

Abstract
Determining which antigen must be included in AIDS vaccines to confer maximum protection is of utmost importance. In primate models, vaccines consisting of or including accessory viral proteins have yielded conflicting results. We investigated the protective potential of the accessory protein ORF-A of feline immunodeficiency virus (FIV) in cats. All three immunization strategies used (protein alone in alum adjuvant, DNA alone, or DNA prime-protein boost) clearly generated detectable immune responses. Upon challenge with ex vivo homologous FIV, ORF-A-immunized cats showed distinct enhancement of acute-phase infection relative to mock-immunized animals given alum or empty vector DNA. This effect was tentatively attributed to increased expression of the FIV receptor CD134 that was observed in the immunized cats. However, at subsequent sampling points that were continued for up to 10 months postchallenge, the average plasma viral loads of the ORF-A-immunized animals were slightly but consistently reduced relative to those of the control animals. In addition, CD4+ T lymphocytes in the circulation system declined more slowly in immunized animals than in control animals. These findings support the contention that immunization with lentiviral accessory proteins can improve the host's ability to control virus replication and slow down disease progression but also draw attention to the fact that even simple immunogens that eventually contribute to protective activity can transiently exacerbate subsequent lentiviral infections.
Failure to develop efficacious AIDS vaccines based on the major structural components Env and Gag of human immunodeficiency virus type 1 (HIV-1) has prompted investigation of a variety of unprecedented strategies, including the use of the small regulatory proteins of the virus as immunogens. Although several such proteins are targeted by immune responses during natural HIV-1 infection (51), the latter efforts have mainly focused on the accessory protein Tat, which has a number of attractive properties under this respect (reviewed in references 6 and 45). These efforts, however, have generated conflicting findings. Indeed, in Tat-vaccinated nonhuman primates, some groups have observed substantial levels of protection and proposed that native or inactivated Tat should be included in future multicomponent HIV-1 vaccines (5, 11, 18, 20, 21, 34, 35), while others have failed to demonstrate appreciable beneficial effects (1, 28, 32, 45).
Among the models being used in the struggle to conceive and validate rational approaches to AIDS immune prophylaxis (14), feline immunodeficiency virus (FIV) is of particular interest because it circulates widely among domestic cats, where it sustains infections and produces pathological effects similar to HIV-1 in humans, thus allowing for vaccine trials in the field as well as in the laboratory (4). FIV lacks a tat gene and its corresponding transactivation response element. It does, however, code for an accessory 77- or 78-amino-acid (aa) protein designated ORF-A or ORF-2, which was initially considered Tat-like because of its ability to transactivate viral transcription at low levels but was subsequently shown to share several properties with HIV-1 Vpr, including nuclear localization and induction of G2 cell cycle arrest (16). Importantly, deletion of the ORF-A gene results in the production of viruses impaired for replication in lymphocytes and with reduced virulence. These characteristics provide ORF-A-deleted constructs with appropriate prerequisites for vaccine candidates (15, 36, 38).
On the basis of these premises and the hypothesis that a tailored immune response against the ORF-A protein would impact replication of wild-type (wt) virus, we evaluated the potential of this protein as a protective immunogen by immunizing cats with recombinant protein in alum or with DNA alone or by a DNA prime-protein boost protocol. All of the immunized animals produced various levels of anti-ORF-A antibodies, and most also generated transiently measurable ORF-A-specific T-cell responses. Upon challenge with a moderate dose of ex vivo homologous FIV, the ORF-A-immunized animals showed an enhancement of acute-phase infection relative to controls who had received parallel courses of alum adjuvant alone or empty vector DNA. Despite this initial paradoxical effect, the ORF-A-immunized animals exhibited reduced average post-acute-phase plasma FIV loads and slower decline of circulating CD4+ T lymphocytes than the control animals did. Attempts to understand the reasons for the exacerbation of acute-phase infection observed following ORF-A immunization are also described.
MATERIALS AND METHODS
Cloning of ORF-A into eukaryotic and prokaryotic expression vectors.
Both the DNA and protein ORF-A immunogens were obtained from p34TF10 (nucleotides [nt] 5992 to 6228, referred to as the Petaluma strain of FIV [FIVPET], GenBank accession number NC_001482), after the stop codon at position 6120 had been replaced with Trp, the residue present at this position in wild-type FIV. ORF-A was amplified by PCR using primers bearing appropriate restriction sites and then inserted into vectors containing eukaryotic or prokaryotic expression promoters.
With regard to the DNA immunogen, two versions were produced. The first, designated CMV-ORF, was obtained by cloning ORF-A into the EcoRV/XhoI site of pcDNA3 (Invitrogen, Milan, Italy) and placing it under the control of the cytomegalovirus (CMV) promoter. The second, designated LTR-ORF, was generated from the previous one by replacing the CMV promoter with the FIV 5′ long terminal repeat (5′LTR) derived from p34TF10. In both, ORF-A translation was rendered Rev/Rev response element independent by introducing the Mason-Pfizer monkey virus constitutive transporting element downstream of ORF-A. Variants of these constructs were also produced which had the green fluorescent protein (GFP; 239 aa, 27 kDa) gene downstream of and in frame with ORF-A, so that the fused product ORF-A-GFP (318 aa, 36.5 kDa) could be used to monitor expression in transfected cells by flow cytometry.
The ORF-A protein was produced with the pQE system (QIAGEN, Milan, Italy). ORF-A was subcloned into a plasmid which encoded an Arg-Gly-Ser-(six)His tag (six-His tag) at the amino terminus. ORF-A cloning was carried out by PCR, using a sense primer having the BamHI restriction site and the ATG start (Met codon) mutagenized to ACG (Thr codon), and an antisense primer that contained the SalI restriction site downstream of the terminal stop codon. Once cleaved with BamHI-SalI, the amplicon was inserted into the vector downstream of the six-His tag. The use of the BamHI site (GGATCC) also resulted in the addition of Gly-Ser codons immediately in front of the six-His tag. The six-His tag and the BamHI restriction site rendered the protein produced (H-ORF-A) slightly larger (89 aa, 11.0 kDa) than the wt ORF-A. The pQE-H-ORF-A vector was then inserted into the Escherichia coli M15 strain made competent by the standard CaCl2 protocol.
H-ORF-A induction, purification, and analysis.
Transformed bacteria were grown at 37°C in Luria-Bertani broth (LB) containing 100 μg/ml ampicillin overnight with vigorous agitation. H-ORF-A expression was induced by adding 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) when the culture reached an optical density at 660 nm (OD660) of approximately 0.6 and extending the incubation for an additional 16 h. Optimal incubation time was determined by collecting at selected times aliquots that were stored at −80°C until tested.
H-ORF-A purification was carried out from IPTG-induced bacteria. Cell pellets from 100-ml cultures were resuspended in 10 ml lysis buffer containing 8 M urea and 50 mM NaH2PO4 (pH 8.0), sonicated with Sonifier B-12 (three impulses of 10 seconds each, 60% maximum output; Branson Benchtop Ultrasonic Cleaners, Danbury, CT), and freeze-thawed three times. Ruptured cells were then centrifuged at 10,000 × g for 30 min, and the cleared lysate was saved. H-ORF-A was then purified by Ni-nitrilotriacetic acid (Ni-NTA) affinity chromatography using a gradient elution method. An HR 16/5 column (Amersham Biosciences, Milan, Italy) was packed with 6 ml of ProBond resin (Invitrogen) and equilibrated with two washes of water and three washes of denaturing binding buffer (DBB) (pH 7.8) (8 M urea, 20 mM NaH2PO4, 500 mM NaCl). The column was then loaded with the cleared lysate and gently agitated in the horizontal position at room temperature (RT) for 1 h to maximize protein binding. The column was washed with ice-cold increasingly acidic DBB (pH 7.8, pH 6.0, and pH 5.4). Bound material was eluted with ice-cold DBB, pH 4.0, and 500-μl fractions were collected and dialyzed against 20 mM Tris-HCl, pH 7.4, at 4°C overnight.
The H-ORF-A-containing fractions were pooled, concentrated to 1 ml in a SpeedVac (Savant, Ramsey, MN), loaded on a preparative 14% polyacrylamide gel, and electroporated in a MiniPrep cell (Bio-Rad, Milan, Italy). Protein bands were checked for purity and integrity by Coomassie blue. Protein concentration was determined with Coomassie Plus (Pierce Biotechnology, Rockford, IL) using the manufacturer's protocol for standard (100 to 1,500 μg/ml) or micro (1 to 25 μg/ml) test tubes. Bovine serum albumin was used as a protein standard. Western blot analysis of H-ORF-A was carried out with the anti-His monoclonal antibody RGS-(His)4 (QIAGEN), followed by horseradish peroxidase (HRP)-conjugated rabbit anti-mouse immunoglobulin G (IgG) (anti-His; 1/1,000) serum (Sigma).
Cell transfections.
Expression of the ORF-A constructs was monitored in Crandell feline kidney fibroblast (CrFK), Chinese hamster ovary epithelial (CHO), and human epithelial 293T cell lines, transfected by a modified calcium phosphate method. The numbers of GFP-positive cells were determined with a FACScan flow cytometer and CELLQuest version 2 software (BD Biosciences, Milan, Italy) at selected times posttransfection.
Animals, immunizations, and challenge.
Twenty-three 6-month-old specific-pathogen-free domestic female cats (IFFA Credo, Lyon, France) were assigned to the five experimental groups in Table 1 at random. All were free from FIV and feline leukemia virus antibodies at the commencement of the study. The animals were housed individually in our climatized animal facility in accordance with European Community guidelines, had ad libitum access to fresh water and a proprietary brand of cat food, and were sedated with ketamine/diazepam intramuscularly prior to any procedure. Two weeks before initiation of the experiment and prior to each immunizing dose, the animals were bled for routine hematochemical analyses and circulating lymphocyte subset counts. The immunizing protocols used are detailed in Table 1. No local reactions or adverse clinical signs were observed during immunization. Animal challenge was carried out intravenously with 1 ml of pooled plasma samples obtained from cats infected with a strain of FIVPet and diluted to contain 10 50% cat infectious doses (CID50). The virus had previously been readapted to in vivo growth by several consecutive passages in cats and, as a result, had reacquired virulence characteristics typical of wt FIV (37).
TABLE 1.
Cat immunization protocols
| Group | No. of animals | Material used for immunization | Total dose of immunogen (mg) | Dose (mg) at wk:
|
||||
|---|---|---|---|---|---|---|---|---|
| 0 | 3 | 6 | 12 | 24 | ||||
| ORF-A-immunized groups | ||||||||
| PA | 5 | Protein immunogen in aluma | 60 | 20 | 10 | 10 | 10 | 10 |
| DA | 5 | DNA immunogenb | 1,050 | 300 | 150 | 150 | 150 | 300 |
| DP | 5 | DNA immunogen | 750 | 300 | 150 | 150 | 150 | |
| Protein immunogen in alumc | 15 | 15 | ||||||
| Mock-immunized groups | ||||||||
| NC | 5 | PBS in alumd | 0 | 0 | 0 | 0 | 0 | 0 |
| DC | 3 | Empty vector DNAe | 1,050 | 300 | 150 | 150 | 150 | 300 |
H-ORF-A was mixed 1:1 with alum (kindly provided by Fausto Titti, Istituto Superiore di Sanità, Rome, Italy), and the mixture was injected subcutaneously into two sites in the animal back (500 μl/site).
CMV-ORF was given at weeks 0, 6, and 24 and LTR-ORF was given at weeks 3 and 12. Each dose was administered in four injections into the tibial muscle of both hind legs (200 μl/site). DNA inoculations were preceded by the injection of 300 μl of 10 μM cardiotoxin at the same sites 4 days earlier in an attempt to maximize plasmid uptake.
CMV-ORF was given at weeks 0 and 6 and LTR-ORF was given at weeks 3 and 12 and administered as described in footnote b. H-ORF-A was given at week 24 and administered as described in footnote a.
PBS mixed with alum and administered as described in footnote a.
CMV plasmid was given at weeks 0, 6 and 24 and LTR plasmid was given at weeks 3 and 12 and administered described in footnote b.
ORF-A antibodies.
Enzyme-linked immunoassay (ELISA) antibodies were determined in microtiter plates (High Binding; Greiner, Lumezzane, Italy) coated overnight at 4°C with H-ORF-A diluted 1:40 (10-μg/ml final concentration) in coupling buffer (10 mM NaHCO3, 1 mM EGTA, pH 9.6). The wells were washed three times with Tris-buffered saline with Tween 20 (TBST) (100 mM NaCl, 50 mM Tris, 0.05% Tween 20, pH 7.6), and any unreacted sites were blocked with TBST supplemented with 2% skim milk at RT for 1 h. Following washing, 100 μl of cat plasma diluted 1:100 in TBST containing 20% normal goat serum was added to the wells and incubated at RT for 2 h. Bound antibodies were detected using a 1:8,000 dilution of biotinylated goat anti-cat IgG (Sigma), followed by 1:4,000 streptavidin-HRP (ExtrAvidin-peroxidase conjugate; Sigma), and developed with diaminobenzidine/peroxidase. The reaction was stopped with 50 μl of 4 M H2SO4 per well, and the OD405 of each well was measured using a multiscan spectrophotometer (BEP processor II; Dade Behring, Milan, Italy). Samples were scored positive whenever readouts were equal to or above the cutoff value (OD of 0.156), set as five times the average OD values obtained from a panel of 10 plasma samples from FIV-naive cats. To ascertain whether the reactivity detected was directed against ORF-A or the six-His tag present in H-ORF-A, an identical ELISA was run using six-His-tagged recombinant calf type 2 5′-nucleotidase (kindly provided by Maria G. Tozzi, University of Pisa) as the test antigen. Due to the lack of specific anti-ORF-A antibodies, the monoclonal antibody RGS-(His)4, developed with anti-mouse IgG, served as a positive control.
ORF-A-specific IFN-γ-secreting T cells.
Quantitation of ORF-A-specific gamma interferon (IFN-γ)-secreting T cells in Ficoll-Paque-purified peripheral blood mononuclear cells (PBMC) was achieved by an enzyme-linked immunospot (ELISPOT) assay using a pool of nine overlapping 15-mer peptides encompassing the entire sequence of the FIVPET ORF-A as the antigen (see Fig. 1). The peptides had been synthesized by Espikem (Florence, Italy) with 9-fluorenylmethoxy carbonyl (fmoc) chemistry and were >95% pure. Multi-Screen 96-well filtration plates (Millipore, Milan, Italy) were coated with 1 μg of the anti-feline IFN-γ monoclonal antibody D9 (19) in 100 μl/well phosphate-buffered saline (PBS) in a humidified atmosphere at 4°C overnight. The wells were then washed four times with sterile PBS, and any unreacted sites were blocked with 100 μl/well RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine, 5 × 10−5 M 2-mercaptoethanol, and 100 IU of penicillin and 100 μg of streptomycin per ml (complete RPMI medium) at 37°C for at least 1 h. The medium was then replaced with 100 μl of complete RPMI medium containing known numbers (20,000 to 50,000 cells/well) of PBMC. Finally, 100 μl of complete RPMI medium containing the peptide pool at a final concentration of either 1 μM or 5 μM or no peptides were added to these cells. The test was performed in duplicate or triplicate, and the plates were incubated overnight at 37°C in a humidified atmosphere with 5% CO2. The wells were then washed four times with PBS containing 0.05% Tween 20 and further incubated with an affinity-purified biotinylated polyclonal sheep anti-feline IFN-γ antibody at RT overnight. Unbound antibody was removed by four washes with PBS-Tween 20, and bound antibody was visualized by the addition of 0.4 μg/ml streptavidin-HRP (Vector Laboratories, Segrate, Italy) in PBS-Tween 20. After a final washing, the assay was developed by the addition of 100 μl/well of amino-9-ethylcarbazole chromogen (Vector Laboratories) according to the manufacturer's instructions, and the reaction was allowed to proceed at RT until spots were visible to the naked eye. The plates were then washed in distilled water and air dried, and the frequencies of ORF-A-specific IFN-γ-secreting cells were determined using a binocular dissecting microscope.
FIG. 1.

Schematic representation of the ORF-A protein with its recognized domains and hydrophobicity profile as calculated with Kyte and Doolittle's algorithm. The nine 15-mer peptides used in the ELISPOT and CTL assays are also shown: as depicted, they encompassed the entire molecule and overlapped by seven residues.
ORF-A-specific CTL.
ORF-A-specific cytotoxic T-cell (CTL) activity of the PBMC was measured using as the target autologous fibroblasts sensitized with the same ORF-A peptide pool used in the ELISPOT assay. Skin fibroblasts, obtained 1 to 2 months before initiation of the experiment, were labeled overnight with 51Cr, washed, and then exposed to the peptide pool (5 μM or 1 μM) at 37°C for 1 h. Similar cells incubated with peptide-free medium served as negative controls. Microcytotoxicity assays were then performed in triplicate as described previously (13) by adding appropriate numbers of PBMC to give effector/target ratios of approximately 50:1, 25:1, and 12.5:1. 51Cr release in the supernatants was measured after 4 h of incubation at 37°C in a humidified atmosphere with 5% CO2.
Plasma viral load.
Viral RNA extracted from plasma using the QIAamp viral RNA kit (QIAGEN), was reverse transcribed and amplified by reverse transcription TaqMan PCR (TM-PCR). Reverse transcription and amplification conditions, and evaluation of the sensitivity of the assay (200 copies/ml plasma) have been previously described (36).
Proviral load in the PBMC.
Genomic DNA was extracted from the PBMC using the QIAamp DNA Blood Mini kit (QIAGEN). Proviral DNA was quantified from 0.4 μg genomic DNA by TM-PCR under the same conditions used for cDNA amplification except that the reaction mixture volume was 25 μl. The sensitivity of the assay was 100 copies/μg genomic DNA (36).
Infectious units in the PBMC.
PBMC (106) were stimulated with concanavalin A, serially 10-fold diluted, and cocultured with 106 MBM cells in RPMI 1640 medium containing 10% fetal bovine serum and 10 U/ml human recombinant interleukin-2 (Roche Diagnostics, Monza, Italy). Culture supernatants were monitored biweekly for p25 and reverse transcriptase production by ELISA for up to 5 weeks as previously described (31).
FIV receptor and coreceptor expression in the PBMC.
Total RNA from 2 × 106 PBMC was extracted with the RNeasy Mini kit (QIAGEN), and the residual coextracted genomic DNA was eliminated with RNase-free DNase set (QIAGEN). The amount and purity of extracted RNA were evaluated by spectrophotometric reading at 260 nm and agarose gel electrophoresis. Two micrograms of total RNA was reverse transcribed into cDNA using 2.5 μM random examers (Roche Diagnostic, Milan, Italy) and 6 U avian myeloblastosis virus reverse transcriptase in a reaction mixture volume of 20 μl. Samples were incubated at 25°C for 10 min (primer annealing), 42°C for 60 min (transcription), and 95°C for 5 min (enzyme inactivation). Each cDNA was amplified for FIV receptor (CD134), coreceptor (CXCR4), and housekeeping glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primers and probes were as follows: fCD134S (5′-CCGTGAACTACGAGCCTTGC-3′; nt 227 to 246; as referred to GenBank accession number AB128982), CD134AS (5′-ATCTCTGCTTGGGCTCGCT-3′; nt 280 to 299), CD134Pr (5′-CCCTGCACACAGTGCAACCAGAGAAGT-3′; nt 250 to 276); CXCR4S (5′-TGCACCTATCTGTGGCAGACC-3′; nt 319 to 339; U63558), CXCR4AS (5′-ACCAGTTTGCCACAGCATCA-3′; nt 369 to 393), CXCR4Pr (5′-CCTCTTTGTCCTCACACTTCCCTTCTGGG-3′; nt 341 to 369); GAPDHS (5′-AAAGTGGACATTGTCGCCATC-3′; nt 73 to 93; AB038241), GAPDHAS (5′-GCCTTGACTGTGCCGTGG-3′; nt 162 to 179), and GAPDHPr (5′-TCAACTACATGGTCTACATGTTCCAGTATGATTCCA-3′; nt 113 to 148). Samples were amplified in triplicate using 5 μl cDNA, 300 to 900 nM primers, and 100 nM probe in 25-μl reaction mixtures. TM-PCR was performed with the ABI Prism 7700 sequence detection system (Applied Biosystems, Monza, Italy). Each run included no-template controls to test for contamination of assay reagents and the corresponding total RNAs to control for residual genomic DNA. The results were reported as threshold cycle.
Miscellanea.
ORF-A electrophoretic mobility, isoelectric point, hydrophobicity, relative solvent accessibility, and chemical-physical properties were calculated using the program packages available at ExPASy (www.expasy.org) and Pôle BioInformatique Lyonnais (http://pbil.univ-lyon1.fr/) software databases. The titers of anti-FIV antibodies were determined by ELISA against whole FIVPET antigen as described previously (31). Plasma found reactive at dilutions of 1:100 or greater was considered antibody positive. Circulating CD4+ and CD8+ T lymphocytes were counted with FE1.7B12 and FE1.10E9 monoclonal antibodies (obtained from Peter F. Moore, Davis, CA), respectively, and analyzed by flow cytometry. Statistical analyses included the Student's t test with two-tailed distribution and two-sample unequal-variance options.
RESULTS
Production and characterization of ORF-A immunogens.
The ORF-A derived from p-Δ00, a clone of FIVPET containing a fully functional gene (36), was cloned in plasmid expression vectors for eukaryotic (DNA immunogen) and prokaryotic cells (protein immunogen).
Previous observations had suggested that promoter choice may affect the outcome of DNA vaccination by influencing gene expression and, possibly, extent and rapidity of gene silencing (10, 12). Because it was plausible that DNA immunization might be rendered more efficacious by alternating the administration of constructs in which immunogen expression was driven by different promoters, ORF-A was cloned into pcDNA3.1 eukaryotic expression vectors that contained either the CMV promoter (construct CMV-ORF) or the FIV LTR (construct LTR-ORF). Both constructs also contained a constitutive transporting element downstream of ORF-A to make its expression independent of the Rev/Rev response element system. Due to the absence of antibodies that effectively recognize the protein, ORF-A expression was monitored using variants of the above constructs that produced an ORF-A-GFP fusion protein. These variants and control GFP constructs devoid of ORF-A were transfected into CrFK, CHO, and 293T cells, which were then monitored for internal fluorescence starting 48 h posttransfection. As shown in Table 2, the CMV promoter-containing constructs functioned well in all three cell types, with proportions of GFP-positive cells that uniformly exceeded 70% at day 2 posttransfection and then declined slowly but were still numerous at day 21, suggesting that the plasmids had become integrated in at least some cells. In contrast, consistent with the inherently low promoter activity of the FIV LTR in human cells (33), the LTR-containing constructs were expressed well in CrFK and CHO cells but very poorly in 293T cells.
TABLE 2.
ORF-A-GFP expression driven by a CMV promoter or FIV LTR in three cell lines
| Cell line | % GFP-positive cells driven by a CMV promoter or FIV LTR at the following time posttransfectiona:
|
|||||
|---|---|---|---|---|---|---|
| 2 days
|
10 days
|
21 days
|
||||
| CMV | LTR | CMV | LTR | CMV | LTR | |
| 293T | 85 | 8 | 66 | 2 | 42 | 0 |
| CrFK | 78 | 75 | 48 | 55 | 38 | 27 |
| CHO | 72 | 71 | 57 | 61 | 32 | 20 |
Percentage of GFP-positive cells (mean of three experiments). Only the results obtained with the constructs expressing GFP-ORF-A fused protein are shown. The constructs expressing GFP alone performed in a comparable manner.
The protein immunogen (H-ORF-A) was produced with the pQE system and purified through a six-His tag cloned at the amino-terminal end. This approach was adopted after repeated attempts to obtain the protein in native form or fused to thyredoxin. Although the latter forms of the ORF-A protein were abundantly produced under optimized culture conditions, proteins manifested a high tendency to associate with bacterial DNA and were toxic to the host bacteria. Moreover, consistent with the high hydrophobicity of the ORF-A protein (Fig. 1), it was soluble only in highly concentrated sodium dodecyl sulfate or urea and was extremely sticky when these were dialyzed out (data not shown). With the pQE system, optimum H-ORF-A production was obtained by growing a 2.5% dilution of overnight bacterial cultures in 100 ml LB under constant agitation at 37°C up to approximately 0.6 OD660. The optimal culture volume, IPTG concentration, and induction time (Fig. 2A) were 100 ml, 1 mM, and 4 h, respectively. Scaling up or using different IPTG concentrations or longer induction times resulted in greater expression of H-ORF-A but also led to substantially augmented bacterial and protein aggregation. In fact, even under conditions optimized for maximum soluble H-ORF-A yield, approximately 70% of the IPTG-induced protein was associated with the insoluble portion of bacterial extracts. H-ORF-A was therefore isolated from sonicated bacteria by Ni-NTA affinity chromatography under denaturing conditions (8 M urea), followed by dialysis and polyacrylamide gel analysis. The H-ORF-A-containing fractions (Fig. 2B) were then pooled and further purified on polyacrylamide gels. As judged by Coomassie blue staining (Fig. 2C), the resulting H-ORF-A was >95% pure, and overall yield was around 10% of total protein. Purified H-ORF-A also had electrophoretic mobility, isoelectric point, hydrophobicity, and solubility values expected on the basis of computational analysis of the ORF-A amino acid sequence. Because the purified protein aggregated very rapidly, and once this had occurred, it was no longer obtainable in soluble form, both vaccination and immunological tests were carried out using H-ORF-A in 8 M urea.
FIG. 2.

ORF-A protein production in E. coli. (A) Time course of IPTG induction evaluated in a polyacrylamide gel with Coomassie blue. (B) Fractions eluted from a Ni-NTA resin column. A sample of each fraction was run in a polyacrylamide gel and analyzed by Coomassie blue staining (top gel) or Western blot with an anti-His monoclonal antibody (bottom gel). The positions of molecular mass markers (in kilodaltons) are indicated to the right of the gels in panels A and B. (C) Fractions eluted from the HR 16/5 column packed with ProBond resin and analyzed in a polyacrylamide gel with Coomassie blue staining. This was the preparation used for cat immunization and immunological tests.
ORF-A-specific immune responses in the immunized cats.
The ORF-A immunogens were used to immunize cats with the protocols in Table 1. Briefly, group PA received five immunizations of protein in alum, group DA received five shots of DNA, and group DP received four shots of DNA followed by a final boost of protein. Control groups NC and DC received alum alone and empty vector DNA alone, respectively. No adverse effects of the immunizations were noted.
Antibodies were monitored by ELISA using H-ORF-A as the antigen at the time of each inoculation and 8 weeks after the last one, when the animals were FIV challenged. As shown by Fig. 3A, the mock-immunized animals remained negative throughout the observation period. Conversely, all the immunized animals developed various levels of antibodies. However, the kinetics and magnitude of response differed considerably depending on immunization protocol. In the PA group, antibodies started to become evident at 3 weeks and readily reached plateau levels that remained relatively unchanged up to the time of challenge. In the DA animals, antibodies started to develop somewhat later and remained low and, on occasion, were undetectable. The DP group showed a pattern similar to that of the DA group, except in two animals that had levels of antibody in the same range as the PA group at challenge. Importantly, the animals tested consistently negative against an unrelated six-His protein, demonstrating that their antibodies were directed against the ORF-A portion of H-ORF-A and not against the six-His tag.
FIG. 3.

ORF-A-specific immune responses in the immunized and mock-immunized cats. Each symbol shows the value from one cat. (A) Antibody response. Plasma samples obtained at the times indicated and diluted 1:100 were examined for ELISA reactivity against H-ORF-A. The dotted line represents the cutoff value calculated by multiplying by 5 the average absorbance (OD405) of plasma samples from 10 FIV-naive animals. (B) IFN-γ-secreting T-cell response. At the times indicated, PBMC were stimulated with 5 μM (left symbols) or 1 μM (right symbols) pooled ORF-A oligopeptides (Fig. 1) and directly examined by ELISPOT analysis. (C) ORF-A-specific CTL activity in the PBMC as determined against autologous skin fibroblasts charged with 5 μM of the same pool of ORF-A oligopeptides used in the ELISPOT assay. Shaded areas are means ± standard deviations of background spot-forming cells (B) or background CTL activity detected in the absence of the ORF-A peptides in the assays (C).
Cell-mediated immune responses were evaluated by determining ORF-A-specific IFN-γ-secreting T cells and CTL activity in the PBMC at week 3, 12, and 32. As determined by ELISPOT tests using a pool of nine partially overlapping 15-mer peptides encompassing the entire ORF-A (Fig. 1) as the antigen, only some immunized animals showed ORF-A-specific IFN-γ-secreting T-cell numbers that were clearly increased relative to background and baseline levels (i.e., the numbers of spots observed in the absence of antigen and in the mock-immunized animals, respectively). This increase was most pronounced in the PA and DP cats, but even in these it was seen only at the first two points sampled: thus, by the time of challenge, IFN-γ-secreting T-cell numbers did not differ appreciably from baseline values (Fig. 3B). As determined with a 51Cr release assay using autologous skin fibroblasts loaded with the same oligopeptide pool used in the ELISPOT assay, ORF-A-specific CTL activity followed an essentially similar pattern. However, at least in the DP animals, some CTL activity was still measurable at the time of challenge (Fig. 3C).
Outcome of FIV challenge.
All animals were challenged intravenously with 10 CID50 of ex vivo FIVPET 8 weeks after the last immunizing dose and then monitored for infection for 10 months.
Consistent with previous findings (37), all eight mock-immunized cats became readily infected, with no appreciable differences between group NC and DC cats. Plasma viremia was already evident in these animals at the first sampling 2 weeks postchallenge (PC) and peaked 1 or 2 months PC with viral RNA copy numbers that varied in individual cats between 106,650 and 1,009,672 per ml. Subsequently, plasma viral loads declined slowly in all animals to reach values of around 60,000 copies per ml 8 and 10 months PC (Fig. 4A). Proviral loads in the PBMC showed similar kinetics, in that they peaked (5,841 to 13,147 copies per μg of genomic DNA) 2 months PC and subsequently declined to values of approximately 2,000 copies per μg of genomic DNA at the end of the observation period (Fig. 4B). Quantitative FIV reisolation from the PBMC was performed at months 1 and 4 PC, with results that confirmed the infected status of all control cats (Table 3); in particular, at 4 months PC, the numbers of infectious units per 106 PBMC ranged between 10 and 104. Anti-FIV antibodies started to become detectable 1 month PC, then steadily increased up to month 4, and eventually stabilized (data not shown). Finally, in these cats, circulating CD4+ T cells declined steadily so that at the end of the experiment their percentages had approximately decreased by 50% (Fig. 4C). In contrast, circulating CD8+ T cells did not show meaningful variations (Fig. 4D).
FIG. 4.

Outcome of FIV challenge in the immunized and mock-immunized cats. Each symbol shows the value from one cat. (A) Plasma viremia determined by reverse transcription TM-PCR. (B) Proviral loads in the PBMC determined by TM-PCR. (C and D) CD4+ and CD8+ T lymphocytes in the PBMC. One and two asterisks indicate significant differences relative to the values for the pooled control groups NC and ND at P ≤ 0.05 and P ≤ 0.02, respectively.
TABLE 3.
FIV infectious units in the PBMC of the ORF-A- and mock-immunized cats PC
| Group | Cat IDa | No. of infectious units/106 PBMC at:
|
|
|---|---|---|---|
| 1 mo PC | 4 mo PC | ||
| Mock-immunized groups | |||
| NC | □ | 10 | 102 |
| ○ | 10 | 103 | |
| ▪ | <1 | 10 | |
| ⋆ | 10 | 103 | |
| • | 10 | 102 | |
| DC | ○ | 10 | 104 |
| • | 1 | 10 | |
| □ | 10 | 102 | |
| ORF-A-immunized groups | |||
| PA | □ | 10 | 102 |
| ○ | 10 | 105 | |
| ▪ | 10 | 103 | |
| ⋆ | 10 | 102 | |
| • | 1 | 103 | |
| DA | □ | 10 | 103 |
| ○ | 10 | 103 | |
| ▪ | 10 | 103 | |
| ⋆ | 10 | 104 | |
| • | 10 | 102 | |
| DP | □ | 10 | 102 |
| ○ | 10 | 103 | |
| ▪ | 10 | 10 | |
| ⋆ | 10 | 103 | |
| ○ | 10 | 104 | |
As determined by all the parameters considered, the ORF-A-immunized animals also became uniformly infected. At the first sampling, 2 weeks PC, these animals exhibited markedly higher viral loads in plasma (Fig. 4A) and proviral loads in the PBMC (Fig. 4B) than the mock-immunized animals, regardless of the immunogen received. In the samplings performed 1 and 2 months PC when infection was at its peak, this difference had completely disappeared. In addition, from month 4 PC onwards, the immunized animals often showed lower plasma viral loads than the mock-immunized animals did (Fig. 4A). This correlated with a rather slow depletion of peripheral CD4+ T cells, which were significantly higher than in the mock-immunized animals by the end of follow-up (P ≤ 0.02; Fig. 4C). In contrast, proviral (Fig. 4B) and infectious unit loads in the PBMC (Table 3) as well as the numbers of circulating CD8+ T cells (Fig. 4D) and kinetics of anti-FIV antibody response (data not shown) were similar to those of the controls.
Effects of FIV challenge on the immune responses to ORF-A.
The study animals were also examined for possible changes in ORF-A-specific immune responses following FIV challenge. In the mock-immunized animals, ORF-A antibodies remained negative or borderline throughout the follow-up except in two cats that were clearly positive, albeit at very low titers (one cat each in groups NC and DC; Fig. 5A). ORF-A-specific cell-mediated immune responses measured 1, 4, and 8 months PC were also negative or borderline (Fig. 5B and C).
FIG. 5.

Postchallenge ORF-A-specific immune responses in the study cats. ORF-A-specific antibodies (A), IFN-γ-secreting cell numbers (B), and CTL activity (C) determined as described in the legend to Fig. 3.
In the immunized animals that already had elevated ORF-A antibodies at challenge (all the cats in PA group and two cats in DP group), average levels of these antibodies first declined somewhat and then went back to prechallenge values from month 4 PC. In the others, ORF-A antibody increased with time PC to reach levels that, at the end of the experiment, were not much lower than in the animals who had responded to ORF-A immunization (Fig. 5A). Immunized cats also underwent generally moderate increase in the number of ORF-A-specific IFN-γ-producing T-cells and in CTL activity relative to prechallenge values, increases which usually were mostly evident 1 month PC (Fig. 5B and C).
No evidence of soluble FIV-enhancing factors in the sera of ORF-A-immunized cats.
To verify whether the enhancement of acute-phase FIV infection seen in the ORF-A-immunized cats might be due to soluble factors, sera collected at challenge from three PA and three DA animals who had exhibited the enhancement were compared to sera from three mock-immunized animals (one DC animal and two NC animals) for the ability to modify FIVPET replication in vitro. The assays used were a standard seroneutralization assay with p25 antigen as a readout, which can reveal the presence of virus-enhancing as well as virus-inhibitory antibody activities (8), and a single-round-of-replication assay using a defective FIV having a GFP reporter gene which becomes expressed after the virus becomes integrated into cell DNA (F. Bonci, E. Zabogli, and M. Pistello, unpublished data). However, neither assays revealed significant traces of FIV-enhancing activity in the study cats (data not shown).
FIV receptor and coreceptor expression in ORF-A-immunized cats.
We also examined whether the enhancement of FIV infection observed in the ORF-A-immunized cats might have reflected an augmented expression of the FIV receptor (CD134) or coreceptor (CXCR4). This was done by testing for CD134 and CXCR4 RNA content in frozen PBMC collected before immunization, 3 weeks after immunization had started, and at the time of challenge (32 weeks after initiation of immunization). As shown by Fig. 6A, before immunization, the levels of expression of the two molecules were comparable and markedly lower than that of the GAPDH reference in all the study groups. As a result of immunization, CD134 expression had increased markedly in the ORF-A-immunized animals and much less so in the mock-immunized animals. The effect was most pronounced at week 3 of immunization, when CD134 expression in the ORF-A-immunized animals reached levels similar to those of GAPDH but was still evident at the time of challenge. In contrast, CXCR4 expression increased only moderately or did not change at all (Fig. 6B).
FIG. 6.

FIV receptor (CD134) and coreceptor (CXCR4) gene expression in the PBMC at different times relative to initiation of immunization. Each symbol shows the value for one cat. (A) CD134 (black boxes), CXCR4 (white boxes), and GAPDH (shaded areas) mRNA content in PBMC and determined by TM-PCR and reported as threshold cycle (CT). (B) Normalized values of individual cats for CD134 and CXCR4, respectively. Data represent changes obtained by subtracting the CT of GAPDH from the CT of CD134 or CXCR4 obtained with PBMC collected at the times indicated and dividing by the corresponding value obtained with PBMC collected before immunization was started (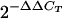 method).
method).
DISCUSSION
The vaccine approaches explored with the FIV model are many (reviewed in references 3, 17, 22, 29, 31, 37, 38, 39, 42, 44, and 48). Yet, no report has dealt with the possible utility of the accessory virus-encoded proteins as protective immunogens in this system. The issue is of interest because vaccines consisting of or including accessory proteins have yielded conflicting results in primate models and because none of the FIV vaccines tested to date can be considered satisfactory, leaving ample room for improvement.
FIV possesses fewer and less well characterized accessory proteins than HIV-1. In particular, the accessory protein ORF-A, investigated as an immunogen in this report, is a small highly basic hydrophobic protein in which a few functional domains can be recognized. The central leucine-rich and C-terminal cysteine-rich domains are essential for viral replication in feline PBMC and T-cell lines (15), and the basic domain is important for cell localization (16). Initially considered the equivalent of the HIV-1 Tat, ORF-A has since been shown to share functions with other accessory proteins of HIV-1 as well (16); thus, no HIV-1 homologue to this protein can be currently identified. It is, however, well documented that an intact ORF-A is essential for FIV replication in CD4+ T cells, a major target of in vivo infection, and an important determinant of FIV virulence, as shown by the markedly attenuated phenotype of viral mutants with the ORF-A gene deleted (15, 36, 38). Thus, we speculated that an immune response elicited against this protein might contain viral replication in CD4+ T cells, hence sparing these cells from depletion and reducing the overall impact of infection on immune functions. The fact that this unique protein had never been tested as an immunogen before also prompted us to pursue this approach.
Primarily, the present study investigated whether it is possible to generate immune responses against ORF-A in cats. This information was necessary because, at least as determined by our ELISA, FIV-infected cats lack detectable ORF-A antibodies (unpublished results). All three rather intensive immunization strategies that were used (protein in alum adjuvant, DNA alone, and DNA prime-protein boost) induced the production of binding antibodies against ORF-A, although, as expected, the titers achieved were most pronounced with the use of ORF-A protein alone, followed by the DNA prime-protein boost, and were very feeble in the animals given DNA alone. On the other hand, the cell-mediated responses generated were less consistent, in that they were evident only in a proportion of the animals regardless of the immunization strategy. Furthermore, consistent with findings in cats immunized with other antigens (12, 22), the numbers of IFN-γ-secreting T cells elicited and CTL activity peaked early after the start of vaccination, proved insensitive to boosting, and had almost completely waned by the time of challenge. Collectively, these results indicated that, albeit poorly immunogenic in the course of FIV infection, ORF-A can generate significant immune responses upon artificial immunization of cats.
We then investigated whether prior ORF-A immunization can impact FIV infection. The ORF-A-immunized cats, along with the controls given alum adjuvant or empty vector DNA, were challenged intravenously with an FIV homologous to the tissue culture-adapted virus from which the immunogens were prepared (FIVPET) but that had been adapted back to grow in vivo. The choice of this challenge was guided by the fear that ORF-A protein polymorphism—which may be as high as 26% intrasubtype and 40% intersubtype on the basis of published sequences—might obscure possible protective effects and by findings that vaccine protection against tissue culture-adapted FIV is not predictive of efficacy against wt virus (9). Although the dose of virus used for challenge (10 CID50/cat) was the minimum known to consistently infect adult cats, there was no evidence that the ORF-A-immunized animals resisted challenge more effectively than the controls. This was expected because immunity to an accessory protein is not likely to bring about a reduction of initial infection (51). On the contrary, immunized animals showed clearly evident enhancement of acute-phase infection. The effect was generalized and rather robust, since 14 of the 15 immunized cats had levels of viral RNA in plasma and proviral DNA in the PBMC which exceeded by approximately 1 log unit those in the controls at 2 weeks PC; however, it was also transient, since viral and proviral loads of immunized and control cats were in the same range by 1 and 2 months PC. At subsequent samplings, which were continued for up to 10 months PC, the average plasma viral loads of all three groups of immunized animals were slightly but significantly reduced relative to those of the controls. This was associated with a reduced impact on circulating CD4+ T lymphocytes, which by the end of follow-up were spared in immunized versus control animals. These findings indicate that, no matter how ORF-A immunization was carried out, it had exerted a limited beneficial effect on the post-acute phase of FIV infection, although infection was enhanced at the beginning.
Of note, while the challenge did not elicit significant levels of anti-ORF-A responses in the mock-immunized animals, ORF-A-immunized cats responded with a modulation of the preexisting levels of immunity to the protein. The modulation consisted in a modest increase in cell-mediated immunity, which was observed at 1 month PC only and readily declined, and then, starting from 4 months PC, by a rebound of ORF-A antibodies to titers as high or higher than those present at challenge. Thus, there was a clear suggestion that ORF-A produced in the course of FIV replication had, on the one hand, absorbed the antibodies present, but, on the other, had behaved as a recall stimulus. If and how this modulation was related to the simultaneous modulation of challenge infection observed in the immunized cats relative to the controls cannot be determined from the present study. It should be noted, however, that the cats who manifested better control of FIV replication exhibited no especially marked modulation of the anti-ORF-A responses. In a recent study, macaques given a simian immunodeficiency virus (SIV) DNA immunogen and then inoculated with an attenuated SIV also showed improved control of the infection despite an initial exacerbation of viral replication, but the mechanisms involved remained undetermined (2).
Vaccine-induced exacerbations of lentiviral infections, similar to the one observed early PC in the present study, have been described in several situations, including cats immunized with FIV Env and Gag protein or DNA and fixed infected cells (17, 23-26, 30, 41, 42, 44), macaques vaccinated with virus-vectored SIV or SIV DNA (2, 46), and ponies immunized with equine infectious anemia virus Env (49). The mechanisms invoked include the production of virus-enhancing antibodies (44), nonspecific stimulation of permissive cell proliferation (46), and a higher than normal permissiveness of FIV-specific T cells (41) but have essentially remained undefined. Importantly, both the initial enhancement and the later control of infection observed in the present study were apparently immunogen related, since both effects were relative to control cats who had received similar courses of inoculations, except for the absence of the ORF-A immunogens.
Similar to what found in other systems (40, 42), we found no evidence of soluble factors that facilitated FIV infection in vitro in the sera of ORF-A-immunized cats. We therefore exploited the recent identification of CD134 as a FIV receptor (43) to examine whether enhancement might result from an up-regulation of this molecule, as postulated by Shimojima et al. (43). Consistent with its nature as a T-cell activation molecule (7, 27), CD134 was found to be expressed at a much higher level in the ORF-A-immunized cats relative to what was seen preimmunization or in mock-immunized animals. The effect reached statistical significance only early after initiation of immunization but was still present at the time of FIV challenge. Moreover, it is plausible that the effect was more pronounced and longer-lasting in lymphoid tissues, such as the periarteriolar sheath, where up-regulation of CD134 following immune stimulation is most evident (47), than in the PBMC used to monitor CD134 expression in the present study. Finally, it is also possible that ORF-A immunization primed for an up-regulation of CD134 postchallenge, an aspect that we did not investigate in the present study. Thus, it seems likely that increased expression of FIV receptor by susceptible cells or an expansion of cells expressing the FIV receptor may have contributed to the observed enhancement. In contrast, expression of CXCR4, the major, if not the only, coreceptor of FIV (50), which was investigated in parallel with CD134, underwent little, if any, change as a result of immunization, ruling out an important role of this molecule in the phenomenon. Analysis of CD134 expression pre- and postchallenge following immunization with additional candidate vaccines might help to elucidate the mechanisms underlying the enhancement of FIV replication that has been repeatedly observed in vaccinated cats and, possibly, provide an additional parameter for determining whether an FIV immunogen is indeed suitable for vaccine use.
Determining which antigen must be included in lentiviral vaccines to confer maximum protection is of utmost importance. The present findings support the contention that immunization with lentiviral accessory proteins can improve the host's ability to control virus replication and slow down disease progression. They, however, also draw attention to the fact that even simple immunogens that eventually contribute to protective activity, as appears to be the case for the FIV ORF-A, can transiently exacerbate subsequent lentiviral infections. The possibility that candidate vaccines result in such a detrimental effect was recognized early in the FIV model but has also recently been verified with SIV (2, 46). Clearly, this is a consequence of lentiviral vaccination that should be further investigated, for example, with regard to the duration postimmunization and possible impact in vaccination trials, and should represent an important concern in the design and testing of candidate AIDS vaccines.
Acknowledgments
This work was supported by the Ministero della Salute Istituto Superiore di Sanità, “IV° Programma Nazionale di Ricerca sull'AIDS” and “Progetto Nazionale AIDS-ICAV,” and the Ministero dell'Istruzione, dell'Università e della Ricerca, FIRB 2001.
REFERENCES
- 1.Allen, T. M., L. Mortara, B. R. Mothé, M. Liebl, P. Jing, B. Calore, M. Piekarczyk, R. Ruddersdorf, D. H. O'Connor, X. Wang, C. Wang, D. B. Allison, J. D. Altman, A. Sette, R. C. Desrosiers, G. Sutter, and D. I. Watkins. 2002. Tat-vaccinated macaques do not control simian immunodeficiency virus SIVmac239 replication. J. Virol. 76:4108-4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amara, R. R., K. Patel, G. Niedziela, P. Nigam, S. Sharma, S. I. Staprans, D. C. Montefiori, L. Chenareddi, J. G. Herndon, H. L. Robinson, H. M. McClure, and F. J. Novembre. 2005. A combination DNA and attenuated simian immunodeficiency virus vaccine strategy provides enhanced protection from simian/human immunodeficiency virus-induced disease. J. Virol. 79:15356-15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broche-Pierre, S., J. Richardson, A. Moraillon, and P. Sonigo. 2005. Evaluation of live feline immunodeficiency virus vaccines with modified antigenic properties. J. Gen. Virol. 86:2495-2506. [DOI] [PubMed] [Google Scholar]
- 4.Burkhard, M. J., and G. A. Dean. 2003. Transmission and immunopathogenesis of FIV in cats as a model for HIV. Curr. HIV Res. 1:15-29. [DOI] [PubMed] [Google Scholar]
- 5.Cafaro, A., A. Caputo, C. Fracasso, M. T. Maggiorella, D. Goletti, S. Baroncelli, M. Pace, L. Sernicola, M. L. Koanga-Mogtomo, M. Betti, A. Borsetti, R. Belli, L. Akerblom, F. Corrias, S. Butto, J. Heeney, P. Verani, F. Titti, and B. Ensoli. 1999. Control of SHIV-89.6P infection of cynomolgus monkeys by HIV-1 Tat protein vaccine. Nat. Med. 5:643-650. [DOI] [PubMed] [Google Scholar]
- 6.Caputo, A., R. Gavioli, and B. Ensoli. 2004. Recent advances in the development of HIV-1 Tat-based vaccines. Curr. HIV Res. 2:357-376. [DOI] [PubMed] [Google Scholar]
- 7.Croft, M. 2003. Costimulation of T cells by OX40, 4-1BB, and CD27. Cytokine Growth Factor Rev. 14:265-273. [DOI] [PubMed] [Google Scholar]
- 8.Del Mauro, D., D. Matteucci, S. Giannecchini, F. Maggi, M. Pistello, and M. Bendinelli. 1998. Autologous and heterologous neutralization analyses of primary feline immunodeficiency virus isolates. J. Virol. 72:2199-2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunham, S., and O. Jarrett. 2005. FIV as a model for AIDS vaccine studies, p. 293-332. In H. Friedman, S. Specter, and M. Bendinelli (ed.), In vivo models of HIV disease and control. Springer, New York, N.Y.
- 10.Ellis, J., and S. Yao. 2005. Retrovirus silencing and vector design: relevance to normal and cancer stem cells? Curr. Gene Ther. 5:367-373. [DOI] [PubMed] [Google Scholar]
- 11.Ensoli, B., A. Cafaro, A. Caputo, V. Fiorelli, F. Ensoli, R. Gavioli, F. Ferrantelli, A. Cara, F. Titti, and M. Magnani. 2005. Vaccines based on the native HIV Tat protein and on the combination of Tat and the structural HIV protein variant DeltaV2 Env. Microbes Infect. 7:1392-1399. [DOI] [PubMed] [Google Scholar]
- 12.Flynn, J. N., M. J. Hosie, M. A. Rigby, N. Mackay, C. A. Cannon, T. Dunsford, J. C. Neil, and O. Jarrett. 2000. Factors influencing cellular immune responses to feline immunodeficiency virus induced by DNA vaccination. Vaccine 18:1118-1132. [DOI] [PubMed] [Google Scholar]
- 13.Flynn, J. N., M. Pistello, P. Isola, L. Zaccaro, B. Del Santo, E. Ricci, D. Matteucci, and M. Bendinelli. 2005. Adoptive immunotherapy of feline immunodeficiency virus with autologous ex vivo stimulated lymphoid cells modulates virus and T cell subsets in blood. Clin. Diagn. Lab. Immunol. 12:736-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman, H., S. Specter, and M. Bendinelli. 2005. In vivo models of HIV disease and control. Springer, New York, N.Y.
- 15.Gemeniano, M. C., E. T. Sawai, C. M. Leutenegger, and E. E. Sparger. 2003. Feline immunodeficiency virus ORF-A is required for virus particle formation and virus infectivity. J. Virol. 77:8819-8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gemeniano, M. C., E. T. Sawai, and E. E. Sparger. 2004. Feline immunodeficiency virus ORF-A localizes to the nucleus and induces cell cycle arrest. Virology 325:167-174. [DOI] [PubMed] [Google Scholar]
- 17.Giannecchini, S., P. Isola, O. Sichi, D. Matteucci, M. Pistello, L. Zaccaro, D. Del Mauro, and M. Bendinelli. 2002. AIDS vaccination studies using an ex vivo feline immunodeficiency virus model: failure to protect and possible enhancement of challenge infection by four cell-based vaccines prepared in autologous lymphoblasts. J. Virol. 76:6882-6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldstein, G., K. Manson, G. Tribbick, and R. Smith. 2000. Minimization of chronic plasma viremia in rhesus macaques immunized with synthetic HIV-1 Tat peptides and infected with a chimeric simian/human immunodeficiency virus (SHIV33). Vaccine 18:2789-2795. [DOI] [PubMed] [Google Scholar]
- 19.Graham, E. M., O. Jarrett, and J. N. Flynn. 2003. Development of novel reagents to measure feline interferon-γ. J. Immunol. Methods 279:69-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gringeri, A., E. Santagostino, M. Muca-Perja, H. Le Buanec, B. Bizzini, A. Lachgar, J. F. Zagury, J. Rappaport, A. Burny, R. C. Gallo, and D. Zagury. 1999. Tat toxoid as a component of a preventive vaccine in seronegative subjects. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 20:371-375. [DOI] [PubMed] [Google Scholar]
- 21.Hel, Z., W. P. Tsai, E. Tryniszewska, J. Nacsa, P. D. Markham, M. G. Lewis, G. N. Pavlakis, B. K. Felber, J. Tartaglia, and G. Franchini. 2006. Improved vaccine protection from simian AIDS by the addition of nonstructural simian immunodeficiency virus genes. J. Immunol. 176:85-96. [DOI] [PubMed] [Google Scholar]
- 22.Hosie, M. J., T. Dunsford, D. Klein, B. J. Willett, C. Cannon, R. Osborne, J. Macdonald, N. Spibey, N. Mackay, O. Jarrett, and J. C. Neil. 2000. Vaccination with inactivated virus but not viral DNA reduces virus load following challenge with a heterologous and virulent isolate of feline immunodeficiency virus. J. Virol. 74:9403-9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hosie, M. J., R. Osborne, G. Reid, J. C. Neil, and O. Jarrett. 1992. Enhancement after feline immunodeficiency virus vaccination. Vet. Immunol. Immunopathol. 35:191-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huisman, W., J. A. Karlas, K. H. J. Siebelink, R. C. Huisman, A. de Ronde, M. J. Francis, G. F. Rimmelzwaan, and A. D. M. E. Osterhaus. 1998. Feline immunodeficiency virus subunit vaccines that induce virus neutralising antibodies but no protection against challenge infection. Vaccine 16:181-187. [DOI] [PubMed] [Google Scholar]
- 25.Karlas, J. A., K. H. J. Siebelink, M. A. van Peer, W. Huisman, A. M. Cuisinier, G. F. Rimmelzwaan, and A. D. M. E. Osterhaus. 1999. Vaccination with experimental feline immunodeficiency virus vaccines, based on autologous infected cells, elicits enhancement of homologous challenge infection. J. Gen. Virol. 80:761-765. [DOI] [PubMed] [Google Scholar]
- 26.Karlas, J. A., K. H. J. Siebelink, M. A. van Peer, W. Huisman, G. F. Rimmelzwaan, and A. D. M. E. Osterhaus. 1998. Accelerated viraemia in cats vaccinated with fixed autologous FIV-infected cells. Vet. Immunol. Immunopathol. 65:353-365. [DOI] [PubMed] [Google Scholar]
- 27.Lane, P. 2000. Role of OX40 signals in coordinating CD4 T cell selection, migration, and cytokine differentiation in T helper (Th)1 and Th2 cells. J. Exp. Med. 191:201-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang, X., D. R. Casimiro, W. A. Schleif, F. Wang, M. E. Davies, Z. Q. Zhang, T. M. Fu, A. C. Finnefrock, L. Handt, M. P. Citron, G. Heidecker, A. Tang, M. Chen, K. A. Wilson, L. Gabryelski, M. McElhaugh, A. Carella, C. Moyer, L. Huang, S. Vitelli, D. Patel, J. Lin, E. A. Emini, and J. W. Shiver. 2005. Vectored Gag and Env but not Tat show efficacy against simian-human immunodeficiency virus 89.6P challenge in Mamu-A*01-negative rhesus monkeys. J. Virol. 79:12321-12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lockridge, K. M., M. Chien, G. A. Dean, C. K. Stefano, R. C. Montelaro, P. A. Luciw, and E. E. Sparger. 2000. Protective immunity against feline immunodeficiency virus induced by inoculation with vif-deleted proviral DNA. Virology 273:67-79. [DOI] [PubMed] [Google Scholar]
- 30.Lombardi, S., C. Garzelli, M. Pistello, C. Massi, D. Matteucci, F. Baldinotti, G. Cammarota, L. Da Prato, P. Bandecchi, F. Tozzini, and M. Bendinelli. 1994. A neutralizing antibody-inducing peptide of the V3 domain of feline immunodeficiency virus envelope glycoprotein does not induce protective immunity. J. Virol. 68:8374-8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matteucci, D., M. Pistello, P. Mazzetti, S. Giannecchini, P. Isola, A. Merico, A. Rizzuto, and M. Bendinelli. 2000. AIDS vaccination studies using feline immunodeficiency virus as a model: immunisation with inactivated whole virus suppresses viral replication following intravaginal challenge with infected cats. Vaccine 18:119-130. [DOI] [PubMed] [Google Scholar]
- 32.Mooij, P., I. G. Nieuwenhuis, C. J. Knoop, R. W. Doms, W. M. Bogers, P. J. Ten Haaft, H. Niphuis, W. Koornstra, K. Bieler, J. Kostler, B. Morein, A. Cafaro, B. Ensoli, R. Wagner, and J. L. Heeney. 2004. Qualitative T-helper responses to multiple viral antigens correlate with vaccine-induced immunity to simian/human immunodeficiency virus infection. J. Virol. 78:3333-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mustafa, F., P. Jayanth, P. S. Phillip, A. Ghazawi, R. D. Schmidt, K. A. Lew, and T. A. Rizvi. 2005. Relative activity of the feline immunodeficiency virus promoter in feline and primate cell lines. Microbes Infect. 7:233-2399. [DOI] [PubMed] [Google Scholar]
- 34.Osterhaus, A. D., C. A. van Baalen, R. A. Gruters, M. Schutten, C. H. Siebelink, E. G. Hulskotte, E. J. Tijhaar, R. E. Randall, G. van Amerongen, A. Fleuchaus, V. Erfle, and G. Sutter. 1999. Vaccination with Rev and Tat against AIDS. Vaccine 17:2713-2714. [DOI] [PubMed] [Google Scholar]
- 35.Pauza, C. D., P. Trivedi, M. Wallace, T. J. Ruckwardt, H. Le Buanec, W. Lu, B. Bizzini, A. Burny, D. Zagury, and R. C. Gallo. 2000. Vaccination with tat toxoid attenuates disease in simian/HIV-challenged macaques. Proc. Natl. Acad. Sci. USA 97:3515-3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pistello, M., M. Moscardini, P. Mazzetti, S. Macchi, F. Bonci, P. Isola, G. Freer, D. Matteucci, S. Specter, and M. Bendinelli. 2002. Development of feline immunodeficiency virus ORF-A (tat) mutants: in vitro and in vivo characterization. Virology 298:84-95. [DOI] [PubMed] [Google Scholar]
- 37.Pistello, M., D. Matteucci, F. Bonci, P. Isola, P. Mazzetti, L. Zaccaro, A. Merico, D. Del Mauro, N. Flynn, and M. Bendinelli. 2003. AIDS vaccination studies using an ex vivo feline immunodeficiency virus model: protection from an intraclade challenge administered systemically or mucosally by an attenuated vaccine. J. Virol. 77:10740-10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pistello, M., F. Bonci, P. Isola, P. Mazzetti, A. Merico, L. Zaccaro, D. Matteucci, and M. Bendinelli. 2005. Evaluation of feline immunodeficiency virus ORF-A mutants as candidate attenuated vaccine. Virology 332:676-690. [DOI] [PubMed] [Google Scholar]
- 39.Pu, R., J. Coleman, J. Coisman, E. Sato, T. Tanabe, M. Arai, and J. K. Yamamoto. 2005. Dual-subtype FIV vaccine (Fel-O-Vax FIV) protection against a heterologous subtype B FIV isolate. J. Feline Med. Surg. 7:65-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raabe, M. L., C. J. Issel, and R. C. Montelaro. 1999. In vitro antibody-dependent enhancement assays are insensitive indicators of in vivo vaccine enhancement of equine infectious anemia virus. Virology 259:416-427. [DOI] [PubMed] [Google Scholar]
- 41.Richardson, J., S. Broche, S. Baud, T. Leste-Lasserre, F. Femenia, D. Levy, A. Moraillon, G. Pancino, and P. Sonigo. 2002. Lymphoid activation: a confounding factor in AIDS vaccine development? J. Gen. Virol. 83:2515-2521. [DOI] [PubMed] [Google Scholar]
- 42.Richardson, J., A. Moraillon, S. Baud, A.-M. Cuisinier, P Sonigo, and G. Pancino. 1997. Enhancement of feline immunodeficiency virus (FIV) infection after DNA vaccination with the FIV envelope. J. Virol. 71:9640-9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shimojima, M., T. Miyazawa, Y. Ikeda, E. L. McMonagle, H. Haining, H. Akashi, Y. Takeuchi, M. J. Hosie, and B. J. Willett. 2004. Use of CD134 as a primary receptor by the feline immunodeficiency virus. Science 303:1192-1195. [DOI] [PubMed] [Google Scholar]
- 44.Siebelink, K. H. J., E. Tijhaar, R. C. Huisman, W. Huisman, A. de Ronde, I. H. Darby, M. J. Francis, G. F. Rimmelzwaan, and A. D. M. E. Osterhaus. 1995. Enhancement of feline immunodeficiency virus infection after immunization with envelope glycoprotein subunit vaccines. J. Virol. 69:3704-3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silvera, P., M. W. Richardson, J. Greenhouse, J. Yalley Ogunro, N. Shaw, J. Mirchandani, K. Khalili, J. F. Zagury, M. G. Lewis, and J. Rappaport. 2002. Outcome of simian-human immunodeficiency virus strain 89.6p challenge following vaccination of rhesus macaques with human immunodeficiency virus Tat protein. J. Virol. 76:3800-3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Staprans, S. I., A. P. Barry, G. Silvestri, J. T. Safrit, N. Kozyr, B. Sumpter, H. Nguyen, H. McClure, D. Montefiori, J. I. Cohen, and M. B. Feinberg. 2004. Enhanced SIV replication and accelerated progression to AIDS in macaques primed to mount a CD4 T cell response to the SIV envelope protein. Proc. Natl. Acad. Sci. USA 101:13026-13031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stüber, E., and W. Strober. 1996. The T-cell B cell interaction via OX40-OX40L is necessary for the T cell-dependent humoral immune response. J. Exp. Med. 183:979-989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uhl, E. W., T. G. Heaton-Jones, R. Pu, and J. K. Yamamoto. 2002. FIV vaccine development and its importance to veterinary and human medicine: a review. FIV vaccine 2002 update and review. Vet. Immunol. Immunopathol. 90:113-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang, S. Z.-S., K. E. Rushlow, C. J. Issel, R. F. Cook, S. J. Cook, M. L. Raabe, Y.-H. Chong, L. Costa, and R. C. Montelaro. 1994. Enhancement of EIAV replication and disease by immunization with a baculovirus-expressed recombinant envelope surface glycoprotein. Virology 199:247-251. [DOI] [PubMed] [Google Scholar]
- 50.Willett, B. J., L. Picard, M. J. Hosie, J. D. Turner, K. Adema, and P. R. Clapham. 1997. Shared usage of the chemokine receptor CXCR4 by the feline and human immunodeficiency viruses. J. Virol. 71:6407-6415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu, X. G., M. Lichterfeld, M. M. Addo, and M. Altfeld. 2005. Regulatory and accessory HIV-1 proteins: potential targets for HIV-1 vaccines? Curr. Med. Chem. 12:741-747. [DOI] [PubMed] [Google Scholar]