

Abstract
To facilitate RNA recombination studies, we tested whether Saccharomyces cerevisiae, which supports brome mosaic virus (BMV) replication, also supports BMV RNA recombination. Yeast strains expressing BMV RNA replication proteins 1a and 2apol were engineered to transiently coexpress two independently inducible, overlapping, nonreplicating derivatives of BMV genomic RNA3. B3Δ3′ lacked the coat protein gene and negative-strand RNA promoter. B3Δ5′ lacked the positive-strand RNA promoter and had the coat gene replaced by the selectable URA3 gene. After 12 to 72 h of induction, B3Δ3′ and B3Δ5′ transcription was repressed and Ura+ yeast cells were selected. All Ura+ cells contained recombinant RNA3 replicons expressing URA3. Most replicons arose by intermolecular homologous recombination between B3Δ3′ and B3Δ5′. Such recombinants were isolated only when 1a and 2apol were expressed and after transient transcription of both B3Δ3′ and B3Δ5′, showing that recombination occurred at the RNA, not DNA, level. A minority of URA3-expressing replicons were derived from B3Δ5′, independently of B3Δ3′, by 5′ truncation and modification, generating novel positive-strand promoters and demonstrating that BMV can give rise to subgenomic RNA replicons. Intermolecular B3Δ3′-B3Δ5′ recombination occurred only when both parental RNAs bore a functional, cis-acting template recognition and recruitment element targeting viral RNAs to replication complexes. The results imply that recombination occurred in RNA replication complexes to which parental RNAs were independently recruited. Moreover, the ability to obtain intermolecular recombinants at precisely measurable, reproducible frequencies, to control genetic background and induction conditions, and other features of this system will facilitate further studies of virus and host functions in RNA recombination.
RNA recombination provides a means for major, immediate changes within viral genomes, thus enabling a virus to modify or to create new genes or regulatory elements, to borrow them from other viruses or the host cell, and to reorganize whole genomes (24, 34). Some of these changes may lead to stable evolutionary jumps. Accordingly, RNA recombination is a major engine of RNA virus evolution and of short-term virus variability and survival (24).
Despite the scientific and practical importance of RNA recombination, many questions remain about its mechanisms. RNA recombination events most commonly have been suggested to occur via template-switching mechanisms during RNA replication (22, 24), but substantial evidence also exists for primer alignment and extension (34) or breakage-rejoining (13, 14) mechanisms. Such alternate mechanisms are not necessarily mutually exclusive, as indicated by evidence for poliovirus RNA recombination by template switching (8, 22), primer alignment and extension (34), and breakage-rejoining (14) mechanisms. Despite extensive studies of some specific classes of RNA recombination events, substantial uncertainties exist about the relative activity of different RNA recombination pathways, the viral and cellular determinants of their activity, and their possible relationships to normal viral replication processes.
To address such fundamental questions and the experimental challenges of low-frequency RNA recombination events, further in vivo experimental systems could be instrumental. We sought to establish a system to induce and to more sensitively and precisely measure intermolecular RNA recombination using brome mosaic virus (BMV) as a model virus and Saccharomyces cerevisiae as a host.
BMV is a member of the alphavirus-like superfamily of positive-strand RNA viruses. The BMV genome is divided into three RNAs. RNA1 and RNA2 encode replication factors 1a and 2apol, respectively. 1a anchors the RNA replication complex to endoplasmic reticulum membranes, induces the invagination of these membranes into 50- to 70-nm replication compartments, recruits viral RNA templates and the polymerase-like 2apol, and provides a helicase-like domain and RNA capping functions for RNA synthesis (38, 42). RNA3 is dispensable for RNA replication in vivo but encodes the noncapsid movement protein 3a and the single capsid protein, which are required for systemic movement in infected plants. 3a is translated from RNA3, while the capsid protein is expressed from a subgenomic mRNA, RNA4 (10). A 250-base intergenic region in RNA3 harbors the promoter for transcribing subgenomic RNA4 from negative-strand RNA3 (Fig. 1A), making expression of the coat protein, or any genes replacing it, dependent on RNA3 replication (10). Initiation of negative-strand RNA synthesis is directed by sequences within a conserved, ∼200-nucleotide (nt) tRNA-like region (TLS) at the 3′ end of all three BMV genomic RNAs (11). Similarly, initiation of positive-strand synthesis requires 5′-proximal noncoding sequences (untranslated regions [UTR]) (11, 15, 17). RNA3 replication additionally requires an ∼150-nt subset of the 250-base intergenic region (42). This template recruitment element (RE) (Fig. 1) is recognized by 1a to recruit RNA3 to the membrane-associated RNA replication compartments (38, 42).
FIG. 1.

(A) DNA-dependent initiation of B3URA3 replication and subgenomic mRNA transcription. The bracketed region represents a cDNA copy of B3URA3. Single lines represent noncoding regions, and the labeled boxes represent the 3a gene, the URA3 gene, the template RE, the subgenomic RNA4 promoter (SG), and the 3′ TLS. Flanking regions represent a 5′-linked GAL1 promoter and 3′-linked hepatitis delta ribozyme. (B) Nonreplicable, overlapping B3URA3 derivatives used as recombination partners. B3Δ3′ and B3Δ5′ RNAs were transcribed in vivo from plasmids carrying the inducible CUP1 or GAL1 promoter, respectively. B3Δ3′ lacks the coat protein gene and the 3′ TLS. The 3′ end is formed by the ADH1 polyadenylation signal (An). B3Δ5′ has the wt RNA3 5′ UTR and the first half of the 3a coding sequence replaced by the GAL1 leader sequence. B3Δ3′ and B3Δ5′ share ∼0.618 kb of overlapping sequence indicated between vertical dashed lines. Homologous recombination within this common region would generate B3URA3. Sequences used for making parental RNA-specific or common probes for Northern blotting are indicated at the bottom.
BMV RNA recombination has been demonstrated (3) and studied in whole plants (29) and in plant cell protoplasts (18) and has been used as a model system to study the mechanisms of both homologous and nonhomologous recombination (29).
BMV directs RNA replication and encapsidation not only in its natural plant hosts but also in yeast (17, 38). As in studies of many cellular processes, yeast provides multiple advantages for studying viral replication, including the ability to study the contributions of viral and host functions (23) and to apply strong selections to large yeast populations to detect low-frequency events such as RNA recombination. To test if yeast also supported BMV RNA recombination and to access yeast advantages for recombination studies, here we report the establishment of a system for controlled induction and precise measurement of BMV RNA3 recombination in yeast, provide the first demonstration of intermolecular RNA recombination in yeast, show that recombination in this system is a by-product of RNA replication, and use this system to reveal the required roles of the template recruitment element in intermolecular RNA recombination. We also demonstrate reproducible recovery of subgenomic RNA replicons from larger parental RNAs a process previously not demonstrated for BMV.
MATERIALS AND METHODS
Yeast.
YPH500 (MATα ura3-52 lys2-801 ade2-101 trp1-63 his3-200 leu2-1) was used in all experiments. Cultures were grown at 30°C in defined synthetic medium containing 2% galactose or 2% glucose. Relevant amino acids were omitted to select for plasmids (19).
Plasmids.
BMV RNA replication proteins 1a and 2apol were expressed from pB1CT19 and pB2CT15, expressing HIS3 and LEU2 selectable markers, respectively (20). When either was omitted, pRS313 (HIS3) or pRS315 (LEU2) “empty” markers (40) were transformed into yeast to allow growth of all cultures in the same medium. Plasmids containing RNA3 derivatives were based on pB3URA3 (Fig. 1A), which contains the TRP1 marker and a full-length RNA3 cDNA between the GAL1 promoter and a self-cleaving hepatitis delta virus ribozyme with the coat gene replaced by URA3 (17). Plasmids expressing RNA3 derivatives from the CUP1 promoter had the LYS2 marker to simultaneously select for two RNA3-encoding plasmids. No empty TRP1 or LYS2 markers were used when RNA3-encoding plasmids were omitted.
Standard procedures were used for all DNA manipulations (37). When necessary, 5′ overhangs were filled in with the Klenow fragment of DNA polymerase I, and 3′ overhangs were blunted with T4 DNA polymerase. Plasmids were verified by restriction digestion and sequencing. Laboratory plasmid designations are given in parentheses.
pB3Δ5′ (pB3HGR1).
The EcoRI-ClaI fragment of pB3URA3 (17) was replaced with the EcoRI-BstBI fragment from pB2YT2 (M. Ishikawa and P. Ahlquist, unpublished results).
pB3Δ3′ (pB3HGR3).
pB3HGR2 was made by inserting the EcoRI-PstI fragment from pB3URA3 into pJBCLYS21 (J. den Boon and P. Ahlquist, unpublished results), creating a plasmid equivalent to pB3URA3 with LYS2 instead of TRP1. To make pB3Δ3′, the EcoRI-SacI fragment of pB3HGR2 was replaced with the EcoRI-BglII fragment from pB3VG1 (23).
pB3ΔBoxB5′ (pB3HGR17).
The BglII-PstI fragment of pB3MS26 (42) was replaced with the equivalent piece from pB3URA3, generating pHGR11. pHGR12 was made by replacing the EcoRI-BglII fragment of pHGR11 with the equivalent piece from pB3Δ5′. To make pHGR13, the PflMI-BglII fragment of pHGR12 was replaced with the equivalent piece from pB3MS46 (42). The BglII-BamHI fragment from pB3MS142 (42) was inserted into the BglII site of pHGR13, generating pHGR16. To make pHGR17, the EcoRI-NcoI fragment of pB3Δ5′ was replaced with the equivalent piece from pHGR16.
pB3ΔBoxB3′ (pB3HGR22).
The EcoRI-PstI fragment from pB3Δ3′ replaced the equivalent piece on pHGR11, generating pHGR19. The ClaI-PpuMI fragment on pHGR19 was replaced with the equivalent piece from pHGR24, generating pHGR21. pHGR24 is a derivative of pB3URA3 with a box B deletion as described for pB3MS142 (42), made by replacing the PflMI-NcoI fragment of pB3URA3 with the equivalent piece from pHGR17. To make pB3ΔB3′, the EcoRI-PstI fragment of pB3Δ3′ was replaced with the equivalent fragment from pHGR21.
pB3Δ3′ΔIR (pB3HGR7).
The ClaI-AfeI fragment of pB3Δ3′ was replaced with a ClaI-AfeI-digested PCR fragment generated using d(GACTACCAACGCAATATGGATTGTCAGAATC) and d(AGCGCTCGGATTTACCTATTTAATTCTAAGCG) with pB3URA3 as a template. Next, the AfeI-PstI fragment was replaced with the AfeI-PstI-digested PCR fragment generated using d(CGACGCAGCGCTAAAAAAAAAAAAAAAAAGATCCGG) and d(GCTATGACCATGATTACGC) with pB3Δ3′ as a template.
Nested deletions of pB3Δ5′.
pHGR110 was made by digesting pHGR12 with HindIII and religating. 5′-terminal deletions were made on pHGR110 and moved into pB3Δ5′. To make pB3Δ817 (pHGR111), the PstI-PflMI fragment of pHGR110 was removed, the plasmid religated, and the resulting EcoRI-AfeI fragment used to replace the corresponding fragment of pB3Δ5′. To make the other deletions, pHGR110 was digested with PstI, blunted, digested with ApaI, and ligated to PCR products made using pB3Δ5′ as a template. Oligonucleotide 1976, containing an ApaI site, [d(GGTTCCTTTGTTACTTCTTCTGCCGCCTGCTTCAAACCGCT)] was a common leftward primer. The rightward primer was specific for each construct. The sequence and restriction site used are as follows: pB3Δ909 (pHGR112), d(GCGCGCGTTTAAACGCTCTCGGCAGAGGTGT) (DraI); pB3Δ947 (pHGR113),d(GCGCGCGTTTAAACCGCGTCATCTGTCGCTGGAC) (DraI); pB3Δ997 (pHGR114), d(GCGCGCTTTAAATAGGTAAATCCGGTCTAACAAG) (DraI); pB3Δ1003 (pHGR115), d(GTCCTACGCTTAGAATTAAATACGTAAATCCG) (SnaBI); pB3Δ1042 (pHGR116), d(GCGCGCTTTAAAGCAAGCTGGGGAGACCCCC) (DraI). Deletions were moved into pB3Δ5′ as EcoRI-AfeI fragments. To make pB3Δ1083 (pHGR117), the PstI-AfeI fragment of pHGR110 was removed and the vector religated. The resulting EcoRI-ApaI fragment was moved into pB3Δ5′.
Internal deletions of pB3Δ5′.
A two-step cloning process was needed. pB3Δ5′Δ1004-1042 (pHGR107) was made by removing the PflMI-AfeI fragment on pB3Δ5′. A PCR-generated fragment [oligonucleotide 2098, d(TAACCACTTTAACTAATACTTTCAACA) and d(CGACGCAGCGCTATTTAATTCTAAGCGTAGGACTGG)] was used as an insert after PflMI-AfeI digestion. On the resulting construct, the AfeI-ApaI fragment was replaced with a PCR-generated insert [oligonucleotide d(GCGCGCTTTAAAGCAAGCTGGGGAGACCCCC) and oligonucleotide 1976] after DraI-ApaI digestion. The same approach was used for making pB3Δ5′Δ971-1042 (pHGR108) and pB3Δ5′Δ931-1042 (pHGR109), except that the oligonucleotides used for making the first PCR fragments were different. Oligonucleotides 2098 and d(GCGCGTAGCGCTTCACACCTCTGCCGAGAGCGG) were used for making the first insert corresponding to pHGR108. Oligonucleotides 2098 and d(GCGCGTAGCGCTAAGCGTAGGACTGGACACAG) were used for making the first insert corresponding to pHGR109. pB3Δ5′ was used as the template.
Transient induction of transcription and screening for Ura+ cells.
Induction cultures (8 ml) and controls were inoculated to an A600 optical density (OD) of 0.002. Transcription of parental RNAs was controlled by the carbon source (2% glucose or 2% galactose) and 0 or 500 μM CuSO4 in uracil-amended liquid media. Transcription was repressed after ∼9 to 10 generations. Faster-growing cultures in glucose were incubated for 36 to 48 h, while slower-growing galactose cultures were incubated for 72 h. Transcription was repressed by diluting the induced cultures to an A600 OD of 0.05 in media lacking uracil and copper and with glucose as the only carbon source. Repression cultures were incubated for 2 h and further diluted 2-fold (OD = 0.025) and 100-fold (OD = 0.0005). Ura+ cells were identified by selecting for colony formation on solid media lacking uracil. For every treatment, 0.1 ml of the twofold-diluted repression culture (OD = 0.025) was spread onto each of 10 plates lacking uracil. In parallel, 200 μl of the final dilution (OD = 0.0005) was spread onto two uracil-amended plates to determine the total number of cells present. In both cases, selection for the plasmids encoding BMV replication proteins 1a and 2apol was maintained.
Frequency of recombination per cell per yeast generation.
The experiment was conducted as described above, except that transcription was transiently induced for 0, 12, 24, 48, or 72 h. At each time point, the optical density of the culture was determined to estimate the number of yeast generations that had been formed. To ensure that all cultures remained in exponential-phase growth throughout, cultures harvested after 12 and 24 h of transcription were started at an OD of 0.3, while those harvested after 48 and 72 h were started at an OD of 0.002.
RNA extraction and analysis.
Induction cultures and all Ura+ colonies were individually grown in 8-ml volumes. RNA extraction and Northern blot analysis were performed as described previously (20). Parental RNAs and RNA recombinants were detected with strand-specific 32P-labeled RNA probes transcribed in vitro. Plasmid MJUR1 (20) was used as a template for making probes to detect positive-strand (linearized with EcoRI and transcribed with T7 RNA polymerase) or negative-strand (linearized with HindIII and transcribed with SP6 RNA polymerase) URA3 sequences. Plasmid HGR80 was used as a template for making probes to detect positive-strand (linearized with HindIII and transcribed with SP6) and negative-strand (linearized with EcoRI and transcribed with T7 RNA polymerase) sequences corresponding to the 3′-terminal half of the BMV 3a open reading frame (ORF) (3′ 3a), which is common to all parental RNAs. A PCR-generated template was used for making probes (5′ 3a) targeting sequences exclusively present on pB3Δ3′ and its derivatives (Fig. 1). Such a DNA fragment was generated using pB3URA3 as a template and oligonucleotides d(CAGAGATGCATAATACGACTCACTATAGGGAGAATGTCTAACATAGTTTCTCCC) and d(CCAAGCCTTCATTAACCCTCACTAAAGGGAGATTCCTACCGCTATCACCGCCG). Probes to detect positive or negative strands were made using T3 or T7 RNA polymerase, respectively.
Reverse transcription (RT)-PCR cloning and sequencing of wild-type-size RNAs.
Three micrograms of DNase I-treated total RNA was used for reverse transcription. Full-length cDNA was made using primers that target the wild-type (wt) 3′ UTR [d(CTTTTAGAGATTTACAGTGT)] and 5′ UTR [d(GCGCTACGTAAAATACCAACTAATTCTCGTTCGATT)] of BMV RNA3. The latter oligonucleotide introduces a SnaBI site for cloning purposes. PCR products were digested with SnaBI and ApaI before being ligated into SnaBI-ApaI-digested pB3HGR5. Thus, cloning and sequencing included the region from the 5′ UTR to the ApaI site of URA3. Plasmid B3HGR5 is a derivative of pB3URA3 lacking the 5′ half of the 3a ORF, the entire intergenic region, and the 3′-terminal quarter of the URA3 ORF. pB3HGR5 was made by removing the ClaI-NcoI fragment from pB3URA3, blunting the ends, and religating.
Total RNA from wt yeast and from yeast replicating B3URA3 were used as negative and positive controls, respectively. To test for the artificial formation of recombinants during RT-PCR, total RNA from yeast expressing either B3Δ3′ or B3Δ5′ and a mixture of such RNA preparations were included in the analysis.
Rapid amplification of cDNA ends (RACE).
Twenty micrograms of total RNA was dephosphorylated and decapped using calf intestinal phosphatase and tobacco acidic pyrophosphatase, respectively, using the Invitrogen GeneRacer kit according to manufacturer's instructions. Anchor RNA oligonucleotide (ACUGACAUGGACUGAAGGAGUAUACGUA) was ligated to decapped RNA using T4 RNA ligase. Reverse transcription was carried out using oligonucleotide d(CTTTTAGAGATTTACAGTGT), which targets the 3′ UTR of BMV RNA3. First-strand DNA was PCR amplified using oligonucleotide d(GGTTCCTTTGTTACTTCTTCTGCCGCCTGCTTCAAACCGCT), which targets the URA3 gene 12 nt downstream of the ApaI site, and an oligonucleotide [d(GAGGACACTGACATGGACTGAAGGAGTATACGTA)] whose target is the RNA anchor sequence on the first-strand DNA and which introduces a SnaBI site. DNA products were digested with SnaBI and ApaI and ligated into pB3HGR5. PCR amplification, cloning, and sequencing included the region from the unknown 5′ end to the ApaI site in URA3.
As negative and positive controls, total RNA from wt yeast and yeast replicating wt B3URA3 were used, respectively. To account for the formation of recombinants during RT-PCR, total RNA from yeast expressing either B3Δ3′ or B3Δ5′ and a mixture of such RNA preparations were analyzed in parallel. An additional set of controls was established by not providing the RNA anchor to the RNAs described above and to the RNA samples of interest. Further details are provided in Results.
Plasmid recovery.
Plasmids were recovered from Ura+ yeast as described previously (35). DNA was treated with SmaI to digest plasmids encoding replication proteins 1a and 2apol while preserving plasmids encoding RNA3 derivatives. Escherichia coli was transformed, and DNA was miniprepped for 4 to 10 independent colonies. Miniprep DNA was digested with EcoRI and PflMI. pB1CT19, pB2CT15, pB3Δ5′, and pB3Δ3′ were used as controls.
Frequencies of recombination and transmission per cell generation.
If F is the fraction of cells per generation that acquired a Ura+ phenotype by recombination and τ is the transmittal efficiency of RNA replicons to daughter cells at each cell division in the absence of selection, then the fraction of cells that harbor recombinants after N generations, R(N), is given by:
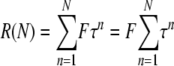 |
(1) |
where each of the N terms in the summation represents the generation of new recombinants and their subsequent survival rate, determined by τ, during the successive n generations of the induction period. Specifically, for each value of n, the corresponding term represents those recombinants that arose at frequency F in the nth generation before the culture was sampled and that survived transmission through n further cell divisions at a cumulative transmission efficiency of τn to be present in the final sample.
For the large cell numbers and unsynchronized cultures of these experiments, equation 1 can be replaced by the continuous expression:
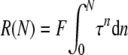 |
(2) |
whose solution is
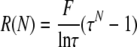 |
(3) |
Based on equation 3, one can solve for τ using
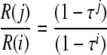 |
(4) |
and
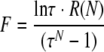 |
(5) |
RESULTS
Inducible BMV RNA recombination in yeast.
Yeast strain YPH500 is unable to grow in medium lacking uracil (Ura−) due to a nonreverting mutation of the URA3 uracil biosynthesis gene (36). However, YPH500 yeast expressing BMV RNA replication proteins 1a and 2apol (hereafter 1a2a+ yeast) become Ura+ when transfection or transient induction of plasmid transcription is used to introduce B3URA3, a BMV RNA3 derivative with the coat protein ORF replaced by the URA3 ORF. In 1a2a+ yeast, B3URA3 is maintained as a plasmid-independent RNA replicon and expresses URA3 via a subgenomic RNA, allowing the formation of colonies on medium lacking uracil (17, 20) (Fig. 1A).
To test for RNA recombination, we generated plasmids that transiently express two overlapping, nonreplicable deletion derivatives of B3URA3, whose homologous recombination could generate B3URA3 and the Ura+ phenotype (Fig. 1B). RNA B3Δ3′ lacked the 3′ half of B3URA3, including the URA3 gene and the promoter for negative-strand RNA synthesis (6). RNA B3Δ5′ retained the 3′ two-thirds of B3URA3, including the URA3 gene and 3′ tRNA-like sequence, but substituted the yeast GAL1 mRNA leader for the 5′ third of RNA3, including half of the 3a gene and the promoter for positive-strand RNA synthesis (11, 17). URA3 in this 3′ position cannot be translated from B3Δ5′ but only from a subgenomic mRNA (RNA4) produced by a replicating RNA3 (Fig. 1A) (17). To provide a target for homologous RNA recombination, B3Δ3′ and B3Δ5′ have identical overlapping sequences of 618 nt (Fig. 1B).
To allow transient expression of B3Δ3′, B3Δ5′, or both on demand, B3Δ3′ was expressed from the copper-inducible CUP1 promoter, while B3Δ5′ was expressed from the galactose-inducible, glucose-repressible GAL1 promoter (Fig. 1B). As intended, treating yeast carrying both plasmids with CuSO4, galactose, or both induced transcription of B3Δ3′, B3Δ5′, or both (Fig. 2). When yeast were transferred to medium lacking copper and with glucose instead of galactose, the nonreplicable B3Δ3′ and B3Δ5′ transcripts rapidly decayed and were lost (data not shown) as described previously for other RNA3 derivatives (42).
FIG. 2.

Accumulation of B3Δ5′ and B3Δ3′ in 1a2a+ yeast after transcription for 72 h in media containing or lacking galactose and copper. Three micrograms of total RNA was fractionated in 1% denaturing agarose gels. Positive- and negative-strand RNA sequences were detected with the indicated 32P-labeled parental RNA-specific or common RNA probes (Fig. 1B). B3URA3 was used as a size marker (∼2,406 nt). Black arrowheads mark the location of B3Δ5′ transcript that has not been cleaved by the ribozyme (∼2,500 nt) (Fig. 1B). Open arrowheads indicate a background band (∼1,200 nt) formed by premature termination of DNA-dependent transcription (42). B3Δ5′ (∼1,822 nt) and B3Δ3′ (∼1,800 nt) transcript RNAs are indicated to the left of each panel. Basal transcription of B3Δ3′ in the absence of copper is just visible in lanes 1 and 3. The bottom panel shows ethidium bromide staining of 18S rRNA in the same samples.
To assay for RNA recombinants, B3Δ3′ and B3Δ5′ transcription were induced separately or together in yeast constitutively expressing BMV RNA replication proteins 1a and 2apol. After inducing transcription for 72 h (9 to 10 yeast generations) in medium containing uracil, B3Δ3′ and B3Δ5′ transcription was repressed and previously accumulated uracil was depleted by incubating the cells for 2 h in liquid medium lacking uracil and copper and with glucose as the only carbon source. An average of 100,000 cells (Fig. 3) was spread onto solid medium lacking uracil to select for cells that had acquired a Ura+ phenotype, as by recombinational generation of B3URA3. All recombination assays were repeated multiple times with consistent results. Figure 3 reports the results of three independent repetitions of the assay per condition tested.
FIG. 3.

Frequency of Ura+ cells, RNA recombination, and formation of sURA3 RNAs. The presence (+) or absence (−) of BMV replication proteins 1a and 2apol and plasmids encoding parental RNAs is indicated. The histogram shows the averages and standard errors corresponding to results from three independent repetitions of the experiment. The number of relevant events found is indicated in parentheses. The corresponding number of cells plated is indicated. Black bars represent the percentage of cells that acquired the Ura+ phenotype and harbored either an intermolecular B3URA3 RNA recombinant (gray bar) or an sURA3 RNA (white bars).
After B3Δ3′ and B3Δ5′ were transiently coinduced in 1a2a+ yeast, 0.11 to 0.16% of total cells became Ura+ (Fig. 3, condition 2). Passaging showed that these cells remained Ura+ indefinitely in the absence of further pB3Δ3′ and pB3Δ5′ transcription and after loss of both pB3Δ3′ and pB3Δ5′ plasmids following growth in the absence of plasmid selection. Below we show that each Ura+ cell line contained a recombinant RNA3 that was maintained as a plasmid-independent RNA replicon and that expressed URA3 via the expected subgenomic mRNA. Even prior to direct RNA analysis, the Fig. 3 results revealed important points about these Ura+ cells. For example, Ura+ cells in principle might have arisen by DNA recombination between the intact URA3 ORF in pB3Δ5′ and the insertionally inactivated yeast chromosomal ura3-52 allele in YPH500 (36), or another DNA site, to generate a functional URA3 allele. However, when 1a and 2apol were omitted, no Ura+ cells were found (Fig. 3, condition 1). Thus, the Ura+ phenotype depended on BMV RNA replication functions, which is inconsistent with direct URA3 expression from DNA. Moreover, when neither pB3Δ3′ nor pB3Δ5′ was induced, no Ura+ cells arose even in the presence of 1a and 2apol (Fig. 3, condition 4), showing that acquisition of the Ura+ phenotype also depended on transient expression of parental RNA transcripts.
Recombination between pB3Δ3′ and pB3Δ5′ (Fig. 1B) could generate an intact B3URA3 cDNA linked to the CUP1 promoter (Fig. 1B) and so transcribed to initiate B3URA3 RNA replication and URA3 expression upon addition of copper. However, no Ura+ cells were found when copper was added to induce the CUP1 promoter without galactose to induce the GAL1 promoter of pB3Δ5′ (Fig. 3, condition 5). Moreover, the recombinant plasmid would be dicentric and unstable (28), and only unaltered pB3Δ3′ and pB3Δ5′ were recoverable from Ura+ cells.
Intriguingly, when galactose was added to induce B3Δ5′ transcription, but copper induction of B3Δ3′ was omitted, Ura+ cells were detected at a frequency of 0.03 to 0.06% of total cells (Fig. 3, condition 3). These Ura+ cells did not result from low basal transcription of B3Δ3′ without added copper (Fig. 2, lanes 1 and 3), since after B3Δ5′ induction, similar levels of Ura+ cells were consistently detected with or without pB3Δ3′ (Fig. 3, conditions 3 and 7). Thus, B3Δ5′ was necessary and sufficient to form Ura+ colonies at a low frequency in 1a2a+ yeast.
Detection of consistent levels of Ura+ cells from yeast expressing B3Δ5′ and B3Δ3′ and lower levels of Ura+ cells from yeast expressing only B3Δ5′ implied two different classes of genetic events. Below we show that these corresponded to homologous intermolecular RNA recombinants and functional but further truncated RNA3 replicons with in vivo-generated 5′ ends that supported positive-strand RNA synthesis.
Homologous intermolecular RNA recombination products.
Ura+ colonies obtained after transient coinduction of pB3Δ5′ and pB3Δ3′ transcription were grown in liquid medium lacking uracil, to maintain selection for Ura+ cells, and lacking galactose or copper, to block further transcription from the GAL1 and CUP1 promoters. Total RNA was extracted and analyzed by Northern blotting with URA3 and 5′ 3a probes specific to parental RNAs B3Δ5′and B3Δ3′, respectively (Fig. 1B). The 430 Ura+ colonies from three independent coinduction experiments fell into one of two classes. One class contained RNAs of a size matching that of B3URA3 and that hybridized with both parental RNA-specific probes (Fig. 4A, lanes 1 and 3). The second class contained RNAs that were only ∼250 nt longer than subgenomic RNA4 and hybridized to a URA3 probe but not to a 5′ 3a probe or to a common probe (Fig. 4A, lane 2). These short URA3-hybridizing RNAs are hereafter designated sURA3 RNAs.
FIG. 4.

Sample Northern blot analyses of URA3-expressing BMV RNA replicons in cells that acquired a Ura+ phenotype. Positive- and negative-strand RNAs were detected with 32P-labeled probes as indicated in Fig. 2. wt B3URA3 was included as a size marker. (A) Examples of the two classes of RNA progeny detected in Ura+ cells obtained after transcription of both B3Δ3′ and B3Δ5′. Homologous intermolecular recombination products (lanes 1 and 3) matched the size of B3URA3 and were detected with both parental RNA-specific probes. sURA3 RNAs (lane 2) were only slightly longer than RNA4 and failed to hybridize to probes targeting 3a sequences. Open arrowheads point to sURA3 of both positive and negative polarity detected with URA3 probes. (B) sURA3 RNAs obtained after transcription of B3Δ5′ alone in the absence of pB3Δ3′. (C) Nucleotide sequences at the 5′ end of sURA3 RNAs aligned with the 3′ end of the 3a ORF in B3URA3 and B3Δ5′. The 3a stop codon is underlined. The in vivo-generated 5′ ends consisted of the last 7 to 11 nucleotides from the 3a ORF and all downstream sequences of B3Δ5′. An additional G, GU, or AU was present at the extreme 5′ end (indicated by a box). For clone number 4, nucleotide 5 (bracketed) was a U-to-A substitution. Sequences derived from the 3a ORF are shown in boldface type. (D) 5′ ends of sURA3 RNAs aligned with the 5′ end of wt RNA3.
Both B3URA3-like and sURA3 RNAs were present in positive- and negative-strand forms (Fig. 4A, top and middle panels), as expected for replicating RNAs and consistent with the 1a plus 2apol dependence of the Ura+ phenotype and RNA persistence after the loss of both pB3Δ3′ and pB3Δ5′ plasmids (above). Consistent with the Ura+ phenotype, both classes of RNAs also produced a positive-strand RNA band coelectrophoresing with the URA3-expressing subgenomic mRNA, RNA4, of B3URA3 (Fig. 4A and B, upper panels).
URA3-hybridizing RNAs similar to wt B3URA3 (Fig. 4A, lanes 1 and 2) were found at a frequency of 0.07 to 0.10% of the total yeast population (Fig. 3, condition 2), representing 60% or more of the Ura+ colonies obtained. Recombinant RNAs in this class from 5 independently selected Ura+ colonies obtained from three independent inductions were cloned and sequenced from the 5′ end through the ApaI site of URA3. Consistent with the hybridization of these RNAs to both B3Δ5′- and B3Δ3′-specific probes (Fig. 4A), sequencing results showed in all cases that the full wt B3URA3 sequence was restored. Thus, such RNAs were the result of intermolecular, homologous recombination between parental RNAs B3Δ5′ and B3Δ3′.
Generation of sURA3 replicons from nonreplicating B3Δ5′.
After transiently inducing B3Δ5′ and B3Δ3′ (Fig. 3, condition 2), 186 of 430 Ura+ colonies analyzed by Northern blotting (0.04 to 0.07% of total cells) contained the sURA3 class of small RNA replicons that hybridized to a URA3 probe but not to probes specific for B3Δ3′ or the 3′ 3a coding region common to both parental RNAs (Fig. 4A, lane 2). Similarly, after transient expression of B3Δ5′ in 1a2a+ yeast lacking pB3Δ3′ (Fig. 3, condition 7), 126 Ura+ colonies were isolated in three independent inductions (0.03 to 0.04% of total cells), and all contained equivalent sURA3 RNA replicons in positive- and negative-strand form (Fig. 4B). These results confirmed that B3Δ5′ was necessary and sufficient to form sURA3 RNAs.
As noted above, B3Δ5′ RNA has the intergenic RE sequences required to recruit RNA templates to the replication complex (42), the 3′ tRNA-like sequence required to initiate negative strand synthesis (6), and the subgenomic mRNA promoter required to express URA3 (17) but cannot replicate because it lacks 5′ sequences required for RNA polymerase-dependent positive-strand RNA synthesis (15) (Fig. 1B). Indeed, negative-strand B3Δ5′ RNA3 accumulated to significant levels when yeast was grown in galactose (Fig. 2, lower panel). Thus, the consistent recovery of Ura+ colonies containing sURA3 RNA replicons after transiently inducing nonreplicable B3Δ5′ RNA suggested that sURA3 RNAs represented B3Δ5′ derivatives whose 5′ ends had been modified to acquire or form a functional positive-strand promoter.
The sequence at the 5′ end of recombinant RNAs was determined by 5′ RACE (43) for five sURA3 RNA-containing Ura+ colonies obtained after transiently inducing both B3Δ5′ and B3Δ3′. cDNA synthesis was initiated with an oligonucleotide primer specific for the BMV 3′ tRNA-like region. To allow subsequent in vivo functional testing of the cloned sequences, the sURA3 cDNA products were cloned into a yeast shuttle vector that fused the 5′ end of each full-length sURA3 cDNA to the GAL1 promoter and reconstituted the full BMV 3′ end linked to a self-cleaving ribozyme for in vivo 3′ end formation as in pB3URA3 (Fig. 1A).
For each of the five sURA3-containing yeast colonies analyzed, five cDNA clones were sequenced, each from a different E. coli colony. In each case, all sURA3 cDNA clones from a single Ura+ yeast colony were identical, while the sequences from different Ura+ yeast colonies were similar but unique (Fig. 4C). Consistent with their size and failure to hybridize to 5′ 3a or 3′ 3a probes, each sURA3 RNA was a B3Δ5′ derivative from which nearly all 3a ORF sequences were deleted. Specifically, each sURA3 RNA contained the last 7 to 13 nt of the 3a ORF and all downstream sequences of B3Δ5′, preceded by an additional 5′ terminal G, GU, or AU (Fig. 4C). Since the clones were obtained using a 5′ RACE protocol (43) that selectively targets 5′-capped RNAs, we conclude that the sURA3 RNAs were capped. Loss of 3a sequences from sURA3 replicons is consistent with prior findings that the 3a ORF is dispensable for RNA3 replication in plant cells and yeast (10, 16).
In conjunction with the ability of these sURA3 5′ ends to support RNA replication, the first 33 nt of the intergenic region share considerable sequence similarity with the wt RNA3 5′ end (Fig. 4D). The same intergenic sequences contain three adjacent regions (overlined in Fig. 4D) with similarity to the box B consensus sequence GGUUCAAUYC. This box B consensus sequence is also present at nt 17 to 26 of the BMV RNA2 5′ end, where its complement functions in the negative strand as an important determinant of positive-strand initiation (41).
Parallel analysis of RNA from yeast expressing B3URA3 or B3Δ5′ produced only cDNA clones with the expected 5′ ends of B3URA3 or the 5′ GAL1 leader, respectively. To rule out generation of recombinant sequences or novel RNA ends in vitro, we applied the same RACE procedure to a mixture of total RNAs from two yeast cultures, one expressing B3Δ5′ and the other expressing B3Δ3′. As expected from the use of a cDNA primer to the BMV 3′ tRNA-like region, this produced only cDNA clones with the 5′ GAL1 leader sequences of B3Δ5′. Additionally, the RNA anchor (see Materials and Methods) was not included in another control set of RNA samples, including wt B3URA3, homologous recombinant RNAs, sURA3, B3Δ5′, and a mixture of B3Δ5′ and B3Δ3′ RNAs. No DNA was obtained after PCR amplification, confirming participation of the ligated RNA anchor and showing that the novel 5′ ends found in the sURA3 cDNA clones (Fig. 4C) were specific to sURA3 replicon-containing Ura+ cells.
Cloned sURA3 RNAs initiated and were maintained through RNA replication.
To test the functionality of the cloned sURA3 RNAs, we measured their ability to replicate and support Ura+ colony formation after being launched by transient transcription from a plasmid vector. Ura− yeast cells were transformed with 1a, 2apol, and plasmids containing cDNA copies of the sURA3 recombinants under control of the GAL1 promoter. For each of the five independently isolated sURA3 sequences (Fig. 4C), three independent yeast transformants were grown in galactose medium to transiently induce sURA3 transcription for 72 h. The frequency of Ura+ cells was determined by colony formation in the absence of uracil as described for recombination experiments, except that only 1,200 cells were plated per induction culture.
As shown in Fig. 5A, 19 to 27% of such cells became Ura+, similar to the 25% efficiency of yeast expressing 1a and 2apol and full-length B3URA3 with the wt RNA3 5′ end. For B3URA3 and each of the five sURA3 sequences analyzed, total RNA from five representative Ura+ colonies was isolated from cultures grown in glucose liquid medium lacking uracil and analyzed by Northern blotting. All maintained positive- and negative-strand forms of the B3URA3 or sURA3 RNA replicons and expressed a subgenomic URA3 mRNA, RNA4 (Fig. 5B). Moreover, after continuing growth in glucose as plasmid-independent RNA replicons, the sURA3 replicon RNA sizes remained stable.
FIG. 5.

(A) Percentage of 1a2a+ yeast cells that acquired a Ura+ phenotype after transcription for 72 h of wt B3URA3 or the sURA3 RNAs indicated in Fig. 4A. The histogram shows the averages and standard errors of results from three independent inductions. (B) Positive- and negative-strand RNA accumulation in yeast replicating wt B3URA3 or sURA3 RNAs. A URA3 probe of both polarities was used. The Northern blot shows only two samples, however, for each sURA3 cloned. Five Ura+ colonies were grown under uracil selection, and the RNA was extracted and analyzed. (C) Relative accumulation of positive (black bars)- and negative (gray bars)-strand RNA3 and positive-strand RNA4 (white bars) averaged over cultures from five independent Ura+ colonies replicating wt B3URA3 or the indicated sURA3 RNA. Shadowed bars indicate the subgenomic promoter strength (ratio of subgenomic RNA4 to positive-strand RNA3) in sURA3 RNAs and wt B3URA3. Genomic positive-strand RNA3 and subgenomic RNA4 accumulation was determined for 50 Ura+ independently isolated colonies harboring wt B3URA3, recombi nant B3URA3 (not shown because there was no difference), or not cloned sURA3 RNAs. Shadowed bars with the same letter are not statistically different.
Yeast containing sURA3 replicons accumulated only 18 to 50% of the level of subgenomic URA3 mRNA of B3URA3 (Fig. 5C). However, cells replicating sURA3 or B3URA3 grew at equal rates on medium lacking uracil, implying that URA3 protein levels were not limiting. Moreover, for all five cloned sURA3 RNAs, the ratio of subgenomic URA3 mRNA to genomic sURA3 RNA was similar to that of B3URA3 (19 to 38%) (Fig. 5C). Since both subgenomic mRNA and genomic RNA are synthesized from the same genomic length negative-strand RNA template, this indicates that the relative strengths of the subgenomic and genomic RNA promoters were closely similar between B3URA3 and sURA3 RNAs. Similar subgenomic-to-genomic RNA ratios were found for 50 additional Ura+ colonies harboring sURA3 RNAs (Fig. 5C).
Formation and selection of sURA3 RNAs.
The 5′ ends of all sURA3 RNAs mapped to a short region (nt 991 to 997) in RNA3. This specificity might arise either in generation or functional selection of these RNAs. Secondary structure 5′ to this region, e.g., might promote premature termination during negative-strand RNA synthesis. To determine what sequences were required for sURA3 RNA formation, we tested a series of 5′ truncations and deletions in B3Δ5′ (Fig. 6A).
FIG. 6.

Formation of sURA3 RNAs from B3Δ5′. (A) Deletions of B3Δ5′. Nucleotide numbers correspond to the wt BMV RNA3 sequence. The number included in the name of each construct indicates the last nucleotide removed. In internal deletions of B3Δ5′, the nucleotides removed are indicated after the Δ. RNAs were transcribed in vivo from the GAL1 promoter, and the GAL1 mRNA leader was present, as on B3Δ5′. The histogram shows the frequency of Ura+ cells after transcription of each construct for 72 h (9 to 10 yeast generations) in the presence of BMV replication proteins 1a and 2apol. Bars show the averages and standard errors of results from six independent repetitions. Bars with the same letter are not statistically significant. Letter D indicates constructs for which no Ura+ cells were detected. (B) Sample Northern blot of cells that acquired a Ura+ phenotype after transient induction of B3Δ909. Positive-strand RNAs were detected with a 32P-labeled probe against URA3. The B3Δ909 progeny consisted of two sizes (open arrowhead): small replicons that comigrated with sURA3 RNAs (lane 4), and RNAs longer than sURA3 (LURA3, lanes 3, 5, and 6). The bottom panel shows ethidium bromide staining of 18S rRNA. (C) 5′ sequence of representative sURA3 RNAs derived from B3Δ997 and B3Δ1003. Both sURA3 RNAs represent 5′ truncations of the initial plasmid transcript, retaining the last 9 nt of the GAL1 mRNA leader (indicated between vertical dashed lines) and bearing a single noncontiguous 5′ G (boxed). GAL1 leader sequences were fused to B3Δ997 sequences from the 3a ORF (bold) or to RNA3 intergenic region in B3Δ1003. Sequence numbering corresponds to wt RNA3. (D) 5′ sequence of three LURA3 RNAs sequences derived from B3Δ909 and their prevalence among 10 independent LURA3 cDNA clones sequenced. All are 5′-truncated derivatives of the parental B3Δ909 transcript, including the last 9 to 10 nt of the GAL1 leader (indicated between vertical dashed lines). An AA linker generated in cloning is over- and underlined. All downstream RNA3 sequences are in boldface type. In two cases, these sequences are fused at the 5′ end to noncontiguous AU repeats (boxed), which might have been derived by duplication of flanking downstream sequences (underlined) or recombinational joining of upstream sequences (overlined). (E) Comparison of the 5′ end of the most prevalent LURA3 sequence from panel B to the 5′ end of wt RNA3. (F) Representative sequence of a sURA3 RNA derived from B3Δ1042. Such sURA3 RNAs contained the complete sequence of the starting B3Δ1042 transcript, initiated at a previously documented (17) alternate start site 9 nt upstream of the usually cited GAL1 transcription start, fused via a single noncontiguous G (boxed) to the last 26 nt of B3Δ1042, which corresponds to the last 26 nt of the 3′ UTR of wt RNA3, and preceded by 1 to 17 nt of A-rich sequence of unknown origin (boxed).
When the remaining 3a coding sequences of B3Δ5′ were progressively deleted in B3Δ817 through B3Δ1003, Ura+ cells were obtained at frequencies similar to B3Δ5′ or, for B3Δ909, at even higher levels (Fig. 6A). Thus, 3a coding sequences upstream of the major sURA3 RNA 5′ ends (Fig. 4C) were not required for sURA3 formation. However, production of Ura+ cells was essentially abolished by deleting nt 1004 to 1042, between the 3a coding region and the RE, either alone or as part of larger deletions (B3Δ1042 and deletions below) (Fig. 6A). This was striking, since nt 1004 to 1042 are dispensable for RNA template recruitment to replication complexes (42).
Northern blotting showed that all Ura+ cells obtained in these experiments contained sURA3 RNA replicons coelectrophoresing with those from B3Δ5′, with one exception. B3Δ909, which produced fivefold more Ura+ cells than B3Δ5′, produced two classes of Ura+ replicons: ∼20% of these comigrated with B3Δ5′ sURA RNAs (Fig. 6B, lanes 2, 4), while ∼80%, corresponding to the excess Ura+ cells, migrated as longer RNAs (LURA3 RNAs) (Fig. 6B, lanes 3, 5, 6).
Cloning and sequencing revealed that the sURA3 RNAs of B3Δ817, −909 and −947, all had 5′ ends equivalent to those of B3Δ5′-derived sURA3 RNAs, i.e., novel 5′-terminal G, GU, or AU linked to B3Δ5′ sequences downstream of RNA3 nt 991 to 997 (Fig. 4C). When the progressive deletions extended into or beyond this region in B3Δ997 and B3Δ1003, the resulting sURA3 RNAs additionally incorporated the last 10 to 15 nt of the GAL1 leader fused to the expected downstream RNA3 sequences (Fig. 6C). Consequently, just as the B3Δ5′ sURA3 RNAs contained 8 to 14 nt 5′ to the RNA3 intergenic region (Fig. 4C), the B3Δ997 and B3Δ1003 sURA3 RNAs contained 10 to 13 nt 5′ to the intergenic region. In both cases, the resulting 5′ ends were AU-rich and initiated with noncontiguous G, GU, or AU nt to create GUA or AUA 5′ ends.
The LURA3 RNAs from B3Δ909 similarly retained the last 9 to 10 nt of the GAL1 leader fused to the expected downstream RNA3 sequences (Fig. 6D). In this case, the extreme 5′ ends were AU repeats either already present in the GAL1 leader (overlined in Fig. 6D) or derivable by duplicating flanking sequences. Like the sURA3 RNA 5′ ends (Fig. 4C), the LURA3 5′ ends also showed significant sequence similarity with the wt RNA3 5′ end (Fig. 6E).
The rare URA3 replicons produced by B3Δ1042 contained the complete GAL1-promoted B3Δ1042 transcript sequence fused via a single additional G downstream of the last 26 nt from the 3′ end of RNA3 (Fig. 6F). Similar dimeric or partially dimeric RNAs are seen with nodaviruses (1) and tombusviruses (5, 33) and have been suggested to arise when RNA polymerase initiates a new round of RNA synthesis without releasing the daughter strand made during the previous round (1). As more URA3 replicons were sequenced for B3Δ5′ and its derivatives, rare examples of similar partially dimeric replicons were seen for other parental RNAs. Such partial dimers thus appear to constitute a low general background that was revealed when the primary B3Δ5′-like sURA3 RNAs were suppressed by the deletion in B3Δ1042.
Recombination required a functional template RE on both parental RNAs.
Bromovirus RNA replication occurs in endoplasmic reticulum membrane-bound replication complexes (38). Recruitment of positive-strand RNA3 templates to these membrane-associated RNA replication complexes is mediated by 1a through a well-defined, cis-acting, template RE located in the intergenic region. This RE is an ∼125-nt sequence that folds into a long, bulged stem-loop called box B (42). Box B sequences are conserved at the 5′ ends of BMV genomic RNAs 1 and 2 as an essential part of a similar 5′-proximal template recruiting element (4).
To determine if the RE was required on both, either, or neither parental RNA for recombination, we made derivatives of B3Δ5′ and B3Δ3′ with the RE inactivated by a 17-nt deletion encompassing the conserved box B sequence (ΔBox B) (Fig. 7A). In the absence of 1a, RE deletion did not reduce B3Δ5′ accumulation and increased B3Δ3′ accumulation (Fig. 7B). Thus, equal or greater levels of the ΔBox B variant RNAs were available to potentially initiate recombination. However, as shown in Fig. 7B, these RE deletions blocked 1a-mediated recruitment of the RNAs into replication, which was evidenced for the intact wt B3Δ5′ and B3Δ3′ by a marked increase in RNA stability and accumulation associated with RNA transfer into the protected environment of the replication complex (38, 42).
FIG. 7.

(A) wt B3URA3 and its derivatives (B3Δ5′, B3Δ3′, and B3Δ3′ΔIR) are shown. Within the RE, the nucleotide sequence of box B is indicated, along with the box B deletion (ΔBox B). The intergenic region was removed from B3Δ3′ to generate B3Δ3′ΔIR. (B) RNA accumulation in the presence (+) or absence (−) of BMV replication protein 1a after transcription for 72 h. For each construct, six independent cultures were analyzed: three in the presence and three in the absence of 1a. B3URA3 and B3Δ5′ were transcribed from the GAL1 promoter, while B3Δ3′ and its derivatives were transcribed from the CUP1 promoter. Three micrograms of total RNA was fractionated in 1% denaturing agarose gels. Positive-strand RNA sequences were detected with a 32P-labeled 3′3a common RNA probe (Fig. 1B). B3URA3 was used as a size marker and positive control. Ethidium bromide staining of 18S rRNA in the same samples is indicated at the bottom of each panel. (C) Frequency of Ura+ cells and RNA recom binants obtained after transcription of parental RNAs with or without a functional RE in 1a2a+ yeast. The histogram shows the averages and standard errors of results from three independent repetitions. Black bars represent the percentages of cells that acquired the Ura+ phenotype and harbored either an intermolecular homologous RNA recombinant (gray bar) or an sURA3 RNA (white bars). Numbers in parentheses indicate the total number of cases observed in three independent repetitions of the experiment. The corresponding total number of cells plated is indicated.
The ability of these derivatives to support intermolecular RNA recombination was tested in various combinations with wt B3Δ5′ and B3Δ3′, using the assay established above, in which transient transcription of B3Δ5′, B3Δ3′, or their derivatives was followed by selection of Ura+ colonies and Northern blot analysis to distinguish intermolecular homologous B3URA3 recombinants from sURA3 RNAs. All experiments were repeated multiple times with similar results. Figure 7C reports the results of three independent repetitions. As controls, we tested unmodified B3Δ5′ and B3Δ3′, reproducing the previously observed production of intermolecular recombinants and sURA3 RNAs (compare Fig. 7C, condition 3, to Fig. 3, condition 2). In contrast, when the RE was inactivated on either parental RNA, intermolecular recombination was nearly abolished (Fig. 7C, conditions 2, 4, and 5). This was perhaps most striking for the combination of wt B3Δ5′ and B3Δ3′-ΔBoxB (Fig. 7C, condition 4), which share 450 nt of common sequence upstream of the mutated box B in B3Δ3′ where homologous recombination would have produced wt B3URA3 (Fig. 1B). To insure that the box B deletion in the RE (ΔBoxB) did not interfere with recombination by any transdominant negative effects, we also tested wt B3Δ5′ with B3Δ3′-ΔIR, in which RE function was abolished by deleting the entire RE and all flanking intergenic sequences (Fig. 7A). This mutation also blocked intermolecular recombination (Fig. 7C, condition 5). Since these combinations expressed wt B3Δ5′, however, they did produce the B3Δ3′-independent sURA3 RNAs at normal frequencies, providing an additional internal control (Fig. 7C, conditions 4 and 5). However, production of these sURA3 RNAs was abolished when the B3Δ5′ RE was inactivated with a box B deletion whether the mutated RNA (B3Δ5′-ΔBoxB) was expressed alone or in combination with wt B3Δ3′ (Fig. 7C, conditions 1 and 2). Possible explanations for the RE dependence of intermolecular recombination are considered in Discussion.
Frequency of recombination and transmission per cell generation.
The experiments above measured the frequencies of recombination after inducing transcription of both B3Δ5′ and B3Δ3′ or B3Δ5′ alone for 72 h, which corresponded to 9 to 10 yeast generations. The accumulation of Ura+ cells over this period will be a combined function of the rates at which recombinant RNAs are formed and lost due to imperfect segregation to daughter cells or other effects. In the absence of selection, free B3URA3 RNA replicons are lost at a rate of 10 to 15% per cell division (16). In our experiments, previously formed recombinant replicons might be lost at a faster rate due to the continuous plasmid-directed production of B3Δ5′ and B3Δ3′ transcripts, which may compete with replicating RNAs for interaction with 1a and 2apol. If recombinant loss due to such competition were significant, the final accumulation of recombinants after several yeast generations might seriously underestimate the actual rate of recombination.
To determine the frequency of RNA recombination and the corresponding formation of Ura+ cells per cell per yeast generation and to determine the efficiency of transmission of recombinant RNA replicons to daughter cells, we carried out a time course experiment. We measured the frequency of Ura+ cells after inducing both B3Δ3′ and B3Δ5′ or B3Δ5′ alone for 0, 24, 48, and 72 h. Figure 8 shows the average of three repetitions for each condition and time point. The frequency of Ura+ cells increased throughout the 72 h tested (Fig. 8), suggesting that the majority of Ura+ recombinants formed early must be present at later time points. Using equation 4 presented in the last section of Materials and Methods and the values shown in Fig. 8 for B3Δ3′ and B3Δ5′ or B3Δ5′ alone, the efficiency of Ura+ replicon transmission to daughter cells at each cell division was ∼88%, in excellent agreement with the prior measurement of 85 to 90% for unselected B3URA3 replicons (16). Similarly, using equation 5 from Materials and Methods and the values shown in Fig. 8, the fraction of cells that acquire a Ura+ phenotype per yeast cell generation (∼8 h) was found to be 2.6 × 10−4 for B3Δ3′ plus B3Δ5′ induction (B3URA3 plus sURA3 replicons) and 0.9 × 10−4 for B3Δ5′ alone (sURA3 replicons), implying a frequency of 1.7 × 10−4 per cell per generation for the formation of homologous intermolecular recombinant B3URA3 replicons. These recombination frequencies are >50% higher than the estimates obtained without considering transmission losses, i.e., simply dividing the recombinant yields (Fig. 3) by the number of yeast generations.
FIG. 8.

Time course of the generation of Ura+ cells after transcription of both B3Δ5′ and B3Δ3′ (black squares) or only B3Δ5′ (white squares) for 72 h, during which 10 yeast generations were formed. The percentage of Ura+ cells that harbored a homologous B3URA3 recombinant (dashed curve) was estimated by subtracting the percentage of Ura+ cells obtained after induction of B3Δ5′ alone (bottom curve) from the percentage of Ura+ cells obtained after induction of both B3Δ5′ and B3Δ3′ (top curve). Please see Results regarding the derivation from these data of the frequencies of formation of RNA recombinants and sURA3 RNAs and their transmittal efficiency to daughter cells per yeast generation.
DISCUSSION
Because of the importance of RNA recombination in virus evolution and short-term variability, we developed here an inducible system for BMV RNA recombination in yeast. Yeast also support the formation of tombusvirus RNA dimers, a form of RNA recombination that has been suggested to be due to reinitiation of RNA synthesis on the same template RNA A (5, 32, 33). Our experiments show that yeast also support homologous, intermolecular recombination between two distinct BMV RNAs as well as formation of truncated RNA3 replicons. After transiently expressing replication-incompetent, overlapping, fragments of B3URA3 (Fig. 1B) in Ura− yeast expressing BMV replication factors 1a and 2apol, replication-competent, URA3-expressing recombinant RNAs were selected by yeast growth in the absence of uracil. Two classes of RNA progeny were identified and characterized: homologous intermolecular recombinants between B3Δ5′ and B3Δ3′, regenerating full-length B3URA3 (Fig. 3 to 4), and sURA3 derivatives of B3Δ5′, whose further truncated 5′ ends were fused to one or a few noncontiguous nucleotides (Fig. 4C), generating novel positive-strand promoters.
In keeping with the much lower rates (102- to 103-fold lower) of DNA recombination for nonlinearized plasmids (9), multiple results showed that recombination occurred at the level of viral RNAs, not DNA (Fig. 3). The need for 1a and 2apol to generate and maintain Ura+ cells (Fig. 3, conditions 1, 2, 6, and 7), e.g., showed that Ura+ cells did not arise by generating a DNA locus expressing URA3. The need to induce both B3Δ5′ and B3Δ3′ RNAs to generate B3URA3 recombinants (Fig. 3, conditions 2, 3, and 5) showed that Ura+ cells did not arise by recombination between pB3Δ5′ and pB3Δ3′ plasmids (Fig. 1B) to generate a full-length B3URA3 cDNA from which RNA replication could have been launched by CUP1 promoter induction alone.
This system offers multiple useful features for studying RNA recombination events due to facile yeast growth, selection, and genetic manipulation; the ability to readily modify, express, and repress the parental BMV RNA derivatives, their supporting BMV replication factors, and host factors; and consistent, precisely measurable recombination frequencies (Fig. 3, 7C, and 8). Below we discuss additional features of the recombinants and their generation.
Homologous intermolecular RNA recombination.
Most of the Ura+ replicons recovered were homologous recombinants between B3Δ5′ and B3Δ3′, yielding B3URA3 (Fig. 3 to 4A). Cloning and sequencing five representative recombinants confirmed restoration of the full B3URA3 sequence, showing that the junction sites were perfect and located in the 618-nt region of sequence identity between these parental RNAs (Fig. 1B).
B3Δ3′ lacks a negative-strand RNA promoter and, even in the presence of 1a and 2apol, exists only as a positive-strand RNA (Fig. 2). Such B3Δ3′ positive strands might recombine with B3Δ5′ through breakage and ligation or template switching. Below we note that breakage and ligation appear inconsistent with requirements for a template recruitment element on both parental RNAs (Fig. 7C). By contrast, template switching during negative-strand synthesis is fully consistent with the results. B3Δ5′ directs substantial synthesis of negative strands (Fig. 2) that could switch to B3Δ3′ templates in the region of shared homology. Template switching is also consistent with the requirement for a major cis-acting RNA replication signal, the RE, for recombination (see the next section), with the influences of RNA replication factors 1a and 2apol on RNA recombination in plants (31) and with evidence for template switching during negative-strand RNA synthesis by poliovirus and other positive-strand RNA viruses (22, 26).
RE requirement for homologous RNA recombination.
Intermolecular, homologous RNA recombination required a functional template RE on both parental RNAs (Fig. 7C). Since RE mutations induce dramatic, parallel defects in template recruitment, negative-strand synthesis, and positive-strand synthesis (42), a recombinant RNA must possess a functional RE to be a stable, selectable replicon. However, since B3Δ5′ and B3Δ3′ share sequence identity on both sides of the RE (Fig. 1B), unrestricted homologous recombination would allow the RE to be contributed by either parental RNA. Even if recombination were polarized so that one parent preferentially donated the RE, RE mutation would be allowed on the other parent. This appears to exclude some mechanisms. If, e.g., homologous RNA recombinants were formed prior to RNA replication, as by breakage and ligation, RE mutation should be allowed on one parent. Similarly, if recombination involved initial association of both parental RNAs in a partial heteroduplex (30), a single RE should suffice to recruit this heteroduplex to the replication complex.
At least two nonexclusive hypotheses might explain the RE requirements. The RE might be required on both parental RNAs for independent recruitment to the RNA replication complex. Alternatively or in addition, the RE might be a preferred recombination site. The RE interaction with 1a (42) might promote replicase dissociation from a donor template or reassociation with an acceptor template.
BMV RNA replication occurs in 50- to 70-nm membrane-bounded compartments that sequester positive- and negative-strand RNA templates. Similar compartments are associated with RNA replication by many other positive-strand RNA viruses (8, 38). BMV negative-strand RNAs are synthesized and retained in these compartments (38), consistent with observations that negative-strand RNAs cannot be transferred between replication complexes (18). Accordingly, template switching during negative-strand synthesis likely requires recruitment of both parental RNAs to a single replication complex. This would explain the requirement for REs on both parents and likely contributes to the relatively low frequency of RNA recombination.
The RE requirement in recombination is not explainable simply as an enhancement of parental RNA accumulation. In the absence of 1a, deleting the RE had no effect on B3Δ5′ accumulation and increased B3Δ3′ accumulation (Fig. 7B). In the presence of 1a, RE deletion inhibited B3Δ5′ and B3Δ3′ accumulation only ∼4-fold (Fig. 7B) but inhibited intermolecular RNA recombination by at least 45- to 250-fold (Fig. 7C). Thus, recombination correlated with an active RE signal functional in recruiting parental RNAs to replication complexes.
Frequency of Ura+ cells and RNA recombination.
When B3URA3 transcripts were induced in 1a2a+ yeast to launch RNA replication without recombination, 25% of cells became Ura+ bearing B3URA3 replicons (Fig. 5A). When B3Δ5′ and B3Δ3′ transcripts were coinduced under the same conditions, Ura+ cells bearing homologous, B3URA3 recombinants were reproducibly isolated at a frequency of ∼8 × 10−4 per cell (Fig. 3 and 8) or ∼3 × 10−3 of the frequency of nonrecombinant, full-length B3URA3. This is similar to the upper frequency of 2 × 10−3 homologous recombinants per nonrecombinant genome recovered from mixed poliovirus infections measuring recombination between markers 600 nt apart (21), similar to the 618-nt overlap between B3Δ5′ and B3Δ3′ (Fig. 1B).
In plants, some forms of intermolecular BMV RNA recombination occur in 10 to 100% of local lesions (29). These rates are difficult to compare to the above frequencies of BMV and poliovirus recombinants per wt genome, since individual local lesions can contain over 100 cells (25), each bearing over 106 BMV genomes (27). In other experiments with parallels to this study, 25% of tobacco protoplasts inoculated with wt RNA1, RNA2, and RNA3 became infected (18), while only 1 per 105 protoplasts inoculated with RNA1, RNA2, and an RNA3 derivative lacking the 3′ noncoding region acquired replicating RNA3 by nonhomologous recombination with RNA1 or RNA2 (18). Thus, while the frequencies of initiating replication by replication-competent RNA3 were similar in tobacco and yeast, the frequency of nonhomologous intermolecular recombinants per wt replicon in tobacco cells was 10- to 100-fold lower than the frequency of homologous recombinants in our experiments, whether measured as ∼8 × 10−4 per cell per 9 or 10 yeast generations (Fig. 3 and 8) or as ∼1.7 × 10−4 per cell per yeast generation (Fig. 8 and associated Results text). This is consistent with general expectations that guiding homology might facilitate homologous over nonhomologous intermolecular recombination.
Generation of truncated RNA replicons from nonreplicating B3Δ5′.
B3Δ5′ RNA did not support BMV positive-strand RNA synthesis but gave rise to Ura+ sURA3 RNA replicons at a frequency of 0.9 × 10−4 per cell per generation (Fig. 3, condition 7; and Fig. 6A). sURA3 RNAs were characterized by nearly complete deletion of 3a gene sequences from B3Δ5′, creating new 5′ ends in which the last 7 to 13 nt of the 3a ORF were fused to one or a few additional 5′ terminal nucleotides not contiguous in the starting sequence (Fig. 4C).
Deletion analysis showed that production of these truncated sURA3 RNAs required no specific nucleotide upstream of their 5′ endpoints of (nt 991 to 997) (Fig. 4C). Thus, generating the truncated 5′ ends of these replicons was not a highly specific limiting process in sURA3 RNA creation. As long as appropriate downstream sequences were provided (next paragraph), such novel 5′ ends were generated with nearly equal frequencies at sites ∼10 to 15 nt upstream of the RNA3 intergenic region whether these upstream sequences were derived from the 3a ORF (B3Δ5′, B3Δ817, B3Δ947) (Fig. 4C and 6A) or GAL1 leader (B3Δ997, B3Δ1003) (Fig. 6C). These 5′-truncated ends may have been generated by RNA template cleavage, polymerase pausing during negative-strand synthesis, or internal initiation of positive-strand RNA. Such 5′-truncated ends usually bore one or more 5′ nucleotides not contiguous in the starting sequence, possibly added by nontemplated synthesis by BMV polymerase (39), local duplications (Fig. 4C, sURA3 RNAs 2 and 4; Fig. 6D, second and third LURA3 RNAs), or recombination with surrounding sequences.
In contrast to upstream flexibility, nt 1003 to 1042, immediately downstream of the 5′ endpoints, were required for sURA3 RNA appearance (Fig. 6A). These sequences appear more likely required for sURA3 RNA replication than generation, since they comprised the replicon 5′ ends (Fig. 4C) and contained similarities to the wt RNA3 5′ end (Fig. 4D). The ability of these 5′ sequences to support positive-strand RNA synthesis was confirmed by the stable maintenance of sURA3 RNA replicons (Fig. 4B), even after loss of the B3Δ5′ plasmid, and by efficient launching of sURA3 replication from cDNA clones (Fig. 5A).
Similar conclusions emerged from the B3Δ909 LURA3 RNAs (Fig. 6D) whose 5′ ends retained the last 9 to 10 nt of the GAL1 leader fused to RNA3 nt 910 onward. Since the LURA3 5′ truncation sites were in the GAL1 leader common to all parental RNAs in Fig. 6A and also served as 5′ truncation sites in B3Δ997 and B3Δ1003 sURA3 RNAs (Fig. 6C), similarly truncated RNAs were likely formed by other parental RNAs but were not functional in replication. Thus, recovery of sURA3 and LURA3 replicons with 5′ ends from only certain sites, such as wt RNA3 nt 991 to 997 or the B3Δ909 junction of GAL1 sequences to RNA3 nt 910, appears due to the ability of only certain sequences to support positive-strand RNA synthesis.
The profound effects of 5′ sequences on BMV positive-strand RNA synthesis are best studied for RNA2, where sequence-function relations suggest contributions by multiple mechanisms (41). wt RNA3 lacks some 5′ features important for RNA2 replication (41) but replicates to higher levels in vivo. The 5′ ends of all sURA3, LURA3, and partially dimeric replicons from B3Δ1042 share several features with RNA3 (Fig. 4D and 6E and F). These include GUA or AUA 5′ ends, which are common in the bromoviridae (2, 7, 12); A- or AU-rich 5′ proximal sequences; and at ∼20 nt from the 5′ end, similarity to wt RNA3, nt 18 to 22 (UCUCG). Similarly, RNA2 nt 17 to 21 are important determinants of positive-strand RNA synthesis (41). Intriguingly, sequence similarities between the BMV RNA3 intergenic region and 5′ end of RNA3 from another bromovirus, cowpea chlorotic mottle virus, suggested that the natural cowpea chlorotic mottle virus RNA3 5′ end might have been generated by processes similar to the derivation of sURA3 RNAs from B3Δ5′ (2).
Acknowledgments
We thank all members of our laboratory for stimulating discussions throughout the course of this work.
This work was supported by the National Institutes of Health through grant GM35072. P.A. is an Investigator of the Howard Hughes Medical Institute.
REFERENCES
- 1.Albarino, C. G., B. D. Price, L. D. Eckerle, and L. A. Ball. 2001. Characterization and template properties of RNA dimers generated during flock house virus RNA replication. Virology 289:269-282. [DOI] [PubMed] [Google Scholar]
- 2.Allison, R. F., M. Janda, and P. Ahlquist. 1989. Sequence of cowpea chlorotic mottle virus RNAs 2 and 3 and evidence of a recombination event during bromovirus evolution. Virology 172:321-330. [DOI] [PubMed] [Google Scholar]
- 3.Bujarski, J. J., and P. Kaesberg. 1986. Genetic recombination between RNA components of a multipartite plant virus. Nature 321:528-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen, J., A. Noueiry, and P. Ahlquist. 2001. Brome mosaic virus protein 1a recruits viral RNA2 to RNA replication through a 5′ proximal RNA2 signal. J. Virol. 75:3207-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalmay, T., G. Szittya, and J. Burgyan. 1995. Generation of defective interfering RNA dimers of cymbidium ringspot tombusvirus. Virology 207:510-517. [DOI] [PubMed] [Google Scholar]
- 6.Dreher, T. W. 1999. Functions of the 3′-untranslated regions of positive strand RNA viral genomes. Annu. Rev. Phytopathol. 37:151-174. [DOI] [PubMed] [Google Scholar]
- 7.Dzianott, A. M., and J. J. Bujarski. 1991. The nucleotide sequence and genome organization of the RNA-1 segment in two bromoviruses: broad bean mottle virus and cowpea chlorotic mottle virus. Virology 185:553-562. [DOI] [PubMed] [Google Scholar]
- 8.Egger, D., and K. Bienz. 2002. Recombination of poliovirus RNA proceeds in mixed replication complexes originating from distinct replication start sites. J. Virol. 76:10960-10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falco, S. C., M. Rose, and D. Botstein. 1983. Homologous recombination between episomal plasmids and chromosomes in yeast. Genetics 105:843-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.French, R., and P. Ahlquist. 1988. Characterization and engineering of sequences controlling in vivo synthesis of brome mosaic virus subgenomic RNA. J. Virol. 62:2411-2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.French, R., and P. Ahlquist. 1987. Intercistronic as well as terminal sequences are required for efficient amplification of brome mosaic virus RNA3. J. Virol. 61:1457-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujisaki, K., F. Hagihara, M. Kaido, K. Mise, and T. Okuno. 2003. Complete nucleotide sequence of spring beauty latent virus, a bromovirus infectious to Arabidopsis thaliana. Arch. Virol. 148:165-175. [DOI] [PubMed] [Google Scholar]
- 13.Gallei, A., A. Pankraz, H. J. Thiel, and P. Becher. 2004. RNA recombination in vivo in the absence of viral replication. J. Virol. 78:6271-6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gmyl, A. P., E. V. Belousov, S. V. Maslova, E. V. Khitrina, A. B. Chetverin, and V. I. Agol. 1999. Nonreplicative RNA recombination in poliovirus. J. Virol. 73:8958-8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grdzelishvili, V. Z., T. Watanabe, H. Garcia-Ruiz, and P. Ahlquist. 2005. Mutual interference between genomic RNA replication and subgenomic RNA mRNA transcription in brome mosaic virus. J. Virol. 79:1438-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishikawa, M., M. Janda, and P. Ahlquist. 2000. The 3a cell-to-cell movement gene is dispensable for cell-to-cell transmission of brome mosaic virus RNA replicons in yeast but retained over 10(45)-fold amplification. J. Gen. Virol. 81:2307-2311. [DOI] [PubMed] [Google Scholar]
- 17.Ishikawa, M., M. Janda, M. A. Krol, and P. Ahlquist. 1997. In vivo DNA expression of functional brome mosaic virus RNA replicons in Saccharomyces cerevisiae. J. Virol. 71:7781-7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishikawa, M., P. Kroner, P. Ahlquist, and T. Meshi. 1991. Biological activities of hybrid RNAs generated by 3′-end exchanges between tobacco mosaic and brome mosaic viruses. J. Virol. 65:3451-3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito, H., Y. Fukuda, K. Murata, and A. Kimura. 1983. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153:163-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janda, M., and P. Ahlquist. 1993. RNA-dependent replication, transcription, and persistence of brome mosaic virus RNA replicons in S. cerevisiae. Cell 72:961-970. [DOI] [PubMed] [Google Scholar]
- 21.Jarvis, T. C., and K. Kirkegaard. 1992. Poliovirus RNA recombination: mechanistic studies in the absence of selection. EMBO J. 11:3135-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirkegaard, K., and D. Baltimore. 1986. The mechanism of RNA recombination in poliovirus. Cell 47:433-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kushner, D. B., B. D. Lindenbach, V. Z. Grdzelishvili, A. O. Noueiry, S. M. Paul, and P. Ahlquist. 2003. Systematic, genome-wide identification of host genes affecting replication of a positive-strand RNA virus. Proc. Natl. Acad. Sci. USA 100:15764-15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai, M. M. 1992. RNA recombination in animal and plant viruses. Microbiol. Rev. 56:61-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawrence, D. M., and A. O. Jackson. 2001. Interactions of the TGB1 protein during cell-to-cell movement of barley stripe mosaic virus. J. Virol. 75:8712-8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li, Y., and L. A. Ball. 1993. Nonhomologous RNA recombination during negative-strand synthesis of flock house virus RNA. J. Virol. 67:3854-3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loesch-Fries, L. S., and T. C. Hall. 1980. Synthesis, accumulation and encapsidation of individual brome mosaic virus RNA components in barley protoplasts. J. Gen. Virol. 47:323-332. [Google Scholar]
- 28.Mann, C., and R. W. Davis. 1983. Instability of dicentric plasmids in yeast. Proc. Natl. Acad. Sci. USA 80:228-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nagy, P. D., and J. J. Bujarski. 1995. Efficient system of homologous RNA recombination in brome mosaic virus: sequence and structure requirements and accuracy of crossovers. J. Virol. 69:131-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagy, P. D., and J. J. Bujarski. 1993. Targeting the site of RNA-RNA recombination in brome mosaic virus with antisense sequences. Proc. Natl. Acad. Sci. USA 90:6390-6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagy, P. D., A. Dzianott, P. Ahlquist, and J. J. Bujarski. 1995. Mutations in the helicase-like domain of protein 1a alter the sites of RNA-RNA recombination in brome mosaic virus. J. Virol. 69:2547-2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Panavas, T., and P. D. Nagy. 2003. Yeast as a model host to study replication and recombination of defective interfering RNA of Tomato bushy stunt virus. Virology 314:315-325. [DOI] [PubMed] [Google Scholar]
- 33.Pantaleo, V., L. Rubino, and M. Russo. 2003. Replication of carnation Italian ringspot virus defective interfering RNA in Saccharomyces cerevisiae. J. Virol. 77:2116-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pierangeli, A., M. Bucci, M. Forzan, P. Pagnotti, M. Equestre, and R. Perez Bercoff. 1999. ‘Primer alignment-and-extension’: a novel mechanism of viral RNA recombination responsible for the rescue of inactivated poliovirus cDNA clones. J. Gen. Virol. 80:1889-1897. [DOI] [PubMed] [Google Scholar]
- 35.Robzyk, K., and Y. Kassir. 1992. A simple and highly efficient procedure for rescuing autonomous plasmids from yeast. Nucleic Acids Res. 20:3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rose, M., and F. Winston. 1984. Identification of a Ty insertion within the coding sequence of the S. cerevisiae URA3 gene. Mol. Gen. Genet. 193:557-560. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 38.Schwartz, M., J. Chen, M. Janda, M. Sullivan, J. den Boon, and P. Ahlquist. 2002. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell 9:505-514. [DOI] [PubMed] [Google Scholar]
- 39.Siegel, R. W., S. Adkins, and C. C. Kao. 1997. Sequence-specific recognition of a subgenomic RNA promoter by a viral RNA polymerase. Proc. Natl. Acad. Sci. USA 94:11238-11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sivakumaran, K., and C. C. Kao. 2000. Genomic plus-strand RNA synthesis by the brome mosaic virus (BMV) RNA replicase requires a sequence that is complementary to the binding site of the BMV-helicase-like protein. Mol. Plant Pathol. 1:337-346. [DOI] [PubMed] [Google Scholar]
- 42.Sullivan, M. L., and P. Ahlquist. 1999. A brome mosaic virus intergenic RNA3 replication signal functions with viral replication protein 1a to dramatically stabilize RNA in vivo. J. Virol. 73:2622-2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang, X. H., and V. L. Chiang. 1996. Single-stranded DNA ligation by T4 RNA ligase for PCR cloning of 5′-noncoding fragments and coding sequence of a specific gene. Nucleic Acids Res. 24:990-991. [DOI] [PMC free article] [PubMed] [Google Scholar]