

Abstract
Infectious bursal disease virus (IBDV) is a birnavirus causing immunosuppressive disease in chickens. Emergence of the very virulent form of IBDV (vvIBDV) in the late 1980s dramatically changed the epidemiology of the disease. In this study, we investigated the phylogenetic origins of its genome segments and estimated the time of emergence of their most recent common ancestors. Moreover, with recently developed coalescence techniques, we reconstructed the past population dynamics of vvIBDV and timed the onset of its expansion to the late 1980s. Our analysis suggests that genome segment A of vvIBDV emerged at least 20 years before its expansion, which argues against the hypothesis that mutation of genome segment A is the major contributing factor in the emergence and expansion of vvIBDV. Alternatively, the phylogeny of genome segment B suggests a possible reassortment event estimated to have taken place around the mid-1980s, which seems to coincide with its expansion within approximately 5 years. We therefore hypothesize that the reassortment of genome segment B initiated vvIBDV expansion in the late 1980s, possibly by enhancing the virulence of the virus synergistically with its existing genome segment A. This report reveals the possible mechanisms leading to the emergence and expansion of vvIBDV, which would certainly provide insights into the scope of surveillance and prevention efforts regarding the disease.
Infectious bursal disease (IBD) is an immunosuppressive disease of young chickens that causes considerable economic loss to the poultry industry worldwide. The causative agent of IBD is a bisegmented, double-stranded RNA virus of the Birnaviridae family named IBD virus (IBDV). IBD was firstly reported in 1957 in broilers of the Delmarva Peninsula of the United States. It had spread rapidly throughout the United States by 1965 but was effectively controlled by vaccinations in the mid-1970s (23). In 1986, vaccination failures were reported and IBDV isolates with enhanced virulence were identified and characterized (18). These new isolates, named very virulent IBDV (vvIBDV), were then described by Chettle and coworkers (4) as the causative agents of the first reported cases of severe and acute IBD in Europe. These very virulent strains have rapidly spread all over Asia and other parts of the world (11) in an explosive manner (42), following their introduction into Japan in the early 1990s (30).
The epidemiological event leading to the emergence and expansion of vvIBDV is an open question. Phylogenetic analyses have revealed independent evolutionary histories of the two genome segments (17, 44), suggesting that a reassortment event may have played a role in the emergence of vvIBDV. Previous reports suggested that both the major capsid protein (VP2) and the RNA-dependent RNA polymerase (VP1), which are located on genome segments A and B, respectively, contribute to the virulence of IBDV (1-3, 26, 43). Questions have been raised regarding the phylogenetic origins and roles of the unique mutations of the two genome segments in relation to the expansion of vvIBDV.
Temporally spaced sequence data from rapidly evolving RNA viruses provide opportunities to address problems regarding their evolutionary behaviors, e.g., past population dynamics and evolutionary rates (7, 9, 10). Here we investigated the phylogenetic origins and demographic behavior of vvIBDV, as well as the time of emergence of the most recent common ancestor (TMRCA) of its genomes. Several approaches, including linear regression (LR), maximum likelihood (ML), and Bayesian Monte Carlo Markov chain (BMCMC), under the assumption of both strict and relaxed molecular clocks, were employed for TMRCA estimations. An alternative technique for estimating the TMRCA of the VP1 data set, site-stripping clock detection (SSCD) (24), was also used, in an attempt to accommodate deviations from the molecular clock. Comparison of TMRCAs inferred with these approaches provides comprehensive estimations of the time of emergence of both genome segments of vvIBDV. The past population dynamics of vvIBDV were reconstructed by using coalescence approaches in both the ML and BMCMC frameworks (10, 35), allowing us to estimate the time of onset of vvIBDV expansion. By reconstructing the chronological order of these epidemiological events, this report provides insights into the possible mechanism leading to the emergence and expansion of vvIBDV.
METHODS AND MATERIALS
Identification of vvIBDV clades in phylogenies of both genome segments.
For genome segment A, all available nonidentical serotype I IBDV nucleotide sequences containing the VP2 region, excluding all known tissue culture-adapted strains, were retrieved from the GenBank database (http://www.ncbi.nlm.nih.gov) and aligned. Due to the limited number of full-length VP2 (fVP2) (1,323 bp) sequences (n = 61), a larger data set (n = 420) consisting of partial VP2 (pVP2) (330 bp) sequences was also analyzed independently. To identify vvIBDV clades in both VP2 data sets, phylogenetic trees were constructed by the BMCMC method and were rooted with serotype II strain OH (U30818). The BMCMC tree is a majority consensus tree summarized across two sets of four tempered MCMC chains of 10 million states sampled every 100th generation, with the initial 1 million states discarded; it was constructed by MRBAYES (38) with the substitution model suggested by MODELTEST evaluated by the Akaike Information Criterion (34). The chain lengths, sampling frequencies, and burn-in proportions of all MCMC chains in this study are the same as described unless specified.
To investigate the phylogenetic origin of genome segment B of vvIBDV, all available nonidentical VP1 sequences of all birnaviruses were retrieved from GenBank. The codons were aligned, and the sites with gaps were removed. A BMCMC tree was constructed for this VP1 data set as described above. The vvIBDV clades of each data set, designated vvfVP2, vvpVP2, and vvVP1, were defined as the clades containing typical vvIBDV isolates, including DV89, UK611, and OKYM (42), with Bayesian posterior clade probability (BP) support of at least 0.85.
Molecular clock analysis and SSCD.
The sampling years of taxa in vvIBDV clades were obtained from references or personal communication with the authors. The alignments are available on request. The phylogenies of the vvVP1, vvfVP2, and vvpVP2 clades were reestimated with PAUP* (Sinauer Associates, Inc.) using an ML heuristic search with a tree bisection-reconnection branch-swapping algorithm under the nucleotide substitution models suggested by MODELTEST. Due to the large number of taxa (n = 156) of the vvpVP2 clade and the computational intensiveness of ML analysis, phylogenetically similar taxa were discarded manually (n = 48 [after removal]). Branch lengths of the phylogenies were reestimated in an ML framework under a generally unconstrained model (no clock [M0]) or the assumption of a single substitution rate (global clock [M1]), which are implemented in PAML (45). To test for the presence of a molecular clock, a likelihood ratio test (LRT) was used to compare the performance of both models in each data set.
In the vvVP1 clade, since M0 showed a significantly better fit to the data than M1 (P = 0.03), i.e., the molecular clock hypothesis was rejected, an SSCD procedure based on likelihood ratio reduction (24) was applied to recover sites exhibiting clocklike behavior. Briefly, the natural log likelihoods (lnL) of each site under both M0 and M1 were estimated, and the relative contribution to the likelihood ratio statistics, i.e., 2ΔlnL, was then calculated. Those sites that contributed most to the rejection of the molecular clock hypothesis, i.e., sites with the largest 2ΔlnL, were removed (five by five) until M0 no longer fit the data significantly better than M1, i.e., P was >0.05 under the LRT. Random site stripping was used as a control to demonstrate that the insignificant result is not simply the result of lack of power due to the removal of data. The JAVA codes of the SSCD procedure can be found in the PAL package (8).
Estimation of TMRCA.
Under the assumption of a molecular clock, TMRCAs of vvVP2 and the site-stripped vvVP1 data set were estimated with ML, BMCMC, and LR approaches. The ML estimations were carried out with PAML by rescaling the branches of the ML topologies into a calendar time scale under a strict global molecular clock (M1) as described above. The BMCMC estimations were carried out with BEAST (http://evolve.zoo.ox.ac.uk/beast/) under the general time-reversible substitution model with gamma-distributed rate variation (four categories) and a proportion of invariant sites. Posterior distribution of TMRCAs was then summarized across four independent MCMC chains with TRACER (http://evolve.zoo.ox.ac.uk/software.html?id = tracer). In TMRCA estimation by the LR method, ancestral states of the vvIBDV clades were first inferred under the ML criterion, and then the pairwise genetic distances (with the optimal substitution model suggested by MODELTEST) of taxa from their inferred TMRCA, which are implemented in PAUP*, were calculated. Correlations of the estimated pairwise genetic distances and sampling years were examined by LR analysis implemented in GraphPad PRISM (San Diego, CA).
To evaluate the possible biases of the SSCD procedure on TMRCA estimations of the vvVP1 clade, its TMRCA was also estimated in a Bayesian framework under a relaxed molecular clock model using PAML and MULTIDIVTIME (22), according to the procedures described by Rutschmann (http://www.plant.ch), which take into account both uncertainty in branch length estimation and lineage-specific rate variation. The mean normally distributed prior substitution rate was set at 0.7 × 10−3 substitutions per site per year with a standard deviation (SD) of 0.1 × 10−3, and the mean normal prior TMRCA was set at 1980 with an SD of 15 years, based on the posterior distribution of the site-stripped vvVP1 data set under the assumption of a molecular clock. It is noted that the SSCD-estimated mutation rate of the site-stripped vvVP1 data set, i.e., 0.7 × 10−3 substitutions per site per year, is within the general mutation rate range of other RNA viral genes (20) and is comparable to that of other viral RNA-dependent RNA polymerases (40). Different prior TMRCAs, including 1985 ± 10 years, 1965 ± 30 years, and 1945 ± 50 years, were used in order to assess the influence of priors on posterior distribution. The posterior distribution of the TMRCA was summarized across four independent MCMC chains.
Inference of past population dynamics.
To describe the epidemic history of vvIBDV in terms of change of effective population size through time, i.e., Ne(t), a Bayesian skyline plot (BSP) was performed on the vvpVP2 data set (n = 153). Ne(t) can be thought of as the number of infections contributing to new infections rather than the total number of prevalent infections (16). BSP is a nonparametric coalescence analysis for estimating past population dynamics through time without dependence on a prespecified parametric model; it has proven very useful as a demographic model selection tool (10). The BSP of the vvpVP2 data set was jointly estimated with the BMCMC TMRCA inference using BEAST, as described above. To select a parametric model that best describes the past population dynamics of vvIBDV, the goodness-of-fit of five demographic models in the time-scaled genealogy was evaluated in terms of likelihoods and LRT, which were calculated with GENIE (35, 36). These models include the piecewise expansion, logistic, constant, piecewise logistic, and exponential models (35). The demographic parameters of the best-fit model were estimated with confidence intervals, in both the BMCMC and ML frameworks, by BEAST and GENIE, respectively. The BMCMC and ML parametric estimates of the chosen demographic model were then evaluated by superimposing its nonparametric estimates through time, i.e., Bayesian and classical skyline plots (CSP) for BMCMC and ML estimations, respectively. CSP is a piecewise-constant model of population size and typically produces “noisy” plots that display the stochastic variability inherent in the coalescence process, while BSP is an advanced version of CSP that naturally produces a smoother estimate due to the “averaging” effect of MCMC sampling (10). Bayesian and classical skyline plots were estimated with BEAST and GENIE, respectively.
RESULTS
Phylogenetic origins of both vvIBDV genome segments.
The BMCMC phylogeny of the full-length VP2 data set (fVP2) revealed five major clades with BP support greater than 0.85; these were designated the classical 1, classical 2, classical 3, variant, and very virulent (vvfVP2) clades, according to the typical classification of IBDV strains (42). These clades formed a monophyletic lineage, designated the major IBDV lineage, with 0.99 BP support, excluding the Australian strain, suggesting that these clades, contributing to the major world epidemics, evolved from the same TMRCA (Fig. 1A). All of the above clades, including the Australian strain, were together classified as serotype I IBDV strains. Analysis of the partial VP2 data set (pVP2) resulted similar phylogeny, with well-supported major IBDV lineage and the very virulent clade (vvpVP2), except that classical and variant clades were no longer supported by high BP (data not shown). In contrast, in the BMCMC phylogeny of VP1, the vvIBDV clade (vvVP1) was excluded from the lineage containing all other serotype I and II IBDV strains (0.87 BP support) (Fig. 1B), revealing an independent phylogenetic origin of vvVP1 that agreed with previous reports (17). The high BP support (0.99) of the monophyletic IBDV VP1 lineage in the birnavirus VP1 phylogeny suggests that vvVP1 may have an avian origin that shares the TMRCA with the other IBDV strains. With Bayesian-scanning analysis implemented in SLIDINGBAYES (http://www.kuleuven.ac.be), no significant sign of intrasegment recombination was detected in the birnavirus VP1 phylogeny (data not shown), suggesting a nonrecombinant origin of vvVP1; therefore, it may have been reassorted from an unidentified avian reservoir.
FIG. 1.
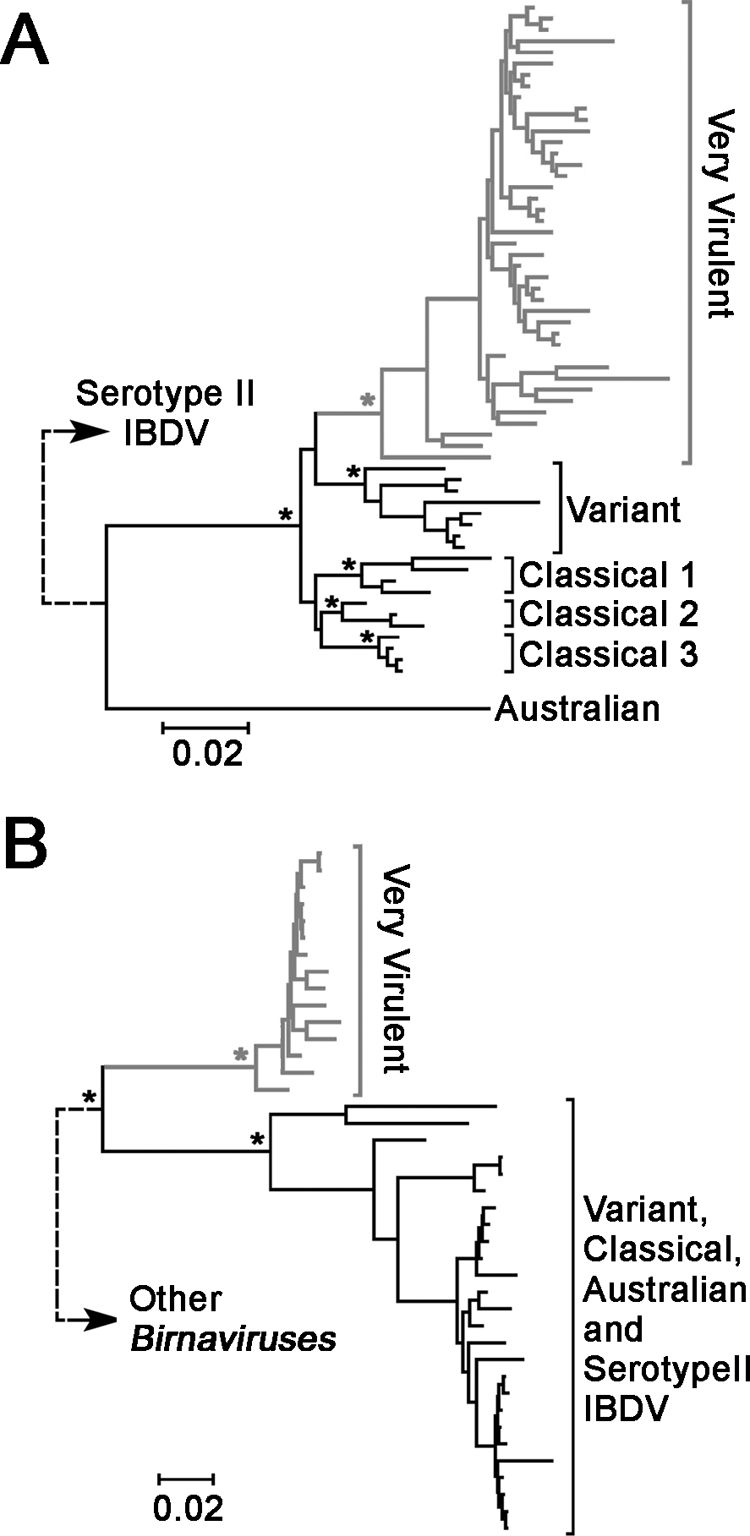
Phylogenetic origins of both genome segments of vvIBDV. (A) BMCMC tree of full-length VP2 of serotype I IBDV, rooted with serotype II strain OH. The branch length of the root (dotted line) is not drawn to scale. (B) BMCMC tree of the full-length VP1 of all birnaviruses. For clarity, only the IBDV lineage is shown, and the branch length of its root to other birnaviruses (dotted line) is not drawn to scale. Asterisks represent clades with BP support of at least 0.85. The scale bars are in units of nucleotide substitutions per site. The vvIBDV clades in both trees are highlighted in gray.
Molecular clock analysis and TMRCA of vvIBDV genomes.
An LRT showed that M0 (no-clock model) does not fit the vvfVP2 data set significantly better than M1 (global-clock model) (P = 0.118), allowing us to time the vvVP2 TMRCA under the assumption of a strict global molecular clock. With the ML, BMCMC, and LR approaches, TMRCA of vvfVP2 was consistently estimated to be around the 1960s (Table 1). It is noted that LR, ML, and BMCMC estimated the evolutionary rate for the clocklike data set vvfVP2 to be 0.64 × 10−3, 0.60 × 10−3, and 0.67 × 10−3 nucleotide substitutions per site per year, respectively, which is comparable to the capsid or envelope proteins of other RNA viruses (20). Comparable results were obtained with the vvpVP2 data set by the BMCMC and LR approaches (Table 1), although the molecular clock was significantly rejected in this case (P = 0.003), suggesting that nonclocklike substitution rates may still be reliable indicators of the average substitution rate if the data set is large enough (20).
TABLE 1.
Molecular clock and TMRCA estimations of both genome segments of vvIBDV based on various approaches
| Genome segment | Length (bp) | No. of taxa | lnLa
|
LRT (P) (clock rejection)b | Yr of TMRCA (range)c
|
|||
|---|---|---|---|---|---|---|---|---|
| M0 | M1 | LR | ML | BMCMC | ||||
| vvfVP2 | 1,323 | 24 | −2,813.171 | −2,828.190 | 0.118 (no) | 1960 (1942-1969) | 1966 (1955-1977) | 1965 (1952-1975) |
| vvpVP2 | 330 | 153 | −3,257.785 | −3,361.408 | 0.003 (yes) | 1968 (1943-1977) | 1946 (1924-1968) | 1962 (1950-1973) |
| vvVP1 | 2,322 | 9 | −4,291.567 | −4,299.338 | 0.03 (yes) | 1981 (NA-1989) | 1979 (1972-1986) | 1974 (1953-1985) |
| Site-stripped vvVP1 | 2,307 | 9 | −4,115.234 | −4,120.421 | 0.168 (no) | 1984 (1972-1988) | 1981 (1976-1986) | 1980 (1973-1988) |
lnL represents the natural log likelihood of the global-clock (M1) and no-clock (M0) models calculated in PAML under the Hasegawa-Kishino-Yano nucleotide substitution model.
The molecular clock is rejected when P is <0.05 under the LRT.
The BMCMC TMRCA refers to the TMRCAs estimated with BEAST under the assumption of a molecular clock. TMRCAs estimated based on the corresponding approaches are presented as the best fit (for LR and ML) or mean (for BMCMC) value, with the 95% CI (for LR and ML) or high-probability density (for BMCMC) in parentheses. NA, not applicable.
Substitution rate variation among sites or lineages causing nonclocklike behavior is a common observation in RNA viruses (20), presenting standard dating applications with uncertainties and perhaps leading to strong dating biases (24), particularly in data sets with limited number of taxa. Although the vvVP1 data set violated the molecular clock (P = 0.03), its clocklike behavior was recovered by stripping 15 sites according to the SSCD procedure (Fig. 2A). The TMRCA of the site-stripped vvVP1 data set was then estimated to be in the early to mid-1980s by the LR, ML, and BMCMC approaches (Fig. 2B and Table 1). Initial dating efforts with the nonclocklike vvVP1 data set generally yielded TMRCA estimations comparable to that of the site-stripped vvVP1 data set, although the confidence intervals (CIs) are relatively large or unable to be estimated (Table 1). The possible biases of SSCD procedures were assessed by reestimating the TMRCA under a relaxed-clock model implemented in MULTIDIVTIME (22). The posterior distribution of the TMRCA of the original vvVP1 data set estimated under the relaxed clock generally overlapped with that of the site-stripped vvVP1 data set under the strict clock at 1981 (1976 to 1984) (Fig. 2C), while altering of the TMRCA prior has a limited effect on its posterior distribution (data not shown). The consistency of TMRCAs estimated from different approaches indicates the robustness of our estimates, and the effect of evolutionary rate variation may be limited in this case. For clarity, LR and ML TMRCA estimations of only the clocklike data sets, i.e., vvfVP2 and site-stripped vvVP1, are presented in Fig. 3.
FIG. 2.

Estimation of vvVP1 TMRCA under both strict and relaxed molecular clock assumptions. (A) Reduction of relative contribution to the likelihood ratio statistics (2ΔlnL) by SSCD. The upper limit of 2ΔlnL for not rejecting the molecular clock hypothesis in the vvVP1 data set is approximately 14. (B) ML estimations of vvVP1 TMRCA with different numbers of sites stripped. (C) Comparison of posterior distributions of the vvVP1 TMRCA estimated under both strict and relaxed molecular clocks.
FIG. 3.

Estimations of TMRCA of both genome segments of vvIBDV and onset of its expansion under different approaches. (A) TMRCA estimations of vvfVP2 and the site-stripped vvVP1 data set with the LR approach. The dotted lines and the solid lines represent 95% CIs and best-fit values, respectively. (B) TMRCA estimations of vvfVP2 and the site-stripped vvVP1 data set, as well as the onset of vvIBDV expansion with the ML approach. The dotted lines and the solid dots represent the 95% CIs and best-fit values, respectively. (C) TMRCA estimations of vvVP2 and vvVP1, as well as the onset of vvIBDV expansion, with the BMCMC approach. The posterior distribution of vvVP2 TMRCA represents pooled BMCMC samples of the vvfVP2 and vvpVP2 data sets (BEAST), while that of vvVP1 TMRCA represents pooled BMCMC samples of the original vvVP1 data set under a relaxed clock (MULTIDIVTIME) and the site-stripped vvVP1 data set under a strict clock (BEAST). The time axes of the three panels are drawn to the same scale.
Estimation of parameters of vvIBDV expansion.
The BSP of the vvpVP2 data set suggests a sudden exponential population growth in the late 1980s, followed by a sharp population drop around the mid-1990s (Fig. 4). Only the vvpVP2 data set was used for demographic behavior estimations, because data sets of less than 25 taxa are unlikely to give reliable coalescence estimates (16). Based on the BSP, none of the existing parametric models adequately describes the sophisticated demographic signals of the vvpVP2 data set. Use of incorrect demographic models will lead to biased and invalid estimates of demographic history (36). In order to describe the sudden exponential population growth with parametric models, the demographic signals of the vvpVP2 data set were simplified by discarding taxa sampled after the estimated sharp population drop at 1995 (designated vvpVP2-95; n = 45). The time-scaled ML genealogy of the vvpVP2-95 data set was then reconstructed with PAUP and PAML (45), and the goodness-of-fit of five demographic models was evaluated with GENIE (35). The piecewise expansion model, i.e., constant population size followed by exponential growth, best describes the vvpVP2-95 ML genealogy in terms of likelihood, and the exponential model was rejected in favor of the piecewise expansion model (P < 0.01). According to model definition, the exponential growth phase start time, i.e., the onset of expansion, is defined as −ln(α)/r, where r is the exponential growth rate and α is the population size prior to change, as a proportion of the population size at present (Fig. 5A). It is noted that the piecewise expansion demographic model is not implemented in BEAST, version 1.3, and the codes for this particular model were developed by the authors; they are available on request. Superimposing the ML and Bayesian parametric estimates under the piecewise expansion model onto their nonparametric skyline plots revealed a good fit to the demographic signal within the vvpVP2-95 data set (Fig. 5B and C). The ML estimation of exponential growth phase start time is 1983 (1970 to 1987), while the BMCMC estimation yielded consistent results with more precise CIs, i.e., 1988 (1986 to 1990). Although ML estimation of demographic parameters based on a single genealogy ignores possible phylogenetic errors, overlapping results were produced with the BMCMC estimation, which takes phylogenetic uncertainty into account (10), reflecting the reliability of our estimations of the onset of vvIBDV expansion.
FIG. 4.

Nonparametric estimation of past population dynamics of vvIBDV with BSP. The BSP was estimated from the vvpVP2 data set and is presented with its 95% high-probability density (HPD) values (gray area).
FIG. 5.

Estimation of the demographic parameters of vvIBDV under the piecewise expansion model. (A) Schematic representation of the piecewise expansion model of population growth. (B) BSP (nonparametric) and BMCMC parametric estimation of Ne of the vvpVP2-95 data set under the piecewise expansion model by using BEAST. (C) CSP (nonparametric) and ML parametric estimation of Ne of the vvpVP2-95 data set under the piecewise expansion model by using GENIE. The BSP is presented with its 95% high-probability density (HPD) values (gray area). Only the mean or best-fit values are presented in the BMCMC and ML parametric estimations, respectively.
DISCUSSION
Coalescence tools for the selection of parameter-rich demographic models allowed us to estimate the onset of vvIBDV expansion at around the late 1980s, which seems to be consistent with its documented explosive transmission pattern. The first documented case of vvIBDV was described by Chettle and coworkers in 1989 (4). In 1995, during the 63rd General Session of the Office International des Epizooties, about 80% of its member countries worldwide reported the occurrence of vvIBDV (11), reflecting the rapid spread of vvIBDV during the early 1990s.
The epidemiological events related to the initiation of vvIBDV expansion have not been investigated systematically. While most of the interest regarding virulence and attenuation of the virus was focused on genome segment A, as it encodes neutralizing epitopes and the putative receptor binding protein (43), our estimations of the vvfVP2 TMRCA indicate that genome segment A of vvIBDV emerged more than 20 years before the first documented case of vvIBDV, suggesting that the vvIBDV expansion may not be directly initiated by the emergence of vvVP2. This observation argues against the hypothesis that mutation of genome segment A is the major contributing factor in the emergence and expansion of vvIBDV (33). Alternatively, the vvVP1 TMRCA was estimated to around the early to mid-1980s, which coincides with the estimated onset of IBDV expansion in a period of approximately 5 years, suggesting a possible role of vvVP1 in the initiation of vvIBDV expansion. The origin of vvVP1 is unknown, but its phylogenetic independence from all other known IBDV strains suggests that it may be reassorted from an unidentified reservoir. A serotype II IBDV strain was successfully isolated from African blacked-footed penguins (Spheniscus demersus) (19), implying the presence of other possible natural hosts of IBDV. Moreover, serological surveys of aquatic fowl (12, 15) and free-living wild birds (13, 31) suggested that several migratory and sedentary avian species, including cattle egrets (Ardeola ibis), pigeons (Columba livia), carrion crows (Corvus corone), and Antarctic penguins (Pygoscelis adeliae), etc., may also be carriers or reservoirs of IBDV based on their seroprevalence.
A number of reports have demonstrated the drastic effects of mutations of VP2 residues on attenuation of the virulence of vvIBDV (3, 43); these residues were proposed to be related to the protein's receptor binding efficiency (5), but they are not the sole determinants of virulence (2). On the other hand, VP1 was also demonstrated to be a determinant of virulence in vivo, possibly by altering the replication efficiency (1, 26). A recent study reported the isolation of a naturally reassorted IBDV with genome segment A of very virulent origin but genome segment B of non-very virulent origin; this isolate showed significantly reduced pathogenicity compared with typical vvIBDVs (25), demonstrating that the determinants on genome segment A may not be enough to induce the hypervirulence of typical vvIBDVs. These observations suggest that the hypervirulence of vvIBDV may be a synergistic effect of mutations on both of its genome segments, although experimental evidence is needed for further elucidation. In addition, examples of hypervirulence of other RNA viruses caused by mutations on their polymerase genes are well documented (6, 21, 27, 41). Taken together with our date estimates, the phylogenetic origin of vvIBDV genomes, the possible roles of both VP1 and VP2 in hypervirulence, and the reported evidence of the existence of possible reservoirs, we hypothesize that vvIBDV expansion may have been initiated by the reassortment of its genome segment B with a mutant VP2 background, which caused a sudden increase in virulence and hence expansion in the mid-1980s, rather than being directly related to the emergence of vvVP2 in the 1960s, as vvVP2 alone may not be enough to induce the hypervirulence of vvIBDV (25). The presence of rare IBDV isolates carrying vvVP2 and non-vvVP1 further supports this hypothesis (25), as these isolates can be considered the descendants of those rare “ancient” vvIBDVs that appeared after the estimated TMRCA of vvVP2 in the mid-1960s but before the estimated reassortment of vvVP1 and the expansion of vvIBDV in the mid-1980s.
Genome reassortment of multisegmented RNA viruses plays important roles in the emergence or expansion of new virus strains with altered antigenicity or pathogenicity (29, 32, 37), best demonstrated by the devastating pandemics of influenza A (39). Investigations of possible evolutionary mechanisms leading to the emergence and expansion of novel pathogenic virus strains would certainly provide insight into the scope of surveillance and prevention efforts (14, 28). In the case of IBDV, while most of the sequencing efforts have been focused on genome segment A of viruses in chickens, larger-scale surveillance in other avian species and more sequence data on genome segment B would certainly provide important clues toward the understanding of the diversity and origin, as well as the evolutionary epidemiology, of IBDV.
Acknowledgments
We thank Susan E. Sommer, of the Ohio Agricultural Research and Development Center, for providing the sampling dates of IBDV isolates; Frank Rutschmann for assistance in relaxed-clock dating; Philippe Lemey and Anne-Mieke Vandamme for valuable comments on the manuscript and the SSCD procedures; and Ken Yan-Ching Chow, of the Molecular Viral Pathogenesis Unit, Institut Pasteur, Paris, France, for useful discussions.
C.-C.H., F.Z., and F.C.C.L. designed the research; C.-C.H. performed all ML analyses and wrote the paper; T.-Y.L. performed SSCD analyses; A.D. and A.R. performed all BMCMC analyses; C.-W.Y. and P.-Y.L. performed LR analyses; and Y.-F.L. and P.T.W.N. performed molecular clock tests. We declare that there were no conflicts of interest relevant to this study.
REFERENCES
- 1.Boot, H. J., A. J. Hoekman, and A. L. Gielkens. 2005. The enhanced virulence of very virulent infectious bursal disease virus is partly determined by its B-segment. Arch. Virol. 150:137-144. [DOI] [PubMed] [Google Scholar]
- 2.Boot, H. J., A. A. ter Huurne, A. J. Hoekman, B. P. Peeters, and A. L. Gielkens. 2000. Rescue of very virulent and mosaic infectious bursal disease virus from cloned cDNA: VP2 is not the sole determinant of the very virulent phenotype. J. Virol. 74:6701-6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandt, M., K. Yao, M. Liu, R. A. Heckert, and V. N. Vakharia. 2001. Molecular determinants of virulence, cell tropism, and pathogenic phenotype of infectious bursal disease virus. J. Virol. 75:11974-11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chettle, N., J. C. Stuart, and P. J. Wyeth. 1989. Outbreak of virulent infectious bursal disease in East Anglia. Vet. Rec. 125:271-272. [DOI] [PubMed] [Google Scholar]
- 5.Coulibaly, F., C. Chevalier, I. Gutsche, J. Pous, J. Navaza, S. Bressanelli, B. Delmas, and F. A. Rey. 2005. The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell 120:761-772. [DOI] [PubMed] [Google Scholar]
- 6.Djavani, M., I. S. Lukashevich, and M. S. Salvato. 1998. Sequence comparison of the large genomic RNA segments of two strains of lymphocytic choriomeningitis virus differing in pathogenic potential for guinea pigs. Virus Genes 17:151-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drummond, A., O. G. Pybus, and A. Rambaut. 2003. Inference of viral evolutionary rates from molecular sequences. Adv. Parasitol. 54:331-358. [DOI] [PubMed] [Google Scholar]
- 8.Drummond, A., and K. Strimmer. 2001. PAL: an object-oriented programming library for molecular evolution and phylogenetics. Bioinformatics 17:662-663. [DOI] [PubMed] [Google Scholar]
- 9.Drummond, A. J., G. K. Nicholls, A. G. Rodrigo, and W. Solomon. 2002. Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161:1307-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drummond, A. J., A. Rambaut, B. Shapiro, and O. G. Pybus. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22:1185-1192. [DOI] [PubMed] [Google Scholar]
- 11.Eterradossi, N. 1995. Progress in the diagnosis and prophylaxis of infectious bursal disease in poultry, p. 75-82. OIE, Paris, France.
- 12.Fabohun, O. A., A. A. Owoade, D. O. Oluwayelu, and F. O. Olayemi. 2000. Serological survey of infectious bursal disease virus antibodies in cattle egrets, pigeons and nigerian laughing doves. Afr. J. Biomed. Res. 3:191-192. [Google Scholar]
- 13.Gardner, H., K. Kerry, M. Riddle, S. Brouwer, and L. Gleeson. 1997. Poultry virus infection in Antarctic penguins. Nature 387:245. [DOI] [PubMed] [Google Scholar]
- 14.Grenfell, B. T., O. G. Pybus, J. R. Gog, J. L. Wood, J. M. Daly, J. A. Mumford, and E. C. Holmes. 2004. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303:327-332. [DOI] [PubMed] [Google Scholar]
- 15.Hollmen, T., J. C. Franson, D. E. Docherty, M. Kilpi, M. Hario, L. H. Creekmore, and M. R. Petersen. 2000. Infectious bursal disease virus antibodies in eider ducks and herring gulls. Condor 102:688-691. [Google Scholar]
- 16.Hue, S., D. Pillay, J. P. Clewley, and O. G. Pybus. 2005. Genetic analysis reveals the complex structure of HIV-1 transmission within defined risk groups. Proc. Natl. Acad. Sci. USA 102:4425-4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Islam, M. R., K. Zierenberg, and H. Muller. 2001. The genome segment B encoding the RNA-dependent RNA polymerase protein VP1 of very virulent infectious bursal disease virus (IBDV) is phylogenetically distinct from that of all other IBDV strains. Arch. Virol. 146:2481-2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackwood, D. H., and Y. M. Saif. 1987. Antigenic diversity of infectious bursal disease viruses. Avian Dis. 31:766-770. [PubMed] [Google Scholar]
- 19.Jackwood, D. J., R. E. Gough, and S. E. Sommer. 2005. Nucleotide and amino acid sequence analysis of a birnavirus isolated from penguins. Vet. Rec. 156:550-552. [DOI] [PubMed] [Google Scholar]
- 20.Jenkins, G. M., A. Rambaut, O. G. Pybus, and E. C. Holmes. 2002. Rates of molecular evolution in RNA viruses: a quantitative phylogenetic analysis. J. Mol. Evol. 54:156-165. [DOI] [PubMed] [Google Scholar]
- 21.Kagiwada, S., Y. Yamaji, K. Komatsu, S. Takahashi, T. Mori, H. Hirata, M. Suzuki, M. Ugaki, and S. Namba. 2005. A single amino acid residue of RNA-dependent RNA polymerase in the Potato virus X genome determines the symptoms in Nicotiana plants. Virus Res. 110:177-182. [DOI] [PubMed] [Google Scholar]
- 22.Kishino, H., J. L. Thorne, and W. J. Bruno. 2001. Performance of a divergence time estimation method under a probabilistic model of rate evolution. Mol. Biol. Evol. 18:352-361. [DOI] [PubMed] [Google Scholar]
- 23.Lasher, H. N., and V. S. Davis. 1997. History of infectious bursal disease in the USA—the first two decades. Avian Dis. 41:11-19. [PubMed] [Google Scholar]
- 24.Lemey, P., M. Salemi, B. Wang, M. Duffy, W. H. Hall, N. K. Saksena, and A. M. Vandamme. 2003. Site stripping based on likelihood ratio reduction is a useful tool to evaluate the impact of non-clock-like behavior on viral phylogenetic reconstructions. FEMS Immunol. Med. Microbiol. 39:125-132. [DOI] [PubMed] [Google Scholar]
- 25.Le Nouen, C., G. Rivallan, D. Toquin, P. Darlu, Y. Morin, V. Beven, C. de Boisseson, C. Cazaban, S. Comte, Y. Gardin, and N. Eterradossi. 2006. Very virulent infectious bursal disease virus: reduced pathogenicity in a rare natural segment-B-reassorted isolate. J. Gen. Virol. 87:209-216. [DOI] [PubMed] [Google Scholar]
- 26.Liu, M., and V. N. Vakharia. 2004. VP1 protein of infectious bursal disease virus modulates the virulence in vivo. Virology 330:62-73. [DOI] [PubMed] [Google Scholar]
- 27.McGoldrick, A., A. J. Macadam, G. Dunn, A. Rowe, J. Burlison, P. D. Minor, J. Meredith, D. J. Evans, and J. W. Almond. 1995. Role of mutations G-480 and C-6203 in the attenuation phenotype of Sabin type 1 poliovirus. J. Virol. 69:7601-7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moya, A., E. C. Holmes, and F. Gonzalez-Candelas. 2004. The population genetics and evolutionary epidemiology of RNA viruses. Nat. Rev. Microbiol. 2:279-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikolakaki, S. V., K. Nomikou, M. Koumbati, O. Mangana, M. Papanastassopoulou, P. P. Mertens, and O. Papadopoulos. 2005. Molecular analysis of the NS3/NS3A gene of Bluetongue virus isolates from the 1979 and 1998-2001 epizootics in Greece and their segregation into two distinct groups. Virus Res. 114:6-14. [DOI] [PubMed] [Google Scholar]
- 30.Nunoya, T., Y. Otaki, M. Tajima, M. Hiraga, and T. Saito. 1992. Occurrence of acute infectious bursal disease with high mortality in Japan and pathogenicity of field isolates in specific-pathogen-free chickens. Avian Dis. 36:597-609. [PubMed] [Google Scholar]
- 31.Ogawa, M., T. Wakuda, T. Yamaguchi, K. Murata, A. Setiyono, H. Fukushi, and K. Hirai. 1998. Seroprevalence of infectious bursal disease virus in free-living wild birds in Japan. J. Vet. Med. Sci. 60:1277-1279. [DOI] [PubMed] [Google Scholar]
- 32.Ohashi, S., Y. Matsumori, T. Yanase, M. Yamakawa, T. Kato, and T. Tsuda. 2004. Evidence of an antigenic shift among Palyam serogroup orbiviruses. J. Clin. Microbiol. 42:4610-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pitcovski, J., D. Goldberg, B. Z. Levi, D. Di-Castro, A. Azriel, S. Krispel, T. Maray, and Y. Shaaltiel. 1998. Coding region of segment A sequence of a very virulent isolate of IBDV—comparison with isolates from different countries and virulence. Avian Dis. 42:497-506. [PubMed] [Google Scholar]
- 34.Posada, D., and T. R. Buckley. 2004. Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst. Biol. 53:793-808. [DOI] [PubMed] [Google Scholar]
- 35.Pybus, O. G., and A. Rambaut. 2002. GENIE: estimating demographic history from molecular phylogenies. Bioinformatics 18:1404-1405. [DOI] [PubMed] [Google Scholar]
- 36.Pybus, O. G., A. Rambaut, and P. H. Harvey. 2000. An integrated framework for the inference of viral population history from reconstructed genealogies. Genetics 155:1429-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramig, R. F. 1997. Genetics of the rotaviruses. Annu. Rev. Microbiol. 51:225-255. [DOI] [PubMed] [Google Scholar]
- 38.Ronquist, F., and J. P. Huelsenbeck. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572-1574. [DOI] [PubMed] [Google Scholar]
- 39.Russell, C. J., and R. G. Webster. 2005. The genesis of a pandemic influenza virus. Cell 123:368-371. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka, Y., K. Takahashi, E. Orito, Y. Karino, J.-H. Kang, K. Suzuki, A. Matsui, A. Hori, H. Matsuda, H. Sakugawa, Y. Asahina, T. Kitamura, M. Mizokami, and S. Mishiro. 2006. Molecular tracing of Japan-indigenous hepatitis E viruses. J. Gen. Virol. 87:949-954. [DOI] [PubMed] [Google Scholar]
- 41.Taubenberger, J. K., A. H. Reid, R. M. Lourens, R. Wang, G. Jin, and T. G. Fanning. 2005. Characterization of the 1918 influenza virus polymerase genes. Nature 437:889-893. [DOI] [PubMed] [Google Scholar]
- 42.Van den Berg, T. P. 2000. Acute infectious bursal disease in poultry: a review. Avian. Pathol. 29:175-194. [DOI] [PubMed] [Google Scholar]
- 43.van Loon, A. A., N. de Haas, I. Zeyda, and E. Mundt. 2002. Alteration of amino acids in VP2 of very virulent infectious bursal disease virus results in tissue culture adaptation and attenuation in chickens. J. Gen. Virol. 83:121-129. [DOI] [PubMed] [Google Scholar]
- 44.Yamaguchi, T., M. Ogawa, M. Miyoshi, Y. Inoshima, H. Fukushi, and K. Hirai. 1997. Sequence and phylogenetic analyses of highly virulent infectious bursal disease virus. Arch. Virol. 142:1441-1458. [DOI] [PubMed] [Google Scholar]
- 45.Yang, Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13:555-556. [DOI] [PubMed] [Google Scholar]