100 μg/ml), and amphotericin B (2.5 μg/ml).
The pCDMRep plasmids contain wild-type and purine nucleotide binding (PNB) site mutant Rep genes cloned in the pCDM8 vector (Invitrogen) and expressed under the control of the cytomegalovirus promoter (25, 39). Plasmids pCDMRep78G and pCDMRep68G contain a methionine-to-glycine mutation in the Rep52/40 initiation codon (9). Plasmid pNTC244 contains the complete AAV2 genome cloned in a pUC derivative, pTZ19U (9).
AAV was generated by pNTC244 transfection of Ad5-infected HeLa monolayers as previously reported (7). AAV was purified over CsCl or by heparin-agarose chromatography and titered by indirect immunofluorescence or by limiting dilution assays (6, 20). Frank Graham generously provided AdlacZ5, an Ad5-based vector in which 1.88 kb of the E3 gene (nucleotides [nt] 28592 to 30470) is replaced by the Escherichia coli lacZ gene (23). Wild-type Ad5, which was originally obtained from the ATCC, and AdlacZ5 were propagated, purified, and titered as previously reported (38). The AAV2-based recombinant vector vAVluc, which contains a luciferase gene in place of the Rep genes, was generated as previously described (31).
Antibodies.
Polyclonal antibody against E2a protein and antiserum specific for E4orf6/7 protein were generously provided by Arnold Berk and Tom Shenk, respectively. Affinity-purified, polyclonal AAV Rep- and Cap-specific antibodies were obtained from rabbits immunized with E. coli-expressed recombinant Rep and Cap proteins and probed with anti-rabbit (31340; Pierce) alkaline phosphatase-conjugated secondary antibody. Commercially available primary antibodies specific for hexon (12-6235-1; American Research) and E1a (DP11; Oncogene) proteins were used in conjunction with anti-goat (SC-2022; Santa Cruz) and anti-mouse (31320; Pierce) alkaline phosphatase-conjugated secondary antibodies.
β-Galactosidase assays.
Assays to detect the β-galactosidase (β-gal) activity of the AdlacZ5 vector were conducted using the Galacto-Star system (Tropix) with minor modifications of the manufacturer's instructions. After AdlacZ5-infected HeLa cells were pelleted and resuspended in Tropix lysis buffer, 30-μl aliquots of the lysates were combined with 100 μl of the Galacto-Star substrate. The samples were incubated for 20 min at room temperature and assayed for β-gal activity using a Lumat LB 9600 luminometer (Berthold Technologies).
AAV and AdlacZ5 primary and secondary coinfections.
Primary coinfections were conducted in 24-well tissue culture (TC) plates containing 70 to 90% confluent HeLa cells. Immediately prior to infection, the medium was replaced with 0.2 ml serum-free medium. The monolayers were infected with increasing infectious units (IU) of AAV and incubated for 1 h at 37°C. Cells were then infected with AdlacZ5 for 1 h, and the infectious medium was replaced with 0.5 ml complete medium per well. Cells were harvested at 48 hpi and assayed for β-gal activity as described above. Aliquots of the harvested cultures were reserved for secondary infections.
Secondary infections were conducted in 48-well TC plates containing 70 to 90% confluent HeLa cells. Prior to inoculation, the medium was replaced with 100 μl of serum-free medium supplemented with 25 mg/ml heparin to block secondary AAV infection. Each well was inoculated with 50 μl of culture lysate from the primary infection. The cells were incubated for 1 h at 37°C, and the medium was replaced with 200 μl of complete medium. Cells were harvested at 24 hpi and assayed for β-gal activity. Three or more replicates of coinfection experiments were performed.
Plasmid transfections.
Approximately 9 × 104 HeLa cells were seeded into each well of 24-well tissue culture plates and incubated overnight at 37°C. On the next day, the cells were transfected with 0.8 μg plasmid DNA and 3 μl of Lipofectamine 2000 (Invitrogen) following the manufacturer's recommendations. During the transfection, each well contained 400 μl serum-free medium supplemented with 0.1% bovine serum albumin. Inclusion of bovine serum albumin minimizes the toxic effects of the Lipofectamine reagent without altering transfection efficiency (12). After 4 h of incubation at 37°C, the cells were infected with AdlacZ5 at a multiplicity of infection (MOI) of 1 and were incubated for 1 h. The inoculum was replaced with 500 μl of complete medium. Cells were harvested at 48 hpi and assayed for β-gal activity. Aliquots of the harvested cultures were used as inocula in secondary infections, which were conducted as described above.
Immunoblot analysis.
HeLa cells were coinfected in 10-cm2 plates with AAV (0, 1, 10, 100, or 1,000 IU) and AdlacZ5 (MOI, 5 or 10) as described above. Cells were harvested at 48 hpi, pelleted by centrifugation, and resuspended in 4 ml phosphate-buffered saline containing 1.5 mM MgCl2. The cell suspension was aliquoted such that 0.5 ml was utilized for β-gal assays to verify AdlacZ5 infections, 1.5 ml was assayed by immunoblotting, and 2 ml was reserved for Southern analysis. Protein levels of the immunoblot aliquots were measured spectrophotometrically using a detergent-competent Bio-Rad system (catalog no. 500-0113). Samples containing equal amounts of protein were resolved by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and probed with the appropriate antibodies.
Southern blot analysis.
Hirt extraction (18) and Southern analysis were performed on the reserved aliquots as previously described (2). Equal levels of DNA, as determined by A260 spectrophotometric measurements, were resolved on 1% agarose gels, transferred to a nitrocellulose membrane, and hybridized overnight at 42°C with 32P-labeled probes specific for Ad or AAV. The AAV Cap hybridization probe was a HindIII restriction fragment from the pNTC244 plasmid from nt 1883 to 4675 of the AAV2 genome. The Ad hexon hybridization probe was generated via PCR amplification of nt 21081 to 21679 of the Ad5 genome. Blots were washed, and DNA levels in each sample were quantified using a Molecular Dynamics Storm 840 PhosphorImager and ImageQuant 5.0 software.
Northern analysis.
Coinfections with AdlacZ5 or Ad5 (MOI, 5) and AAV (0, 1, 10, 100, or 500 IU) were conducted as described above in 10-cm2 TC plates containing 80% confluent HeLa cells. Transfections were conducted with 12 μg pCDM8 or pCDMRep78G and 30 μl Lipofectamine 2000 in 10-cm2 TC plates containing nearly confluent HeLa cells. After being incubated for 20 to 24 h to permit Rep expression, transfected cells were infected with AdlacZ5 or Ad5 for 1 h in serum-free medium. Both coinfected and transfected/infected cells were harvested at 24 hpi. When AdlacZ5 was used, β-gal readings were taken to verify that the infections were successful. Total RNA was isolated using TRIzol reagent (Gibco-BRL) following the manufacturer's protocol and digested with RNase-free DNase to eliminate genomic DNA contamination. A260 spectrophotometric readings were taken to determine RNA levels. Northern analysis was conducted as previously described (2). The E1a hybridization probe was a 714-bp XbaI-to-PvuII restriction endonuclease fragment from nt 626 to 1339 in the Ad5 genome. The E1B BstEII/HindII fragment corresponded to Ad5 nt 1916 to 2804. The E2a hybridization probe was a SfiI/PvuI restriction endonuclease fragment containing nt 23000 to 23505 in the Ad5 genome. The E3 probe was a 1,877-bp PCR product generated from Ad5 by using the following primers: 5′-CTAGAATCGGGGTTGGG-3′ and 5′-TCTAGGGTGTCAGTCATCTCC-3′. The E4 hybridization probe was the XmaI restriction endonuclease fragment containing nt 33092 to 35354 of the Ad5 genome. The VA probe was an XbaI/NruI fragment containing nt 10580 to 11338 of Ad2. Equal amounts of RNA were separated on a 1% formaldehyde gel, transferred to a nitrocellulose membrane, hybridized to 32P-labeled probes, and exposed to film. Relative RNA levels were determined using a Molecular Dynamics Storm 840 PhosphorImager and ImageQuant 5.0 software.
RESULTS
Wild-type AAV-mediated inhibition of AdlacZ5 replication.
A number of assays in this study were conducted with the Ad5-based vector AdlacZ5, which contains the E. coli lacZ gene in place of 1.88 kb of the E3 gene. It has not yet been verified that the E3 promoter directs transcription of the lacZ gene in AdlacZ5. However, the amount of β-gal activity should be directly proportional to viral genome amplification. To verify that β-gal activity correlates with the amount of AdlacZ5 in a sample, we infected HeLa cells with AdlacZ5 at various MOIs and measured β-gal activity at 48 hpi using the luminometer-based Galacto-Star system (data not shown). The results confirmed that β-gal activity is directly proportional to AdlacZ5 levels. Thus, the β-gal assay is a rapid, quantitative indicator of relative AdlacZ5 amounts.
The β-gal system was used to quantify the effects of increasing levels of AAV on Ad propagation. We conducted primary coinfections by inoculating HeLa cells with AAV (0, 1, 10, 100, 500, or 1,000 IU) and AdlacZ5 (1, 5, 10, or 100 MOIs). The cells were harvested at 48 hpi, and cell extracts were assayed for β-gal activity. At all tested multiplicities of AdlacZ5, AAV exerts a dose-dependent inhibition of Ad-directed β-gal activity (Fig. 1A). The largest decrease in β-gal activity was observed with between 10 and 100 IU of AAV.
FIG. 1.

Increasing amounts of AAV resulted in decreased AdlacZ5 β-gal activity and virus production. (A) Primary AAV and AdlacZ5 coinfections were conducted in HeLa cells, harvested at 48 hpi, and assayed for β-gal activity. (B) Secondary infections were performed by inoculating fresh HeLa cells with 50-μl aliquots of primary coinfection cultures. Heparin was included to prevent infection with AAV. Cultures were harvested at 24 hpi and tested for β-gal activity. In both panels, the numbers on the z axis refer to the MOIs of AdlacZ5 used in the coinfections. The numbers on the x axis indicate the IU of AAV. The β-gal activity is expressed on the y axis as the percentage of β-gal activity relative to that of cells infected with AdlacZ5 alone. Coinfections were conducted on 12 separate occasions.
Although β-gal activity correlates with titers of AdlacZ5 in the absence of AAV, coinfection with AAV may alter expression from the E3 promoter and/or the lacZ cassette. Thus, the decrease in β-gal activity described above may reflect either inhibition of lacZ gene expression or inhibition of AdlacZ5 amplification. To measure the level of virus production, we conducted secondary infections by inoculating a fresh plate of HeLa cells with aliquots of infectious media from the experiments whose results are shown in Fig. 1A. If amplification of the virus were blocked in the primary infection, there would be less AdlacZ5 virus in the inoculum for secondary infection. To eliminate potential AAV-mediated inhibition of β-gal activity, the secondary infections were performed in the presence of heparin, which blocks the uptake of AAV produced in the primary infection. Thus, the amount of β-gal activity in the secondary infections is directly proportional to the amount of AdlacZ5 produced in the primary infection, regardless of AAV's effects on the E3 promoter or lacZ expression. Figure 1B shows a similar dose-dependent inhibition of Ad production. During coinfection with 1,000 IU of AAV, maximum 10-fold and 50-fold decreases in β-gal activity were observed in primary and secondary infections, respectively. Together, these results demonstrate that AAV-mediated inhibition of Ad replication is dose dependent at various multiplicities of Ad and that near-maximum suppression of β-gal activity occurs in the first infection with 100 IU of AAV.
We next used the β-gal system to determine whether AAV is able to inhibit Ad replication in the absence of Rep protein expression or AAV amplification. We coinfected HeLa cells with AdlacZ5 and either UV light-inactivated virus (UV-AAV), which is incapable of DNA amplification and protein expression, or the recombinant AAV vector vAVluc, which does not express AAV Rep or Cap (31). The β-gal activity in the presence of UV-AAV or vAVluc was equivalent to that of samples lacking an AAV vector (data not shown). The inability of UV-AAV or vAVluc to decrease β-gal activity reveals that the AAV virion alone is unable to inhibit Ad propagation. It also indicates that AAV gene expression or DNA amplification is required for the reduction of Ad replication.
AAV-mediated inhibition of Ad DNA synthesis.
Previous studies indicate that Ad DNA synthesis is inhibited up to 10-fold in the presence of AAV. To determine whether inhibition of Ad DNA synthesis is also dose dependent, we conducted Southern analysis of viral DNA isolated from coinfections. This also served as verification that the reduced β-gal activity above was due to decreased Ad production, which would yield fewer genomes, and not inhibition of lacZ gene expression. HeLa cells were coinfected with increasing IU of AAV and 1, 5, or 10 MOIs of AdlacZ5. The cultures were harvested at 48 hpi, and viral DNA was isolated. Southern hybridization analysis revealed dramatic effects of AAV on Ad DNA replication, as shown in Fig. 2. PhosphorImager analysis revealed a 10- to 40-fold inhibition of Ad DNA synthesis. The maximum level of inhibition is evident when AAV is present at 100 IU, regardless of the amount of Ad (Fig. 2, top panels, lanes 4). One possible explanation is that AAV may target a cellular factor(s) that is titrated out when the amount of AAV reaches the level of 100 infectious units per cell. The increasing levels of AAV also resulted in a modest decrease in AAV replicative-form DNA. This phenomenon, known as autoinhibition, has been observed previously and is believed to result from defective interfering particles that are more abundant at higher viral concentrations (6).
FIG. 2.

Steady-state AdlacZ5 DNA levels decreased during coinfections. Southern analysis was conducted using HeLa cells coinfected with increasing amounts of AAV and 1, 5, or 10 MOIs of AdlacZ5, as denoted above the panels. Whole-cell DNA was isolated at 48 hpi and analyzed by Southern hybridization. The membranes shown above the lane numbers were surveyed with an Ad-specific probe, while the lower membranes were assayed with an AAV-specific probe. Autoradiography was conducted for visualization. In all panels, the DNA in lane 1 was harvested from cells infected with AdlacZ5 alone. Lanes 2 to 6 contain DNA from cells that were coinfected with AdlacZ5 and 1, 10, 100, 500, and 1,000 IU of AAV, respectively. RFd indicates AAV replicative-form dimer. RFm indicates replicative-form monomer, and SS indicates single-stranded AAV genome.
AAV inhibits Ad early and late gene expression.
Previous studies in our lab demonstrated that AAV exerts a two- to fourfold suppression of Ad E2a protein expression and steady-state levels of E2a mRNA (19, 25). To determine whether AAV alters the transcription of other Ad genes, Northern analyses were performed with equal amounts of total RNA obtained from HeLa cells coinfected with AdlacZ5 (MOI, 5) and AAV (0, 1, 10, 100, or 500 IU). The cells were harvested at 24 hpi and assayed for β-gal activity to confirm that inhibition was observed (data not shown). The 28S and 18S RNA bands confirmed equal loading of RNA (Fig. 3). Following hybridization of probes specific for Ad E1A, E1B, E2A, E4, VA, and hexon, we observed a dose-dependent response to coinfecting AAV. However, not all transcription units were affected equally. According to PhosphorImager analysis, E1a mRNA levels declined approximately twofold in the presence of 500 IU of AAV with respect to those of samples infected with Ad alone. E2a levels declined three- to fourfold. Coinfection with 500 IU of AAV caused a fourfold decrease in E1B mRNA. However, we often observed a modest increase in E1B levels in the presence of 1 IU of AAV compared with that observed in the presence of Ad alone. E4- and hexon-probed Northern blots revealed a striking 8-fold and 12-fold respective decrease in RNA.
FIG. 3.

Increasing titers of AAV reduced Ad steady-state transcripts to various degrees. Northern analysis was conducted using total mRNA harvested at 24 hpi from HeLa cells infected with AdlacZ5 (MOI, 5) and AAV (0, 1, 10, 100, or 500 IU). To assay E3 expression, Ad5 was used in place of AdlacZ5. Equal amounts of RNA were separated by 1% formaldehyde agarose gel electrophoresis, analyzed by Northern hybridization, and visualized by autoradiography. To confirm equal loading, ribosomal 28S and 18S bands were visualized by ethidium bromide staining. A representative gel is shown. Locations of size standards in kilobases are labeled on the right. AAV transcripts are labeled according to their promoter of origin.
We conducted parallel Northern analyses, substituting wild-type Ad5, which contains the E3 gene, for AdlacZ5 and probing for Ad E2A, E4, or hexon. There was no observed difference between gene expression of Ad5 and that of AdlacZ5 (data not shown). Next, we examined the effects of increasing doses of AAV on steady-state levels of Ad5 E3 transcripts. Unlike the other Ad transcription units, AAV did not affect all E3 transcripts equally. There was a dramatic decrease in the smaller E3 mRNAs in the presence of only 1 IU of coinfecting AAV. Increasing the AAV inoculum had little additional effect. In contrast, AAV induced only minor inhibition on the larger E3 mRNAs at 500 IU of AAV. A small dose-dependent decrease in the larger E3 transcripts was occasionally observed.
The unique effects of AAV on E3 expression suggest that AAV induces Ad-specific inhibition and not global effects within the cell. For example, AAV could generate a degrading cellular environment or exert widespread effects on cellular and viral transcription. To confirm that AAV did not mediate global effects on gene expression, we probed the Northern membranes above for cellular GAPDH (glyceraldehyde-3-phosphate dehydrogenase) transcripts. As shown in a representative blot in Fig. 3, GAPDH levels remained constant in the presence of increasing amounts of AAV, indicating that the inhibition of Ad transcription is not global in nature.
The levels of AAV Rep and Cap mRNAs were also examined. There was a slight increase in Rep and Cap mRNAs as IU of AAV increased from 1 to 10. However, we observed a decrease in AAV gene expression when input AAV was 100 IU or greater. Similar to the reduction in AAV replicative-form DNA described above, the decrease in AAV transcripts is likely due to autoinhibition (6).
Ad protein levels in the presence of AAV.
Previous reports indicate that Rep proteins are capable of inhibiting Ad protein translation during plasmid transfections (25, 33, 34). We therefore conducted an immunoblot analysis to determine whether AAV exerts translational effects on Ad gene expression during coinfection. HeLa cell monolayers were coinfected with increasing levels of wild-type AAV and 5 or 10 MOIs of AdlacZ5. The cultures were harvested at 24 hpi and tested for β-gal activity to verify that inhibition was observed. After cellular extracts were prepared, equal amounts of total protein were analyzed by immunoblot analyses to determine the effects of AAV on Ad protein expression. As shown in Fig. 4, increasing amounts of AAV resulted in a dose-dependent decrease in Ad protein expression. E1a and E2a protein levels were reduced one- to twofold and two- to threefold, respectively. The effects of AAV on E4orf6/7 resulted in a more pronounced 10- to 12-fold decrease. We also looked at hexon levels as a representative protein that is produced from the Ad major late promoter. Ad hexon levels were reduced approximately 17-fold. Similar to AAV mRNA levels, Rep and Cap protein levels decreased in coinfections with high titers of AAV. For all Ad genes tested, the steady-state protein levels corresponded with mRNA levels. This suggests that AAV Rep proteins do not exert significant translational modulation during coinfection.
FIG. 4.
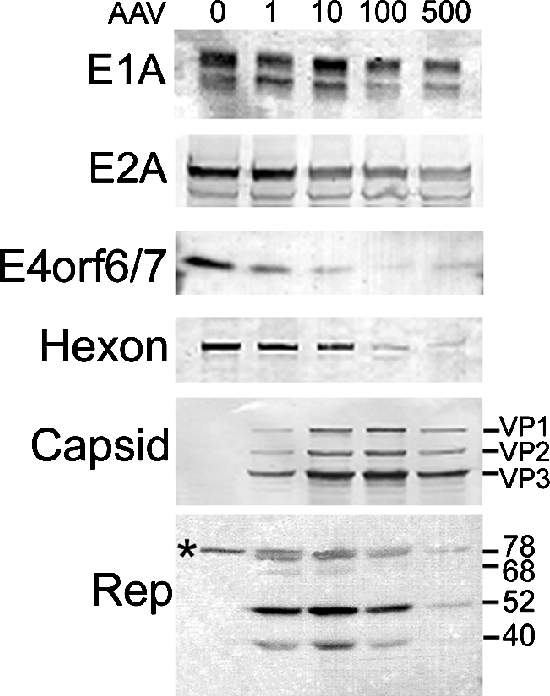
Levels of Ad proteins during coinfection with AAV parallel mRNA levels. Immunoblot analyses were conducted using lysates from HeLa cells coinfected with AAV and AdlacZ5. Cultures were harvested at 48 hpi, and infection was verified by β-gal activity. Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and probed with antibodies specific to Ad or AAV proteins, as denoted to the left of the images. Lane 1 is from AdlacZ5-infected cells (MOI, 5 or 10). Lanes 2 to 5 are from cells coinfected with AdlacZ5 and AAV (1, 10, 100, and 500 IU, respectively). The uppermost band in the Rep panel, which is labeled with an asterisk, indicates a nonspecific interaction.
Temporal expression of Ad mRNA in the presence of AAV.
The assays described above depict steady-state viral gene expression at 24 hpi. To determine when coinfecting AAV exerts its effects on Ad gene expression, we performed a temporal analysis using HeLa cells infected with Ad5 (MOI, 5) in the presence or absence of AAV (100 IU). Cultures were harvested at 3, 6, 9, 12, or 24 hpi. Equal amounts of total RNA were analyzed by Northern analyses using radiolabeled probes specific for Ad, AAV, or cellular transcripts.
The temporal effects of AAV on Ad RNA levels are presented in Fig. 5. We were unable to identify any viral mRNAs at 3 hpi. Although E1A is the first Ad gene to be expressed during an infection, E1a mRNAs do not accumulate early in the infection due to a short half-life (37). As a result, E1A mRNA was visible at 6 hpi only following overexposure of the blot. Coinfecting AAV induced a significant decrease in E1B mRNA beginning at 9 hpi. E3 also demonstrated a slight decrease in mRNA at 9 and 12 hpi. However, this early E3 inhibition was minor compared to the pronounced effects observed at 24 hpi. E2A and E4 mRNA levels were unaffected prior to the 24-hpi time point. Expression of E1A and VA transcripts was inhibited throughout the course of the study. The levels of GAPDH transcripts remained constant throughout the infection, indicating that AAV does not exert global effects within the cell. Similar to what we observed with Ad, the earliest time point at which we observed AAV mRNA transcripts was 6 hpi.
FIG. 5.
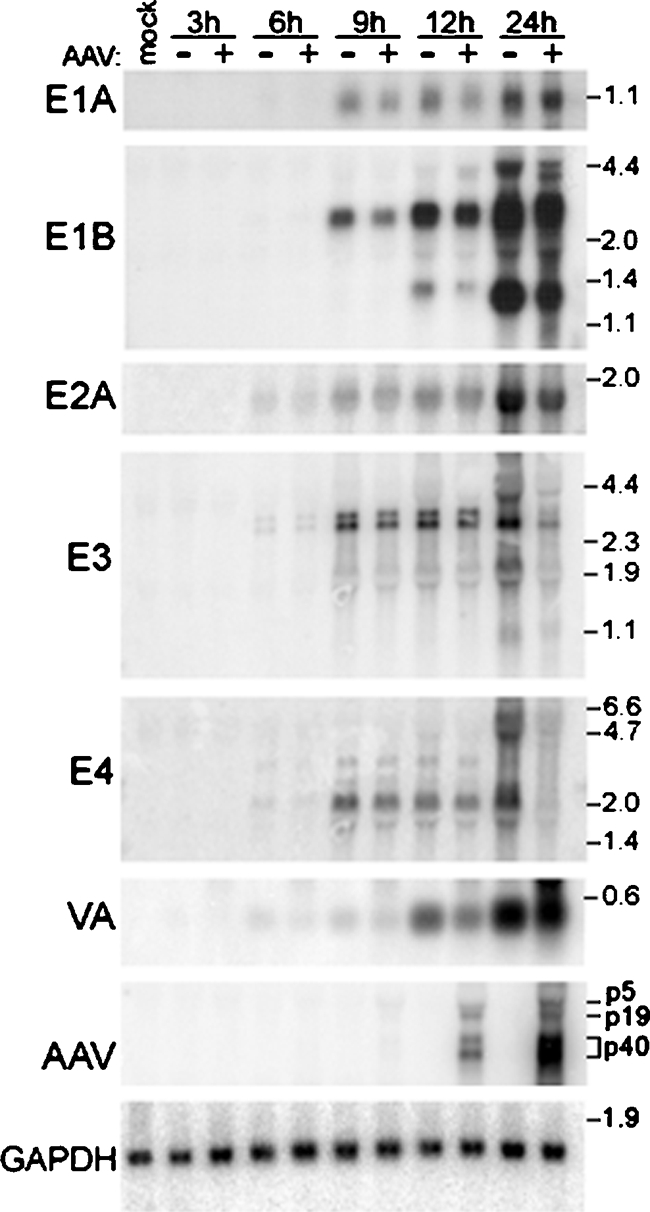
The temporal expression of mRNA for Ad transcription units varied in response to coinfecting AAV. HeLa cells were infected with Ad5 (MOI, 5) in the presence or absence of AAV (100 IU). Cultures were harvested at various times ranging from 3 to 24 hpi. Equal amounts of total RNA were resolved by electrophoresis in 1% formaldehyde gels and transferred to nitrocellulose membranes. Northern hybridization analysis was conducted using probes specific for the indicated Ad or AAV transcripts. Visualization and quantitation were conducted with autoradiography and PhosphorImager analysis. Membranes were subsequently stripped and probed with a GAPDH-specific probe to confirm equal loading. A representative GAPDH blot is shown. Molecular size markers (kb) are shown to the right of the blots. AAV transcripts are labeled according to their promoter of origin.
These results demonstrate that the effects of AAV on individual transcription units vary both in time and in degree of inhibition. In general, inhibition of Ad mRNA levels was more pronounced later in the coinfections. One explanation is that the AAV inhibition of Ad DNA replication may play a large role in reducing Ad gene products since lower Ad DNA levels would provide fewer transcriptional templates. Alternatively, the less dramatic effects at 9 and 12 hpi may be the result of low early-phase Rep expression under the conditions of these assays. We were able to visualize Rep mRNA as early as 6 hpi, and an immunoblot analysis from a previous report identified low levels of Rep78 protein at 4 hpi (28). It is therefore likely that Rep is expressed prior to the onset of Ad replication. However, the level of Rep expression early in the coinfection may be insufficient to substantially inhibit transcription of most Ad genes.
To determine whether Rep expression alone is able to inhibit Ad early-phase gene expression, we induced Rep expression by plasmid transfection, initiated Ad infection, and analyzed the effects on Ad mRNA levels by using Northern analysis. To this end, we first used the β-gal assay described above to verify that transfected Rep proteins are capable of inhibiting Ad production. HeLa cells were transfected with the pCDMRep series of plasmids, which express the Rep proteins from the cytomegalovirus promoter (25, 39). We also tested plasmids that express mutant Rep proteins lacking the PNB site. The cultures were infected with AdlacZ5, incubated for 48 h, harvested, and tested for β-gal activity. Immunoblot analysis confirmed that equal amounts of all Rep proteins were expressed following plasmid transfections (data not shown). Aliquots of these cultures were also used to inoculate fresh HeLa cells in order to eliminate Rep-mediated effects on β-gal activity and measure the amount of infectious virus produced during the primary transfection assay. The β-gal activity of pCDM8-transfected control cells was set at 100%. The β-gal activity of Rep-transfected cells was expressed as the percentage relative to that of pCDM8-transfected cells. The results of both the primary transfection and secondary infection indicate that Rep78 expression mediated the largest reduction in Ad replication (Fig. 6). Rep68 and Rep52 had an intermediate effect, and Rep40 caused no inhibition. The PNB mutant versions of the Rep proteins were as effective as the wild-type proteins, suggesting that AAV-mediated inhibition of Ad production may not require ATPase or helicase activity. These results also indicate that Rep expression alone is sufficient to inhibit Ad propagation. The differences in inhibition of Ad replication mediated by Rep78 and AAV may be due to the inherent differences between plasmid transfections and virus infections. In a plasmid transfection, there are no AAV replication or transcription centers that may alter Ad replication indirectly. Another possibility is that full inhibition of Ad replication may require one or more of the other Rep proteins.
FIG. 6.

AAV Rep proteins inhibit AdlacZ5 production to different degrees. (A) HeLa cells were transfected with pCDMRep constructs, as indicated on the x axis, and subsequently infected with AdlacZ5. Cell lysates were tested for β-gal activity at 48 hpi. (B) Aliquots (50 μl) from transfected cultures were used to inoculate fresh HeLa cells. The secondary infections were harvested at 24 hpi and tested for β-gal activity. The β-gal activity of the pCDM8 empty vector control (not shown) was set at 100%. Relative β-gal activities of samples that were transfected with wild-type Rep and PNB mutant plasmids are shown in black and gray, respectively. Error bars represent the standard deviations from six experiments conducted in triplicate (n = 18).
Since E2a and E4 proteins are essential for efficient Ad DNA amplification, we considered the possibility that Rep-mediated E2a and E4 transcriptional regulation contributes to decreased Ad DNA replication. Previous studies suggested that AAV Rep proteins interact with the E2A promoter and regulate its activity (7). Also, our studies indicate that E4 mRNA levels are dramatically reduced at 24 hpi during coinfection with AAV (Fig. 3). However, temporal Northern analyses suggested that observable AAV-mediated transcriptional effects on E2A and E4 did not occur prior to 12 hpi and therefore do not precede Ad DNA replication. This may be the result of low early-phase Rep expression in our coinfections. Therefore, after determining that expression of Rep proteins alone can inhibit Ad, we conducted Northern analyses to determine whether high levels of Rep proteins suppress Ad E2A and E4 transcription prior to DNA replication. HeLa cells were transfected with CDM8 or CDMRep78 and incubated for 20 to 24 h to permit Rep expression. The cells were then infected with Ad5 (MOI, 5) and harvested at various times after infection. Immunoblot analysis confirmed Rep protein expression at the time of Ad infection (not shown). Northern analyses indicated that even in the presence of high levels of Rep78, inhibition of E2A and E4 transcription was not observed until the 24-h time point (Fig. 7). This suggests that Ad DNA replication precedes AAV modulation of early gene expression. It is therefore possible that the decrease in E2A and E4 transcripts is the result of decreased Ad template and not transcriptional regulation.
FIG. 7.

AAV Rep proteins decreased E2A and E4 mRNA transcript levels during the late phase of Ad infection. HeLa cells were transfected with pCDMRep78G or the empty vector pCDM8, as indicated at the top of the figure. After being incubated for 20 to 24 h to permit Rep expression, the cells were infected with 5 MOIs of Ad5. Infected cultures were harvested at 6, 9, 12, or 24 hpi. Uninfected (Uninf) cells were harvested at 24 hpi. Total RNA was prepared, and equal amounts were analyzed by Northern analysis. Equal loading was confirmed by ethidium bromide staining of rRNA as well as GAPDH analysis (not shown). Western analysis was conducted with samples harvested at 6 hpi to confirm Rep78 expression (not shown). Locations of size standards are noted in kilobases.
Ad E2A and E4 gene expression in the absence of DNA synthesis.
To further test the hypothesis that decreased Ad template numbers precede the reduction in E2A and E4 mRNA, we examined the expression of these early genes in the absence of DNA replication. Hydroxyurea (HU) is an inhibitor of ribonucleotide reductase that is commonly used to inhibit DNA synthesis. As a result of preventing the early-to-late phase shift, HU prolongs the Ad early phase and promotes the accumulation of early transcripts. To determine the effects of AAV on E2A and E4 mRNA in the absence of Ad template amplification, we coinfected HeLa cells with AAV (100 IU) and Ad5 (MOI, 5) and treated the cells with HU at 6, 9, or 12 hpi. The cells were harvested at 24 hpi, total RNA was isolated, and Northern analysis was conducted with E2A- and E4-specific probes. Since Ad DNA replication commences at approximately 6 to 10 hpi, HU treatment at 6 hpi essentially blocks all viral DNA replication. Delaying HU treatment until 12 hpi permits modest levels of DNA replication to occur prior to its inhibition (Fig. 8B). No decrease in E2A or E4 mRNA was observed (Fig. 8A) in the absence of DNA replication (Fig. 8B). Delaying or eliminating HU treatment restores AAV-mediated inhibition. These results suggest that it is unlikely that AAV-mediated inhibition of Ad E2A and E4 gene expression is solely responsible for the observed suppression of Ad DNA synthesis. Instead, it appears that a decrease in Ad template may substantially contribute to the late-phase decrease in Ad early gene expression.
FIG. 8.
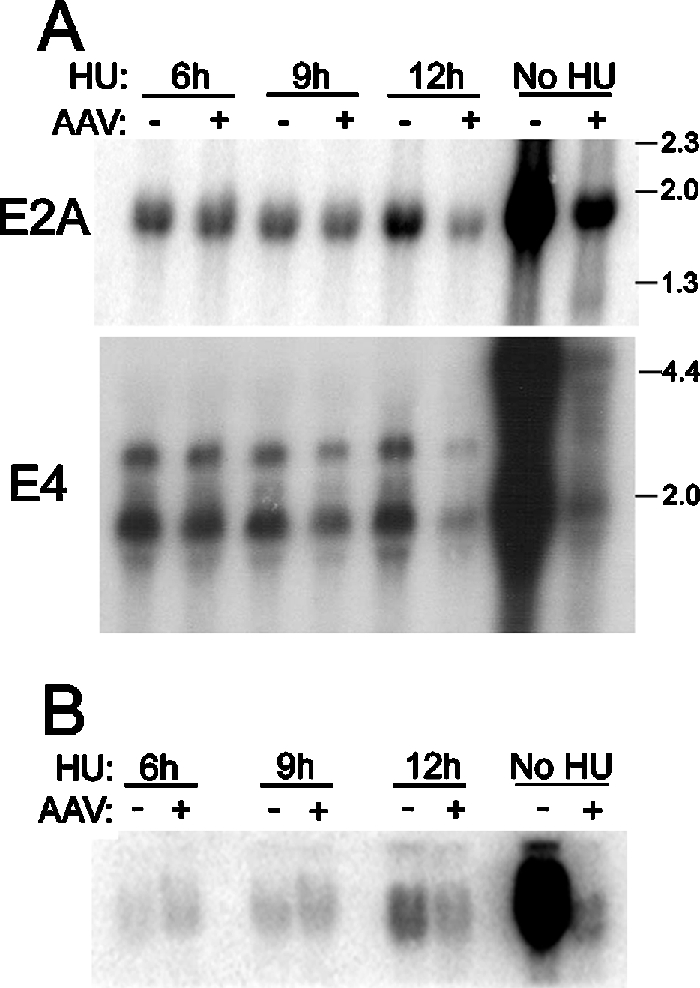
Coinfection with AAV did not reduce E2A or E4 transcript levels in the absence of DNA replication. HeLa cells were infected with Ad5 in the presence (+) or absence (−) of coinfecting AAV, as indicated above the top panel. HU was added to the medium at 6, 9, or 12 hpi. Untreated controls were also included. All cultures were harvested at 24 hpi, total RNA and viral DNA were prepared. (A) Equal amounts of RNA were subjected to Northern analysis using Ad E2A- and E4-specific probes. GAPDH and rRNA analyses confirmed equal loading (not shown). Molecular size markers are given in kilobases. (B) Viral DNA was isolated and analyzed by Southern hybridizations with a radiolabeled Ad DNA probe.
DISCUSSION
In 1984, Binger and Flint published a detailed study of Ad early and intermediate mRNA expression (3). However, a similar investigation of Ad gene expression in the presence of AAV has yet to be published. The work presented here provides the most comprehensive examination yet of the effects of AAV on Ad propagation, DNA synthesis, and gene expression.
Using a readily assayable Ad5 vector that expresses the E. coli lacZ gene in place of the E3 gene, we demonstrate that coinfection of 1 to 1,000 IU of AAV limits Ad replication and virus production. Depending on viral titers, Ad DNA levels in coinfected cells were decreased 10- to 40-fold. This inhibition was greater than what we have previously observed (19). The difference is likely due to the larger AAV-to-Ad ratios used in this work. Control experiments demonstrated that Ad replication was not inhibited by UV-inactivated AAV or a recombinant AAV vector lacking Rep and Cap genes. Therefore, either an AAV gene product or viral DNA amplification is responsible for the inhibition.
Northern and immunoblot analyses of Ad gene expression in the presence of increasing levels of AAV indicate that expression of all early transcription units was affected by coinfecting AAV, albeit to different degrees. We observed a modest decrease in E1A RNA and protein levels and confirmed a previously observed two- to fourfold diminution of E2a expression during coinfection (19). E1B and VA RNAs were inhibited at an intermediate level. However, E1B transcripts commonly increased with 1 IU of AAV and decreased linearly with additional AAV. The largest decrease in early gene expression was observed for the E4 gene. The eightfold decrease in total E4 mRNA levels is corroborated by a comparable decrease in E4orf6/7 protein, which we have used as a representative protein from the E4 gene. Although the aforementioned genes are inhibited to various degrees, they all display a linear dose dependency. E3 mRNA transcripts, however, exhibit a distinctive pattern in which the smaller transcripts are more strongly inhibited than the larger ones. The mechanisms responsible for this unique response to AAV remain to be explored.
Rep-mediated inhibition of protein translation has previously been reported (25, 33, 34). However, these effects were observed using in vitro assays or plasmid transfections. The role of AAV-induced translational effects during coinfection has not been described. Although our experiments were not designed to quantitatively match Ad protein and mRNA levels, the correlation in levels shown here suggests that Rep-mediated posttranscriptional regulation does not play a prominent role in Ad gene expression during coinfection.
Modulation of Ad early gene expression during coinfection may occur at the level of transcription. Previous reports suggesting that Rep proteins may alter Ad transcription are bolstered by evidence that coinfecting AAV does not equally affect the expression of the early genes. Since early gene expression is required for DNA replication, inhibition of Ad early gene promoters could be responsible for decreased DNA synthesis. Neither the 10- to 12-fold decrease in E4 protein levels nor the 2- to 3-fold decrease in E2A levels alone would likely be sufficient to cause the dramatic decrease in Ad DNA synthesis. However, their additive effects could be substantial. Given the central roles of E2a and E4 proteins in Ad DNA replication, we considered the possibility that modest effects on the E2a promoter in concert with more-significant inhibition of the E4 promoter could result in reduced Ad DNA synthesis. In order for this to be true, transcriptional effects on early gene expression would have to precede the onset of DNA replication.
Time course Northern analyses (Fig. 5) indicated that only E1B was inhibited by AAV prior to 12 hpi and that this inhibition was minor compared to the dramatic late-phase inhibition of E4. We considered the possibility that under the conditions of our assays, we achieved only low levels of Rep expression during the Ad early phase. This could prevent potential AAV transcriptional effects from occurring until Rep levels increase. Therefore, we transfected HeLa cells with the pCDMRep plasmids, allowed up to 24 h to permit Rep expression, and then infected the cells with AdlacZ5. Subsequent Northern analysis indicated that even in the presence of overexpressed Rep proteins, inhibition of E2A and E4 transcription does not precede DNA replication. It is therefore unlikely that decreased early gene expression is responsible for AAV-mediated inhibition of Ad DNA synthesis. However, this does not imply that Rep does not inhibit Ad at the transcriptional level. If inhibition of DNA replication were the only means by which AAV inhibited Ad, the decrease in template would induce a proportional decrease in all transcripts. Instead, Ad early promoters are inhibited by coinfecting AAV to different degrees and at different times. The unique effects of AAV on E3 mRNAs are even more striking. Together, these data support the hypothesis that AAV also regulates Ad transcription during coinfection.
Hydroxyurea treatment of infected cultures indicated that no inhibition of E2A and E4 early gene expression was observed in the absence of DNA replication. This could imply that the reduced gene expression that we observed at 24 hpi was exclusively the result of lower levels of Ad template. Although these hypotheses appear to contradict one another, the role of AAV's effects on transcription and the reduction in DNA template may not be mutually exclusive. In blocking DNA synthesis, HU also prevents the onset of the late phase of Ad infection. The early-to-late switch in infection induces numerous changes in viral regulation. For example, there are phase-specific mechanisms that reduce early gene expression in trans during the late phase when transcription is measured per genome (15). These effects require expression from the Ad major late promoter, which does not occur in the presence of HU. It is therefore possible that coinfecting AAV exerts transcriptional effects only under late-phase conditions. Thus, although DNA synthesis likely precedes E2A and E4 inhibition, decreased template amplification may not be solely responsible for decreased early gene expression.
The purpose of this study was to observe the effects of AAV on Ad propagation, DNA replication, and gene expression. It has long been known that AAV inhibits Ad during coinfection, but the details of this interaction have not yet been closely examined. Conducting dose-response and temporal analyses revealed that the effects of AAV during coinfection vary based on the AAV-to-Ad ratio and the time point of the infection. In addition, this study suggests that AAV modulation of DNA replication occurs prior to, and independently of, the inhibition of early gene expression. While this observation invites speculation, unraveling the mechanisms of AAV-mediated inhibition remains outside the scope of this study. Nonetheless, the data presented here provide essential, fundamental knowledge that will support and guide future mechanistic studies.
Acknowledgments
We thank Frank Graham for providing us with the AdlacZ5 virus, Tom Shenk for the E4 antisera, and Arnold Berk for the E2a antisera. We thank Roy F. Collaco for helpful consultation and preparation of Rep and Cap antibodies.
This work was supported by the National Institutes of Health (AI51471 to J.P.T., GM64765 to J.P.T., and AI64129 to J.M.T.).
REFERENCES
- 1.Atchison, R. W. 1970. The role of herpesviruses in adenovirus-associated virus replication in vitro. Virology 42:155-162. [DOI] [PubMed] [Google Scholar]
- 2.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.). 1989. Current protocols in molecular biology, vol. 2. John Wiley and Sons Inc., New York, N.Y.
- 3.Binger, M. H., and S. J. Flint. 1984. Accumulation of early and intermediate mRNA species during subgroup C adenovirus productive infections. Virology 136:387-403. [DOI] [PubMed] [Google Scholar]
- 4.Blacklow, N. R., M. D. Hoggan, and M. S. McClanahan. 1970. Adenovirus-associated viruses: enhancement by human herpesviruses. Proc. Soc. Exp. Biol. Med. 134:952-954. [DOI] [PubMed] [Google Scholar]
- 5.Buller, R. M. L., J. E. Janik, E. D. Sebring, and J. A. Rose. 1981. Herpes simplex virus types 1 and 2 completely help adenovirus-associated virus replication. J. Virol. 40:241-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carter, B. J., C. A. Laughlin, L. M. de la Maza, and M. Myers. 1979. Adeno-associated virus autointerference. Virology 92:449-462. [DOI] [PubMed] [Google Scholar]
- 7.Casper, J. M., J. M. Timpe, J. D. Dignam, and J. P. Trempe. 2005. Identification of an adeno-associated virus Rep protein binding site in the adenovirus E2a promoter. J. Virol. 79:28-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casto, B. C., R. W. Atchison, and W. M. Hammon. 1967. Studies on the relationship between adeno-associated virus type 1 (AAV1) and adenoviruses. I. Replication of AAV1 in certain cell cultures and its effect on helper adenoviruses. Virology 32:52-59. [DOI] [PubMed] [Google Scholar]
- 9.Chejanovsky, N., and B. J. Carter. 1989. Mutagenesis of an AUG codon in the adeno-associated virus rep gene: effects on viral DNA replication. Virology 173:120-128. [DOI] [PubMed] [Google Scholar]
- 10.Chiorini, J. A., B. Zimmermann, L. Yang, R. H. Smith, A. Ahearn, F. Herberg, and R. M. Kotin. 1998. Inhibition of PrKX, a novel protein kinase, and the cyclic AMP-dependent protein kinase PKA by the regulatory proteins of adeno-associated virus type 2. Mol. Cell. Biol. 18:5921-5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coenjaerts, F. E., and P. C. van der Vliet. 1995. Adenovirus DNA replication in a reconstituted system. Methods Enzymol. 262:548-560. [DOI] [PubMed] [Google Scholar]
- 12.Collaco, R., and J. P. Trempe. 2003. A method of helper virus-free production of adeno-associated virus vectors, p. 237-254. In C. A. Machida (ed.), Virus vectors for gene therapy: methods and protocols, vol. 76. Humana Press, Totowa, N.J. [DOI] [PubMed] [Google Scholar]
- 13.Costello, E., P. Saudan, E. Winocour, L. Pizer, and P. Beard. 1997. High mobility group chromosomal protein 1 binds to the adeno-associated virus replication protein (Rep) and promotes Rep-mediated site-specific cleavage of DNA, ATPase activity and transcriptional repression. EMBO J. 16:5943-5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Pasquale, G., and J. A. Chiorini. 2003. PKA/PrKX activity is a modulator of AAV/adenovirus interaction. EMBO J. 22:1716-1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fessler, S. P., and C. S. Young. 1998. Control of adenovirus early gene expression during the late phase of infection. J. Virol. 72:4049-4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Halbert, D. N., J. R. Cutt, and T. Shenk. 1985. Adenovirus early region 4 encodes functions required for efficient DNA replication, late gene expression, and host cell shutoff. J. Virol. 56:250-257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hermonat, P. L., A. D. Santin, R. B. Batchu, and D. Zhan. 1998. The adeno-associated virus Rep78 major regulatory protein binds the cellular TATA-binding protein in vitro and in vivo. Virology 245:120-127. [DOI] [PubMed] [Google Scholar]
- 18.Hirt, B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 26:365-369. [DOI] [PubMed] [Google Scholar]
- 19.Jing, X. J., V. Kalman-Maltese, X. Cao, Q. Yang, and J. P. Trempe. 2001. Inhibition of adenovirus cytotoxicity, replication, and E2a gene expression by adeno-associated virus. Virology 291:140-151. [DOI] [PubMed] [Google Scholar]
- 20.King, J. A., R. Dubielzig, D. Grimm, and J. A. Kleinschmidt. 2001. DNA helicase-mediated packaging of adeno-associated virus type 2 genomes into preformed capsids. EMBO J. 20:3282-3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laughlin, C. A., M. Myers, D. L. Risin, and B. J. Carter. 1979. Defective-interfering particles of the human parvovirus adeno-associated virus. Virology 94:162-174. [DOI] [PubMed] [Google Scholar]
- 22.McPherson, R. A., L. J. Rosenthal, and J. A. Rose. 1985. Human cytomegalovirus completely helps adeno-associated virus replication. Virology 147:217-222. [DOI] [PubMed] [Google Scholar]
- 23.Mittal, S. K., A. J. Bett, L. Prevec, and F. L. Graham. 1995. Foreign gene expression by human adenovirus type 5-based vectors studied using firefly luciferase and bacterial beta-galactosidase genes as reporters. Virology 210:226-230. [DOI] [PubMed] [Google Scholar]
- 24.Muzyczka, N., and K. I. Berns. 2001. Parvoviridae: the viruses and their replication, p. 2327-2359. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams and Wilkins, Philadelphia, Pa. [Google Scholar]
- 25.Nada, S., and J. P. Trempe. 2002. Characterization of adeno-associated virus Rep protein inhibition of adenovirus E2a gene expression. Virology 293:345-355. [DOI] [PubMed] [Google Scholar]
- 26.Pereira, D. J., and N. Muzyczka. 1997. The cellular transcription factor SP1 and an unknown cellular protein are required to mediate Rep protein activation of the adeno-associated virus p19 promoter. J. Virol. 71:1747-1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prasad, C. K., C. Meyers, D. J. Zhan, H. You, M. Chiriva-Internati, J. L. Mehta, Y. Liu, and P. L. Hermonat. 2003. The adeno-associated virus major regulatory protein Rep78-c-Jun-DNA motif complex modulates AP-1 activity. Virology 314:423-431. [DOI] [PubMed] [Google Scholar]
- 28.Redemann, B. E., E. Mendelson, and B. J. Carter. 1989. Adeno-associated virus Rep protein synthesis during productive infection. J. Virol. 63:873-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shenk, T. 2001. Adenoviridae: the viruses and their replication, p. 2265-2300. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams and Wilkins, Philadelphia, Pa. [Google Scholar]
- 30.Shepard, R. N., and D. A. Ornelles. 2004. Diverse roles for E4orf3 at late times of infection revealed in an E1B 55-kilodalton protein mutant background. J. Virol. 78:9924-9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith, A. D., R. F. Collaco, and J. P. Trempe. 2003. Enhancement of recombinant adeno-associated virus type 2-mediated transgene expression in a lung epithelial cell line by inhibition of the epidermal growth factor receptor. J. Virol. 77:6394-6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su, P. F., S. Y. Chiang, C. W. Wu, and F. Y. Wu. 2000. Adeno-associated virus major Rep78 protein disrupts binding of TATA-binding protein to the p97 promoter of human papillomavirus type 16. J. Virol. 74:2459-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeuchi, T., T. Kozuka, K. Nakagawa, Y. Aoki, K. Ohtomo, K. Yoshiike, and T. Kanda. 2000. Adeno-associated virus type 2 nonstructural protein Rep78 suppresses translation in vitro. Virology 266:196-202. [DOI] [PubMed] [Google Scholar]
- 34.Trempe, J. P., and B. J. Carter. 1988. Regulation of adeno-associated virus gene expression in 293 cells: control of mRNA abundance and translation. J. Virol. 62:3356-3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walz, C., A. Deprez, T. Dupressoir, M. Durst, M. Rabreau, and J. R. Schlehofer. 1997. Interaction of human papillomavirus type 16 and adeno-associated virus type 2 co-infecting human cervical epithelium. J. Gen. Virol. 78:1441-1452. [DOI] [PubMed] [Google Scholar]
- 36.Weger, S., M. Wendland, J. A. Kleinschmidt, and R. Heilbronn. 1999. The adeno-associated virus type 2 regulatory proteins Rep78 and Rep68 interact with the transcriptional coactivator PC4. J. Virol. 73:260-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson, M. C., J. R. Nevins, J. M. Blanchard, H. S. Ginsberg, and J. E. Darnell, Jr. 1980. Metabolism of mRNA from the transforming region of adenovirus 2. Cold Spring Harbor Symp. Quant. Biol. 44:447-455. [DOI] [PubMed] [Google Scholar]
- 38.Winters, W. D., and W. C. Russell. 1971. Studies on the assembly of adenovirus in vitro. J. Gen. Virol. 10:181-194. [DOI] [PubMed] [Google Scholar]
- 39.Yang, Q., and J. P. Trempe. 1993. Analysis of the terminal repeat binding abilities of mutant adeno-associated virus replication proteins. J. Virol. 67:4442-4447. [DOI] [PMC free article] [PubMed] [Google Scholar]