

Abstract
Herpes simplex virus type 1 (HSV-1) infection of the corneal stroma remains a major cause of blindness. Primary cultures of corneal fibroblasts (CF) were tested and found susceptible to HSV-1 entry, which was confirmed by deconvolution imaging of infected cells. Plaque assay and real-time PCR demonstrated viral replication and hence a productive infection of CF by HSV-1. A role for glycoprotein D (gD) receptors in cultured CF was determined by gD interference assay. Reverse transcription-PCR analysis indicated expression of herpesvirus entry mediator and 3-O-sulfated (3-OS) heparan sulfate (HS)-generating enzyme 3-O sulfotransferase 3 (3-OST-3) but not nectin-1 or nectin-2. Subsequently, HS isolated from these cells was found to contain two distinct disaccharides (IdoUA2S-AnMan3S and IdoUA2S-AnMan3S6S) that are representative of 3-OST-3 activity. The following lines of evidence supported the important role of 3-OS HS as the mediator of HSV-1 entry into CF. (i) Blockage of entry was observed in CF treated with heparinases. The same enzymes had significantly less effect on HeLa cells that use nectin-1 as the entry receptor. (ii) Enzymatic removal of cell surface HS also removed the major gD-binding receptor, as evident from the reduced binding of gD to cells. (iii) Spinoculation assay demonstrated that entry blockage by heparinase treatment included the membrane fusion step. (iv) HSV-1 glycoprotein-induced cell-to-cell fusion was inhibited by either prior treatment of cells with heparinases or by HS preparations enriched in 3-OS HS. Taken together, the data in this report provide novel information on the role of 3-OS HS in mediating infection of CF, a natural target cell type.
Herpes simplex virus type 1 (HSV-1) infection is the most common cause of infectious blindness in developed countries (33, 34, 49, 62). Following initial infection of epithelial cells, HSV establishes latency in the sensory nerve ganglia of the host (26, 58). The virus emerges sporadically from latency and causes lesions on mucosal epithelium, skin, and the cornea. Prolonged or multiple recurrent episodes of corneal infections can result in vision impairment or blindness due to the development of herpetic stromal keratitis (HSK) (8, 23, 28, 42, 44, 71). The HSK condition is typically characterized by inflammation leading to scarring, thinning, and vascularization of the corneal stroma (12, 15, 19, 24, 33, 34). It accounts for 20 to 48% of all recurrent ocular HSV infection cases (33).
Primary infection begins with the entry of HSV into host cells. It is a complex process initiated by specific interaction of viral envelope glycoproteins and host cell surface receptors (10, 18, 52, 58-60). Both HSV-1 and HSV-2 use glycoproteins B and C (gB and gC, respectively) to mediate their initial attachment to cell surface heparan sulfate (HS) proteoglycans (25, 53, 67). Binding of herpesviruses to HS proteoglycans likely precedes a conformational change that brings viral gD to the binding domain of host cell surface gD receptors (29-31). Thereafter, a concerted action involving gD, its receptor, three additional HSV glycoproteins (gB, gH, and gL), and possibly an additional gH coreceptor triggers fusion of the viral envelope with the plasma membrane of host cells (43, 45, 50). Subsequently, viral capsids and tegument proteins are released into the cytoplasm of the host cell.
The gD receptors include cell surface molecules derived from three structurally unrelated families. These include a member of the tumor necrosis factor receptor family (40), two members of the nectin family of receptors (21), and the product of certain 3-O-sulfated (3-OS) sulfotransferases (3-OSTs), 3-OS HS (41, 55, 57-60). Herpesvirus entry mediator (HVEM or TNFRSF14) principally mediates entry of HSV-1 and HSV-2 (40, 66) into human T lymphocytes and trabecular meshwork cells and is expressed in many fetal and adult human tissues, including the lung, liver, kidney, and lymphoid tissues (27, 32, 37, 40, 62). Nectin-1 and nectin-2, also known as herpesvirus entry proteins C B, respectively, belong to the immunoglobulin superfamily (7, 13, 21). Both nectin-1 and nectin-2 mediate entry of HSV-1 and HSV-2, but only nectin-1 mediates bovine herpesvirus type 1 entry (9, 21, 36, 38, 39, 65). The HSV-1 entry-mediating activity of nectin-2 is limited to some mutant strains only (65). Nectin-1 is extensively expressed in human cells of epithelial and neuronal origin (22, 48, 56, 58), while nectin-2 is widely expressed in many human tissues, but with only limited expression in neuronal cells and keratinocytes (5). The nonprotein receptor 3-OS HS is expressed in multiple human cell lines (e.g., neuronal and endothelial cells) and mediates entry of HSV-1 but not HSV-2 (16, 17, 55, 59, 60).
Although the corneal stroma is a potential target of HSV-1 infection, the role of HSV-1 gD receptors in viral entry into the cells of the stroma remains poorly understood (1, 3). It has been proposed that HSV-1 travels from the corneal epithelium to sensory ganglia and then returns to the stroma to cause disease (47). To study HSV entry and the role of the entry receptor(s), we used in vitro primary cultures of corneal fibroblasts (CF) derived from human corneal stroma. We hypothesized that one or more of the known entry receptors would mediate entry of HSV-1 into CF. Our objectives were (i) to investigate the susceptibility of cultured human CF to productive HSV-1 entry and replication, (ii) to determine the expression of HSV-1 gD receptors in cultured CF, and (iii) to identify a gD receptor(s) crucial for HSV-1 entry into cultured CF. This study, the first of its kind, demonstrated a significant role for, and the physiological significance of, modified HS (3-OS HS) as a receptor for HSV-1 entry into a natural-target human cell type.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
P. G. Spear (Northwestern University) provided wild-type Chinese hamster ovary (CHO-K1) cells, viruses, and the anti-Myc, anti-HVEM (40) antibodies used throughout this study. Recombinant cytomegalovirus (CMV; Towne strain RC256) was obtained from the American Type Culture Collection. Wild-type CHO-K1 cells and African green monkey kidney (Vero) cells were grown as previously described (40). CHO-K1 cells stably expressing HVEM plasmid pBec10 (40) and plasmid 3-OST-3 (68) were grown in Ham's F12 medium (Invitrogen) under G418 sulfate (400 μg/μl; Cellgro) selection. Human lung fibroblasts (LF) provided by Steven J. Ackerman (University of Illinois at Chicago) were grown in Dulbecco's modified Eagle medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS). Cultures of human CF were derived from the corneas of donors 24, 38, 47, 49, and 51 years of age obtained from the Illinois Eye Bank, Chicago, IL. The procurement of tissues was in accordance with the Declaration of Helsinki. As previously described (70), CF were grown in l-glutamine-containing DMEM (Invitrogen) supplemented with 15% FBS. Cells were trypsinized and passaged after reaching confluence. CF from the third to the sixth passages used in this study showed nearly identical results in HSV-1 entry, replication, receptor expression, and antibody-blocking assays. Recombinant β-galactosidase-expressing HSV-1(KOS) tk12 (65) and HSV-1(KOS) gL86 (40) were used. Green fluorescent protein (GFP)-expressing HSV-1(K26GFP) (11) was provided by P. Desai (Johns Hopkins University, Baltimore, MD). The viral stocks were propagated at a low multiplicity of infection (MOI) in complementing cell lines, their titers were determined on Vero cells, and they were stored at −80°C.
Viral entry assays.
Viral entry assays were based on quantitation of β-galactosidase expressed from the viral genome or by CHO-IEβ8 cells, in which β-galactosidase expression is inducible by HSV infection (40). Cells (CF, LF, wild-type CHO-K1 cells, and CHO-K1 cells stably expressing HVEM and 3-OST-3) were plated at 2 × 10 4 per well in 96-well plates at least 16 h prior to infection. HSV entry into CF was determined by o-nitrophenyl-β-d-galactopyranoside (ONPG) assay and also by 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) as previously described (40). Microscopy was performed with the 20× objective of an inverted microscope (Axiovert 100 M; Zeiss). All experiments were repeated a minimum of three times unless otherwise noted.
Deconvolution microscopy.
Confluent monolayers of cultured CF (0.5 × 106) on cover glass (20 by 20 mm; Fisher Scientific, Pittsburgh, PA) were infected at 37°C with GFP-tagged HSV-1(K26GFP) (11) at 50 PFU/cell. After 90 min, cells were washed in phosphate-buffered saline (PBS) and mounted upside down on the microscope slides in Vectashield mounting medium (Vector Laboratories, Inc., Burlingame, CA). Stack microscopy was carried out with Metamorph imaging software and a Zeiss Axiovert 100 M system equipped with 63×, 1.4 numerical aperture oil immersion objectives. The images were deconvolved with Auto Deblur software (AutoQuant Imaging, Inc.). Orthogonal sections were made in a z stack of 25 images (0.4-μm interval). A histogram of an individual stack taken as a part of z series was plotted for maximum light intensity. The picture was produced in Adobe Photoshop 7.0.
Plaque assay.
Virus replication was examined by plaque assay. A monolayer of cultured CF (approximately 4 × 106 cells per 25-ml flask) was infected (MOI, 0.01) with HSV-1(KOS) or mock infected with PBS alone for 2 h at 37°C. Wild-type CHO-K1 cells and CHO-K1 cells expressing 3-OST-3-modified 3-OS HS infected under similar conditions were used as negative and positive controls, respectively. After removal of the inoculum, monolayers were overlaid with DMEM containing 2.5% heat-inactivated calf serum and incubated at 37°C until the time of harvest (12 to 48 h). Infectious virus titers were determined on Vero cells cultured in triplicate by using an overlay of medium containing methylcellulose. In order to block secondary plaque formation, human immunoglobulin G (IgG; Sigma) was added to the inoculum. The cells were washed with PBS buffer, fixed in alcohol, and stained with Giemsa stain. Infectivity was recorded as the number of PFU.
HSV-1 gD interference assay.
CF were transfected with TransIT-TKO reagent (Mirus Corporation) with an HSV-1 gD expression plasmid (pPEP99) (20) or a control plasmid (pCDNA3) in six-well dishes (1.0 μg of plasmid DNA total per well). The interference assay was performed as described elsewhere (20).
Detection of gD receptors by reverse transcription (RT)-PCR analysis.
Total RNA was isolated from cultured human CF and HeLa cells with a QIAGEN RNeasy kit (QIAGEN Corp., Valencia, CA). SUPERSCRIPT II reverse transcriptase (Invitrogen Corp.) was used for RT. PCR amplification of cDNAs was done with primers 5′-TCTCTGCTGCCAGACA-3′ and 5′-GCCACAGCAGAACAGA-3′ for HVEM, primers 5′-TCCTTCACCGATGGCACTATCC-3′ and 5′-TCAACACCAGCAGGATGCTC-3′ for nectin-1, and primers 5′-AGAAGCAGCAGCACCAGCAG-3′ and 5′-GTCACGTTCAGCCAGGA-3′ for nectin-2. The 3-OST-3 sequences were amplified with primers 5′-CAGGCCATCATCATCGG-3′ and 5′-CCGGTCATCTGGTAGAA-3′. RT-PCR analysis was performed as described previously (54). The expected sizes of the PCR products were 1,270 bp for HVEM, 738 bp for nectin-1, 616 bp for nectin-2, and 736 bp for 3-OST-3.
Determination of the presence of 3-OS HS in cultured CF. (i) Metabolic labeling and preparation of 35S-HS.
CF were grown to 90% confluence in T-25 flasks. After removal of growth medium, cells were incubated with 5 ml labeling medium (F-12 medium, 10% dialyzed FBS, 1 mCi/ml Na235SO4) for 10 h at 37°C. One milliliter of 1 mg/ml pronase (Sigma) in a buffer containing 240 mM sodium acetate (NaAcO, pH 6.5) and 1.92 M NaCl was then added to lyse the cells. After incubation at 37°C overnight, the resultant samples were subjected to DEAE-Sephacel column chromatography. The column was washed with 10 ml buffer A (20 mM NaAcO [pH 5], 150 mM NaCl) and eluted with 5 ml buffer B (20 mM NaAcO [pH 5], 1 M NaCl). The eluent was dialyzed against 50 mM ammonium bicarbonate with a membrane with a molecular weight cutoff of 14,000 and dried. The samples were reconstituted in H2O and subjected to β elimination under alkaline conditions at 46°C for 16 h. The samples were then subjected to phenol-chloroform extraction to remove proteins. 35S-HS was precipitated by 75% ethanol. Chondroitin sulfate was removed by incubation with chondroitinase ABC (Sigma) at 37°C for 3 h.
(ii) Disaccharide analysis of 35S-HS.
The 35S-HS isolated from CF and CHO-K1 cells was degraded with nitrous acid at pH 1.5, followed by reduction with sodium borohydride (35). The resultant 35S-labeled disaccharides were desalted on a Bio-Gel P-2 column (0.75 by 200 cm) that was equilibrated with 0.1 M ammonium bicarbonate at a flow rate of 4 ml/h. The disaccharides were resolved with a C18-reversed phase column (0.46 by 25 cm; Vydac) under reverse-phase ion-pairing high-performance liquid chromatography (RPIP-HPLC) conditions. Briefly, the column was eluted with acetonitrile at 8% for 30 min, followed by 15% for 15 min and then by 19.5%, in a solution containing 38 mM ammonium phosphate monobasic, 2 mM phosphoric acid, and 1 mM tetrabutylammonium phosphate monobasic (Fluka) at a flow rate of 0.5 ml/min. The identities of the disaccharides were determined by coelution with appropriate 3H-labeled and 35S-labeled disaccharides (35, 57).
Binding of 3-OS HS derived from cultured CF to HSV-1 gD.
To assay for the binding of 3-OS HS to gD, immunoprecipitation was carried out with anti-gD monoclonal antibody (57). Enzyme-modified HS (100,000 to 200,000 cpm) was incubated in a buffer containing 50 mM Tris-HCl, 150 mM NaCl, and 0.01% Triton (pH 7) (binding buffer) and 2 mg/ml gD at room temperature for 30 min. Anti-gD monoclonal antibody DL6 was added, and the mixture was incubated at 4°C for 1 h, followed by addition of protein A-agarose gel (80 μl of a 1:1 slurry), and agitated at 4°C for an additional hour. The HS was eluted from the gel with 1 ml of 1 M NaCl in the binding buffer.
Virus-free cell-to-cell fusion assay.
In this experiment, the CHO-K1 cells designated effector cells were cotransfected with plasmids expressing four HSV-1(KOS) glycoproteins, pPEP98 (gB), pPEP99 (gD), pPEP100 (gH), and pPEP101 (gL), along with plasmid pT7EMCLuc, which expresses the firefly luciferase gene under the control of the T7 promoter (61). In a parallel experiment, human herpesvirus 8 (HHV-8) glycoproteins gB, gH, and gL (46) were used as controls. Wild-type CHO-K1 cells express cell surface HS but lack functional gD receptors, including 3-OS HS (60). As a result, they are resistant to both HSV entry and virus-induced cell fusion (40, 53, 57). Cultured CF considered target cells were cotransfected with pCAGT7, which expresses T7 RNA polymerase with the chicken actin promoter and the CMV enhancer (46). Effector cells expressing pT7EMCLuc and pCDNA3 (devoid of any glycoproteins) and target CF transfected with T7 RNA polymerase alone were used as negative controls. Activation of the reporter luciferase gene, as a measurement of cell fusion, was examined by reporter lysis assay (Promega) at 24 h postmixing as previously described (61).
Heparinase treatment.
CF and CHO-K1 cells transfected with plasmid 3-OST-5 plated on 96-well plates were washed with Mg2+- and Ca2+-free PBS and incubated at room temperature for 90 min with several dilutions of heparinases II and III (4 U/ml; Sigma) or PBS alone and incubated at room temperature. The cells were then washed with PBS and used for viral entry, cell-to-cell fusion, and gD-Fc binding assays.
gD-Fc binding assay.
Cultured CF and HeLa cells were plated overnight into 96-well dishes (approximately 4 × 104 cells/well). They were treated with heparinases II and III or with PBS buffer as described above for 2 h. This was followed by incubation with gD-Fc (1 μg/ml) for 1 h and fixation at room temperature with 2% formaldehyde and 0.02% glutaraldehyde for 15 min. The cells were washed with PBS containing 3% BSA (Sigma) and then incubated at room temperature sequentially with a biotinylated secondary antibody against rabbit IgG (Sigma) at a 1:500 dilution in PBS-3% BSA for 45 min and AMDEX streptavidin-conjugated horseradish peroxidase (Amersham) at a 1:20,000 dilution for 30 min. Following washing in PBS, the cells were incubated with 50 μl 3,3′,5,5′-tetramethylbenzidine (Sigma) in 50 mM phosphate-citrate buffer. Products were quantitated by use of a spectra MAX 190 enzyme-linked immunosorbent assay reader to measure optical density at 650 nm (OD650).
Surface immunofluorescence assay.
To detect the binding of gD-Fc to the surface of CF and HeLa cells, cultured monolayers of cells (approximately 106) were grown overnight on glass bottom culture dishes (Mat Tek Corp.). The cells were then treated with heparinases II and III (at 4 U/ml) or with PBS, exposed to gD-Fc for 1 h, and fixed with acetone. They were then treated with fluorescein isothiocyanate (FITC)-conjugated anti-rabbit IgG (Sigma) and mounted on glass slides before examination under a confocal microscope.
Spinoculation assay.
Subconfluent CHO-K1 cells were transfected with 1.0 μg of DNA of HVEM (pBEC10) (40), 3-OST-5 (68), or nectin-1 (pBG38) (21). Transfected CHO-K1 cells and cultured CF along with Vero cells were plated on 96-well dishes (approximately 104 cells/well) and allowed to grow for an additional 16 h at 37°C and 5% CO2. The cells were treated at room temperature with heparinases II and III (4 U/ml) or PBS before spinoculation (51). Briefly, a dose of HSV-1(KOS) gL86 and or CMV (35 PFU/cell) was added to the cells for 2 h of incubation and the plates were transferred to 37°C. One set of heparinase-treated cells was spun at 1,200 × g at 37°C, and another set was kept at 37°C for 2 h in an incubator as previously described (51). The plates were then washed with PBS to remove exogenous virus and incubated at 37°C for 4 h. They were further washed with PBS, 50 μl ONPG reagent was added to each well, and viral entry was measured as described above.
RESULTS
CF are susceptible to HSV-1 entry.
To determine HSV-1 entry, confluent monolayers of cultured CF were infected with serial dilutions of recombinant HSV-1(KOS) tk12 (65), which expresses β-galactosidase upon entry into cells. CHO-K1 cells that are naturally resistant to HSV-1 entry were used as a negative control. Entry of HSV-1 was measured after 6 h of viral infection (60). As shown in Fig. 1A, compared to CHO-K1 cells, HSV-1 entered CF in a dose-dependent manner while none entered CHO-K1 cells. In comparison assays, human CF showed an HSV-1 dose response similar to those of human LF cells and gD receptor (HVEM or 3-OS HS)-containing CHO-K1 cells. By contrast, CHO-K1 cells without any receptor remained resistant to HSV-1 entry. A representative entry result obtained with a single virus dose of 5 PFU/cell is shown in Fig. 1B.
FIG. 1.

Entry of HSV-1 into cultured CF. (A) Dose-response analysis of HSV-1 entry into CF. Cultured CF (•), along with wild-type CHO-K1 cells (▪), were plated in 96-well plates and inoculated with twofold serial dilutions of β-galactosidase-expressing recombinant HSV-1(KOS) tk12 ranging from 1 to 180 PFU/cell. After 6 h, the cells were permeabilized and incubated with ONPG substrate for quantitation of β-galactosidase activity expressed from the input viral genome. Enzymatic activity was measured by determining OD410. Data represent the mean ± the standard deviation of results in triplicate wells in a representative experiment. The experiment was repeated three times with similar results. (B) Entry of HSV-1 into cultured CF, human LF cells, and CHO-K1 cells expressing either HVEM or 3-OS HS as a gD receptor. The cells were plated in 96-well dishes and then treated with serial dilutions of β-galactosidase-expressing recombinant HSV-1(KOS) tk12 as described for panel A. The values shown represent the amount of reaction product detected spectrophotometrically at a single input dose of 5 PFU/cell. Data represent the mean ± the standard deviation of results in triplicate wells in a representative experiment. The experiment was repeated three times with similar results. (C) Dark-field image of cultured CF infected with GFP-tagged HSV-1. (D) A section of the deconvolved stack showing GFP-tagged capsids near or at the periphery of DAPI-stained blue nuclei. The vertical line to the right indicates the YZ plane of z sections. The horizontal line at the bottom indicates the XZ plane of z sections. Images were collected on a Zeiss Axiovert 100 M microscope equipped with a 63× objective. (E) Three-dimensional rendering of the entire deconvolved stacks. (F) Optical plane of sections presented at 90°. (G) Optical plane of sections presented at 110°. (H) Plot of maximum light intensity due to GFP-tagged HSV-1 observed per stack going from the top to the bottom of a cell.
Visualization of GFP-tagged HSV-1 internalization in cultured CF by deconvolution microscopy.
GFP-tagged HSV-1(K26GFP) (11) was used to infect cultured CF, and high-resolution deconvolution microscopy was employed to visualize fluorescent virus particles inside infected CF. As shown in Fig. 1C, a majority of the cultured CF were infected with fluorescent HSV-1 virions. Next, to localize the virions inside an infected cell, the cells were stained with 4′,6′-diamidino-2-phenylindole (DAPI) and examined under two separate channels (FITC and DAPI). The punctate green fluorescence of virions was observed surrounding the DAPI-stained blue nucleus. To analyze this further, deconvolution microscopy was used. Optical sections (0.2 μm thick) were collected, and the stacks were deconvolved and analyzed by Metamorph software. The orthogonal section of deconvolved images sliced at two selected planes (Fig. 1D, XZ and YZ) showed the distribution of punctate green fluorescence or virus particles in planes surrounding the nucleus. The subsequent three-dimensional views, which cover the entire depth of a cell with detailed visualization of the GFP-labeled capsid at different angles within the cell, further demonstrate the presence of the virus surrounding the nucleus (transverse view in Fig. 1E and longitudinal views in Fig. 1F to G). Analysis of individual optical sections (z stacks) revealed that maximum florescence was recorded for the midsections, indicating that a majority of the virions were located in the middle (cytoplasmic) portion of the cell. A histogram of this result is shown in Fig. 1H.
HSV-1 replicates in cultured CF.
Because HSV-1 was able to enter cultured CF, we next evaluated if entry of HSV-1 into cultured CF led to productive replication of the virus. The infectious yields of virus were determined by plaque assays with Vero cells. As shown in Fig. 2A, cultured CF exposed to HSV-1(KOS) and CHO-K1 cells expressing 3-OS HS modified by 3-OST-3 at an MOI of 0.01 produced a larger number of plaques over time. In contrast, CHO-K1 cells infected with identical doses of the same virus failed to produce infectious virions. These results, together with those of the entry assay, show that entry of HSV-1 into cultured CF leads to productive infection.
FIG. 2.

Infectious yields of HSV-1 and involvement of gD receptors in viral infection. (A) HSV-1 replicates in cultured CF. Confluent monolayers of CF (○) and wild-type CHO-K1 cells (•) and CHO-K1 cells expressing 3-OS HS modified by 3-OST-3 (▾) were infected with HSV-1 at 0.01 PFU per cell for 90 min at 37°C. Inoculums were harvested at 12 to 48 h postinfection. The infectious-virus titer (PFU per milliliter) determined in triplicate in Vero cells by plaque assay indicates that the viral titer in cultured CF increased over time. Data represent the mean ± the standard deviation of results in triplicate wells in a representative experiment. Experiments were repeated three times, yielding similar results. (B) Receptors for gD are important for infection of CF. Cultured CF were transfected with an HSV-1 gD expression plasmid and infected either with HSV-1 (○) or with CMV (▿). In parallel, CF infected with the empty vector pCDNA3 were infected with either HSV-1 (•) or CMV (▾). Reporter enzyme activity, as a measurement of viral entry, was measured with a spectrophotometer at 410 nm.
Expression of HSV-1 gD in cultured CF renders resistance to HSV-1 entry.
In order to determine if the gD receptors play a role in human CF infection by HSV-1, a gD-mediated interference assay was used (20). This assay is based on the principle that cells that are normally susceptible to viral entry become resistant upon expression of viral gD because of sequestration of gD receptors by cell-expressed gD (20). To carry out the assay, human CF were transiently transfected with an HSV-1 gD-expressing plasmid (or an equal amount of the empty vector, pCDNA3, as a control), followed by infection with serial dilutions of β-galactosidase-expressing HSV-1(KOS) gL86. We used recombinant human CMV as a control that enters independently of gD receptors into cells. As shown in Fig. 2B, HSV-1 entry into gD-expressing CF was approximately 2.0-fold lower compared to the pCDNA3 control. Meanwhile, the expression of gD in cultured CF did not affect CMV entry. Thus, entry into CF is likely mediated by gD receptors expressed on the CF surface in a gD-dependent manner. The transfection efficiency in CF was tested with a nectin-1-GFP-expressing plasmid. We estimated that approximately 80% of cultured CF turned green as a result of transfection under fluorescence microscopy (data not shown).
Identification of gD receptors expressed in cultured CF.
RT-PCR analysis was performed to determine the identity of gD receptors potentially expressed in CF. Specific primers for HVEM, nectin-1, nectin-2, and 3-OST-3 were used. As shown in Fig. 3A, products of the expected sizes for cDNAs encoding HVEM and 3-OST-3, but not nectin-1 and nectin-2, were detected. The cDNA obtained from HeLa cells was used as a positive control. The absence of a nectin-1-specific product was unexpected since it is generally thought to be well expressed in human tissues and cell lines (21).
FIG. 3.

Expression of HVEM and 3-OST-3 mRNAs in cultured CF. (A) RT-PCR analysis of the expression of HVEM- and 3-OST-3 (as a surrogate marker for 3-OS HS)-specific messages in CF and HeLa cells (served as a control). The cDNAs were produced from total RNA isolated from the cells. The PCR products were separated by electrophoresis on an agarose gel and stained with ethidium bromide. Molecular size markers are indicated on the left (sizes are in kilobases). (B) RPIP-HPLC chromatograms of the disaccharide analysis of HS isolated from CF and CHO-K1 cells. 35S-HS was extracted from CHO-K1 cells (top) and CF (bottom) after they were incubated with Na235SO4. Extracted HS was then depolymerized by nitrous acid at pH 1.5, followed by sodium borohydride reduction. The resultant 35S-labeled disaccharides were resolved by RPIP-HPLC. The elution positions of disaccharides are indicated by arrows, where arrow 1 represents IdoUA2S-AnMan3S, arrow 2 represents IdoUA2S-AnMan6S, and arrow 3 represents IdoUA2S-AnMan3S6S.
Demonstration of 3-OS HS on cultured CF and its ability to bind to HSV-1 gD.
Since RT-PCR analysis showed that 3-OS HS-producing enzyme 3-OST-3 is expressed in cultured CF, we decided to verify the presence of its product, 3-OS HS, in CF. A disaccharide analysis of the total HS isolated from the surface of CF grown in medium containing 35S-labeled sodium sulfate was performed. The resultant 35S-labeled disaccharides were resolved by RPIP-HPLC, and the chromatograms are shown in Fig. 3B. By comparing the profiles of the degraded 35S-HS between wild-type CHO-K1 cells and cultured CF, we found two unique 35S-labeled disaccharides in cultured CF. By coelution with the appropriate disaccharide standards on RPIP-HPLC, we identified the two 35S-lableled disaccharides to be 3-O-sulfated disaccharides with the structures of IdoUA2S-AnMan3S (eluted at 32 min) and IdoUA2S-AnMan3S6S (eluted at 65 min). Of note is that IdoUA2S-AnMan3S are the representative disaccharides of 3-OST-3-modified HS believed to be parts of the gD-binding site in 3-OST-3-modified HS that contribute to the activity in assisting HSV-1 entry (57).
Next, we examined the isolated HS for its gD-binding ability (57). The binding of HS and gD was determined by incubating modified 35S-HS with gD, followed by immunoprecipitation with anti-gD monoclonal antibody DL6 to precipitate the complex of 35S-HS and gD. When CHO-K1 cells were the source of HS, binding to gD was 5.3% ± 0.6%, and when the HS source was CF, binding to gD was 9.8% ± 0.4%. These results showed that HS isolated from cultured CF exhibited about a twofold increase in gD binding compared to the control HS isolated from CHO-K1 cells, which naturally lack 3-OS HS (14). A similar increase in gD binding was reported for 3-OS HS modified in vitro by 3-OST-3 (57). Taken together, these results suggest that cultured CF expresses 3-OST-3-modified HS disaccharides capable of HSV-1 gD binding.
Determination of the significance of HS as the mediator of entry and gD binding to cultured CF.
Expression of 3-OS HS raised the possibility that it could be a mediator of HSV-1 entry into CF. Since cells that use 3-OS HS as the major entry receptor become significantly more resistant to HSV-1 upon removal of HS from the surface (57), we decided to compare heparinase-treated CF with HeLa cells, which use nectin-1 as the major mediator of entry (7). As an additional control, we also tested 3-OST-5-expressing CHO-K1 cells in a parallel assay. As shown in Fig. 4A, heparinase-treated CF and 3-OST-5-expressing CHO-K1 cells were resistant to HSV-1 entry compared to the untreated cells and this effect was considerably less pronounced (only about 25% reduction) in identically treated HeLa cells. Moderate resistance was expected since HS plays a role in virus attachment, and this observation is similar to what has been shown before (57). Next, we tested by cell enzyme-linked immunosorbent assay if a soluble recombinant form of HSV-1 gD or gD-Fc (20, 41, 57, 63) would bind cultured CF and whether this binding could be adversely affected by removal of HS from the surface (41, 57, 63). Cells either treated with heparinases or mock treated were allowed to bind equal amount of gD, and binding was detected with a spectrophotometer. Figure 4B shows that gD binding to HeLa cells was not affected by heparinase treatment; however, the binding of gD to CF was reduced by about 75%. This finding strongly suggests that gD-binding receptors were specifically removed upon heparinase treatment in CF but not in HeLa cells. To generate further evidence for this phenomenon, binding of gD to cells was also examined by immunofluorescence. Once again, virtually no fluorescence (gD binding) was detected in heparinase-treated CF and this effect was not seen in identically treated HeLa cells (Fig. 4C). This indicated that heparinase treatment specifically removed receptors in CF but not in HeLa cells.
FIG. 4.

Effect of heparinase treatment on HSV-1 entry and gD binding to CF. (A) Effect of heparinase treatment on HSV-1 entry. Cultured CF, HeLa cells, or control CHO-K1 cells expressing 3-OST-5 were treated with heparinases II and III (4 U/ml) in 96-well tissue culture dishes. A separate set of cultured cells were mock treated with PBS alone. The cells were then exposed to HSV-1(KOS) gL86 at 35 PFU/cell, and viral entry was quantitated 6 h later, as described in the legend to Fig. 1A. Data represent the mean ± the standard deviation of results in triplicate wells in a representative experiment. The experiment was repeated three times with similar results. (B) Effect of heparinase treatment on gD binding. Cultured CF and control HeLa cells in 96-well dishes were treated with heparinases II and III (4 U/ml) or incubated with buffer. The cells were then incubated with gD-Fc for 30 min, followed by fixation and incubation with a secondary antibody and a horseradish peroxidase detection system. The values shown (means of triplicate determinations plus standard deviations) represent the amounts of binding product detected spectrophotometrically (OD650). (C) Confocal imaging of gD binding to heparinase-treated cells. Cultured HeLa cells and CF were plated in glass bottom culture dishes and grown overnight. The cells were treated with heparinases II and III (4 U/ml) or mock treated with buffer. After incubation with gD-Fc, the cells were washed, fixed with acetone, and incubated with FITC-conjugated anti-rabbit IgG. Micrographs of representative HeLa cells and CF are presented to show gD-Fc binding with or without heparinase treatment with a Leica confocal microscope.
Spinoculation fails to restore HSV-1 entry into heparinase-treated cultured CF or CHO-K1 cells expressing 3-OST-5.
To verify that the significance of HS is not limited to the attachment of virions to cultured CF, we made use of the spinoculation assay (51). This assay has recently been used to determine whether the effect of enzymatic removal of HS is limited to the initial attachment of the virus to cells alone or includes membrane fusion as well (51). The spinoculation assay is based on the principle that in cells lacking HS, the attachment but not fusion that is mediated by gD receptors can be substituted for by spinning cells and virions together at 1,200 × g (51). Thus, for each cell type (CF, Vero, nectin-1, HVEM, or 3-OST-5-expressing CHO-K1 cells), a set of heparinase-treated cells was spinoculated while another was infected with an identical viral dose in an incubator (1 × g) as a control. A third set of cells was mock treated (with buffer alone) and infected with the same viral dose in the incubator. As shown in Fig. 5A, in all groups the mock-treated cells (black bars) exhibited the highest entry, which was reduced in cells with heparinase treatment (white bars). The most noteworthy results were seen with heparinase-treated and spinoculated cells (gray bars). While entry was recovered to almost the mock-treated levels in most cases, CF and 3-OST-5-expressing CHO-K1 cells showed no recovery of entry into those cells upon spinoculation. The lack of recovery of entry upon spinoculation has been attributed to loss of the gD receptor upon heparinase treatment (51). Thus, similar to 3-OST-5-expressing CHO-K1 cells, HSV-1 likely uses 3-OS-HS for fusion of its envelope with the plasma membrane of CF during entry. In a separate experiment, CMV was used as a control that enters CF via gD-independent receptors. Spinoculation of heparinase-treated CF restored CMV entry but not HSV-1 entry (Fig. 5B).
FIG. 5.

Spinoculation of heparinase-treated cells recovers entry into Vero cells and nectin-1- or HVEM-expressing CHO-K1 cells but not cultured CF or 3-OST-5-expressing CHO-K1 cells. (A) CHO-K1 cells were transiently transfected with a nectin-1, HVEM, or 3-OST-5 plasmid. gD receptor-expressing cells, along with naturally susceptible Vero cells and cultured CF, were plated and treated with heparinases II and III (4 U/ml) or PBS buffer for 2 h and infected with HSV-1(KOS) gL86 at 35 PFU/cell for 2 h. During the 2-h infection, cells were either spun at 37°C or kept in an incubator. Infection was then continued for an additional 4 h, the plates were rinsed, and viral entry was measured by determining OD410. Results from heparinase-untreated and unspun cells (considered positive controls) are shown as black bars, while those from heparinase-treated and unspun or spun cells are shown as white and gray bars, respectively. Data represent the mean ± the standard deviation of results in triplicate wells in a representative experiment. The experiment was repeated three times with similar results. (B) Spinoculation of heparinase-treated CF recovers entry of CMV but not HSV-1. Cultured CF plated in a 96-well plate were treated with heparinases II and III (4 U/ml) or PBS buffer for 3 h and infected with reporter viruses HSV-1(KOS) gL86 and CMV at 35 PFU/cell for 2 h. During the 2-h infection, cells were spun at 37°C or kept in an incubator unspun. Infection was then continued for an additional 4 h, and viral entry was measured by determining OD410. Results from heparinase-untreated and unspun cells (considered positive controls) are shown as black bars, while those from heparinase-treated unspun or spun cells are shown as white and gray bars, respectively. Data represent the mean ± the standard deviation of results in triplicate wells in a representative experiment. The experiment was repeated three times with similar results.
3-OS HS is required for HSV-1 glycoprotein-induced cell-to-cell fusion of cultured CF.
To verify that 3-OS-HS is indeed the mediator of the membrane fusion that occurs during HSV-1 entry into CF, we used a quantitative and efficient cell-to-cell fusion assay (4, 61). It has been shown that, similar to the membrane fusion that occurs during entry, cells expressing HSV-1 glycoproteins gB, gD, gH, and gL fuse with cells expressing gD receptors (46), and thus cell-to-cell fusion mimics the minimum requirement for entry. First, we studied the significance of HS in the fusion of CF with glycoprotein-expressing CHO cells. HS dependence is a way to determine whether 3-OS HS is the principal mediator of membrane fusion since, unlike nectin-1 and HVEM, which do not require HS for membrane fusion (61), 3-OS HS-mediated cell fusion is highly dependent on the expression of HS on the cell surface (61). As expected (Fig. 6A), fusion of 3-OST-3-expressing CHO-K1 cells with effector cells (expressing the glycoproteins) were significantly (about 90%) reduced upon heparinase treatment, and a similar result was seen with CF but not with HeLa cells that use nectin-1 as the gD receptor (7, 63). In a separate set of experiments, we used cells expressing HHV-8 glycoproteins gB, gH, and gL to test whether the heparinase treatment effect on CF is HSV-1 specific. In this case, heparinase treatment had no effect on cell fusion (Fig. 6B).
FIG. 6.
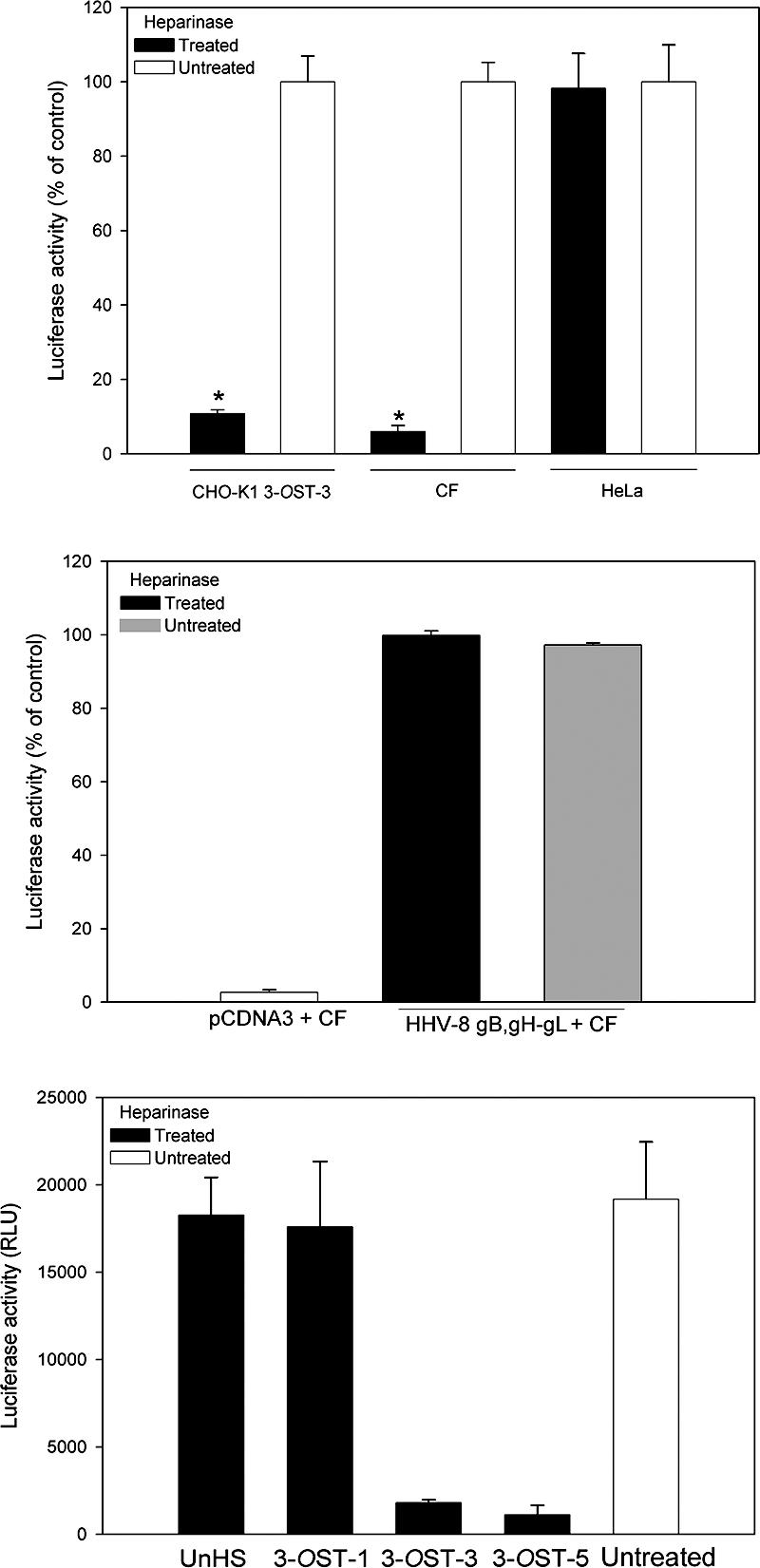
Role of 3-OS HS in membrane fusion. (A) Enzymatic removal of cell surface HS blocks HSV-1-induced cell fusion in cultured CF. CHO-K1 cells expressing 3-OST-3 and naturally susceptible HeLa cells were used as controls. Target cells (CHO-K1 3-OST-3, CF, and HeLa) were treated with heparinase (black bars) or left untreated (white bars) prior to cocultivation with effector cells expressing four essential HSV-1 glycoproteins (gB, gD, gH, and gL). The luminometer readings (relative light units [RLU]) for untreated cells of each kind were determined three or more times, and the mean value was assigned a value of 100%. The number of relative light units obtained for untreated cells was then normalized accordingly to obtain the percentages shown. Significant difference compared to the heparinase-untreated controls, P < 0.01. (B) Enzymatic removal of cell surface HS does not block HHV-8 envelope glycoprotein-induced cell-to-cell fusion in CF. CHO-K1 cells, effector cells that express HHV-8 envelope glycoproteins (gB, gH, and gL) or control plasmid pCDNA3 were mixed at a 1:1 ratio with heparinase-treated or untreated CF. The percentage values were calculated as described above. (C) Soluble 3-OS HS generated by 3-OST-3 and 3-OST-5 but not 3-OST-1 or unmodified HS (UnHS) inhibits cell-to-cell fusion in cultured CF. Effector cells were treated with 1.0 μg of unmodified HS (UnHS) or purified 3-OSTs generated by specific enzymes (3-OST-1, 3-OST-3, and 3-OST-5) or mock treated as a positive control (white bar) prior to being mixed with target cells. Untreated effector cells mixed with cultured CF or 3-OST-3-expressing target cells served as a control. Luciferase activity was measured 24 h after cocultivating the effector and target cells. The number of relative light units shown is from one experiment performed in triplicate.
To verify this result, a competition assay was performed with soluble forms of HS. Approximately 1.0 μg/ml soluble HS from bovine kidney either left unmodified (UnHS) or modified in vitro with one of the purified enzymes (3-OST-1, 3-OST-3A, or 3-OST-5) (6, 35) was added to glycoprotein-expressing effector cells prior to mixing with cultured CF (used as target cells). 3-OST-1 is known not to generate gD receptors, while 3-OST-3 and 3-OST-5 generate gD receptors that are almost identical in structure and are essentially interchangeable for most experimental purposes (57, 61, 68). The extent of 3-O sulfation to the HS was monitored by determining the incorporation of 35S-sulfate into the polysaccharide after in vitro modification by 3-OSTs as previously described (57). The estimated numbers of 3-O-sulfate groups per HS polysaccharide chain was 1.1 sulfate groups per chain for 3-OST-1-modified HS, while for both 3-OST-3- and 3-OST-5-modified HS the value was 1.3 sulfate groups per chain. The concentration of unlabeled HS was measured with alcian blue (2). The glycoprotein expressing effector cells that received mock HS treatment (buffer alone) were considered a control (untreated). As shown in Fig. 6C, the membrane fusion was impaired when HS was 3-O sulfated in vitro by either 3-OST-3 or 3-OST-5. In contrast, unmodified HS (UnHS) or 3-OST-1-modified HS had no significant negative effects on the fusion. Since the cell-to-cell fusion leads to polykaryocyte (multinucleated giant cell) formation, we also examined the effects of soluble forms of HS in blocking this phenomenon. The unmodified and 3-OST-1-modified forms of HS failed to block but 3-OST-3- and 3-OST-5-modified HS effectively blocked polykaryocyte formation (data not shown). Taken together, our results not only demonstrate the specificity of different forms of in vitro-modified soluble 3-OS HS in blocking fusion but also implicate 3-OS HS as the major mediator of membrane fusion for CF.
DISCUSSION
Although 3-OS-HS is known to be a receptor for HSV-1 gD (41, 57, 61, 63, 68, 69), its role in the infection of a human target cell type has remained unclear. It is well documented that during infection of its natural host, HSV-1 encounters different types of cells. Viral entry into these cells is critical for the survival of the virions and for viral spread from the initial sites of infection to neurons and cells of the immune system. The viral receptors on these cells could determine which cells are productively infected, which cells establish latent infections, and the nature of the immune response. Therefore, to study the pathogenesis of HSV-1 infections, it is crucial to identify the viral receptors that function on various target cells.
Despite the fact that HSK is a blinding disease, the cellular receptors for HSV-1 entry into the cells of the stroma remain unknown. We cultured human CF from a 47-year-old donor's eye to develop an in vitro model for studies of HSV-1 entry and replication and for identification of cellular mediators of entry. Quite unexpectedly, we did not detect expression of a well-studied HSV-1 entry receptor, nectin-1, in CF or in the stroma of the murine cornea (64). Thus, it seemed logical that entry into CF was likely mediated by the other two entry receptors, namely, HVEM and/or 3-OS HS. Since anti-HVEM entry-blocking antibodies (40) failed to significantly block entry into CF, our focus switched to 3-OS HS as the potential entry receptor. Adding further strength to this possibility, heparinase enzymes almost completely abolished HSV-1 entry into cultured CF (Fig. 4A), gD binding exhibited sensitivity to enzyme treatment (Fig. 4B and C), and the entry-blocking effect of an anti-HS antibody (10E4) was more pronounced on CF compared to HeLa cells (data not shown).
Since HS can help with attachment and fusion, to verify that the fusion step was indeed affected by HS removal, a spinoculation assays was used (51). It was clear that, similar to HSV-1 entry into 3-OST-5-expressing CHO-K1 cells, HSV-1 entry into heparinase-treated CF could not be restored by spinoculation (Fig. 5), implying that HS removal from CF was affecting fusion as well. By contrast, Vero and CHO-K1 cells expressing HVEM or nectin-1 still showed marked enhancement of entry during spinoculation after the cell surface HS was removed. The centrifugal force of spinoculation is known to enhance virus attachment in the absence of HS and apparently can fully or partially restore entry into HVEM- or nectin-1-expressing cells (51). The lack of recovery by spinoculation in CF suggests that, unlike HVEM-expressing CHO-K1 cells, HVEM is not a major receptor in cultured CF. Expression of 3-OS HS on CF was also demonstrated by disaccharide analysis, and its gD-binding ability was verified (see Results). This further strengthens the notion that 3-OS HS could act as a major mediator of HSV-1 entry into CF. Collectively, the present study, the first of its kind, demonstrates a significant role for a modified form of HS (3-OS HS) as an entry receptor in CF, a cell type that is naturally susceptible to HSV-1 entry.
The knowledge that 3-OS HS is the receptor for entry into CF is useful for many reasons. It is expected to pave the way for the development of anti-3-OS HS compounds and antibodies that can potentially block infection of CF in vitro with cultured cells and ultimately in vivo with an animal model(s). It is also likely to provide new information related to the pathogenesis of HSK. It is still unknown why, despite recurrent episodes of epithelial keratitis, many individuals do not develop HSK, and the virus is rarely detected in the stroma. One possibility relates to the availability of receptors on the cell surface. Since expression of 3-OS HS in vivo is very tightly regulated (16, 17), its availability on the cells of the stroma could be a limiting factor resulting in relatively rare occurrence of infection of the stroma. Conceivably, trauma to the cells of the stroma, in certain cases, could induce the expression of 3-OS HS, which in turn could lead to HSV-1 entry and infection of the stroma. It appears that differential expression of various HSV-1 receptors may indeed influence the course of HSV-1 infection in human cell types. For instance, HVEM is expressed in lymphoid cells, trabecular meshwork cells, and fibroblasts but only weakly in human brain tissues (37, 40, 62). On the other hand, nectin-1 is highly expressed in central nervous system, corneal epithelium and endothelium, and neuronal cell lines (8, 32, 64). Therefore, it is suggested that nectin-1 probably plays an important role during HSV-1 entry and spread in neuronal cells and associated latency while HVEM is important for HSV-1 invasion of the cells of lymphoid and trabecular tissues (37, 40, 66). By analogy, it is also likely that 3-OS HS can facilitate the entry of virions into the stroma and thereby allow the virus or the host's immune system to initiate HSK. In any case, more in vivo data are needed to establish the significance of 3-OS HS during infection of the stroma.
Acknowledgments
This work was supported by NIH grants R01 AI057860 (D.S.), R01 AI050050 (J.L.), and R01 EY03890 (B.Y.J.T.Y.); a core grant (EY01792); a Research to Prevent Blindness career award (D.S.); and a research award from the Illinois Society for the Prevention of Blindness (V.T.) V.T. was supported by a postdoctoral fellowship from the American Heart Association (AHA0525768Z).
We thank Patricia G. Spear (Northwestern University, Chicago, IL) and Prashant Desai (Johns Hopkins University, Baltimore, MD) for reagents; Ruth Zelkha for fluorescence and confocal microscopy; Myung-Jin Oh, Xiang Shen, and Martin Tibudan for tissue culture establishment; and Steven T. Olson (all at the University of Illinois at Chicago) for critical reading of the manuscript.
REFERENCES
- 1.Biswas, P. S., and B. T. Rouse. 2005. Early events in HSV keratitis—setting the stage for a blinding disease. Microb. Infect. 7:799-810. [DOI] [PubMed] [Google Scholar]
- 2.Bjornsson, S. 1998. Quantitation of proteoglycans as glycosaminoglycans in biological fluids using an alcian blue dot blot analysis. Anal. Biochem. 256:229-237. [DOI] [PubMed] [Google Scholar]
- 3.Brandt, C. R. 2005. The role of viral and host genes in corneal infection with herpes simplex virus type 1. Exp. Eye Res. 80:607-621. [DOI] [PubMed] [Google Scholar]
- 4.Browne, H., B. Bruun, and T. Minson. 2001. Plasma membrane requirements for cell fusion induced by herpes simplex virus type 1 glycoproteins gB, gD, gH and gL. J. Gen. Virol. 82:1419-1422. [DOI] [PubMed] [Google Scholar]
- 5.Campadelli-Fiume, G., F. Cocchi, L. Menotti, and M. Lopez. 2000. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 10:305-319. [DOI] [PubMed] [Google Scholar]
- 6.Chen, J., M. B. Duncan, K. Carrick, R. M. Pope, and J. Liu. 2003. Biosynthesis of 3-O-sulfated heparan sulfate: unique substrate specificity of heparan sulfate 3-O-sulfotransferase isoform 5. Glycobiology 13:785-794. [DOI] [PubMed] [Google Scholar]
- 7.Cocchi, F., L. Menotti, P. Mirandola, M. Lopez, and G. Campadelli-Fiume. 1998. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 72:9992-10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colin, J. 2002. Corneal herpes: what's new? Pathol. Biol. 50:445-451. [DOI] [PubMed] [Google Scholar]
- 9.Connolly, S. A., J. C. Whitbeck, A. H. Rux, C. Krummenacher, S. van Drunen Little-van den Hurk, G. H. Cohen, and R. J. Eisenberg. 2001. Glycoprotein D homologs in herpes simplex virus type 1, pseudorabies virus, and bovine herpes virus type 1 bind directly to human HveC (nectin-1) with different affinities. Virology 280:7-18. [DOI] [PubMed] [Google Scholar]
- 10.Cullen, B. R. 2001. Journey to the center of the cell. Cell 105:697-700. [DOI] [PubMed] [Google Scholar]
- 11.Desai, P., and S. Person. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563-7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deshpande, S. P., S. Lee, and M. Zheng. 2001. Herpes simplex virus-induced keratitis: evaluation of the role of molecular mimicry in lesion pathogenesis. J. Virol. 75:3077-3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eberle, F., P. Dubreuil, M. G. Mattei, E. Devilard, and M. Lopez. 1995. The human PRR2 gene, related to the human poliovirus receptor gene (PVR), is the true homolog of the murine MPH gene. Gene 159:267-272. [DOI] [PubMed] [Google Scholar]
- 14.Eisenberg, R. J., D. Long, M. Ponce de Leon, J. T. Matthews, P. G. Spear, M. G. Gibson, L. A. Lasky, P. Berman, E. Golub, and G. H. Cohen. 1985. Localization of epitopes of gD of herpes simplex virus type 1 glycoprotein D. J. Virol. 53:634-644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ellison, A. R., L. Yang, A. V. Cevallos, and T. P. Margolis. 2003. Analysis of the herpes simplex virus type 1 UL6 gene in patients with stromal keratitis. Virology 310:24-28. [DOI] [PubMed] [Google Scholar]
- 16.Esko, J. D., and U. Lindahl. 2001. Molecular diversity of heparan sulfate. J. Clin. Investig. 108:169-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Esko, J. D., and, S. B., Selleck. 2002. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 71:435-471. [DOI] [PubMed] [Google Scholar]
- 18.Fuller, A. O., and P. Perez-Romero. 2002. Mechanisms of DNA virus infection: entry and early events. Front. Biosci. 7:D390-D406. [DOI] [PubMed] [Google Scholar]
- 19.Gangappa, S., P. Deshpande, and B. T. Rouse. 1999. Bystander activation of CD4 T cells can represent an exclusive means of immunopathology in a virus infection. Eur. J. Immunol. 29:3674-3682. [DOI] [PubMed] [Google Scholar]
- 20.Geraghty, R. J., C. R. Jogger, and P. G. Spear. 2000. Cellular expression of alphaherpesvirus gD interferes with entry of homologous and heterologous alphaherpesviruses by blocking access to a shared gD receptor. Virology 268:47-158. [DOI] [PubMed] [Google Scholar]
- 21.Geraghty, R. J., C. Krummenacher, G. H. Cohen, R. J. Eisenberg, and P. G. Spear. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618-1620. [DOI] [PubMed] [Google Scholar]
- 22.Haarr, L., D. Shukla, E. Rødahl, M. C. Dal Canto, and P. G. Spear. 2001. Transcription from the gene encoding the herpesvirus entry receptor nectin-1 (HveC) in nervous tissue of adult mouse. Virology 287:301-309. [DOI] [PubMed] [Google Scholar]
- 23.Hendricks, R. L. 1997. An immunologist's view of herpes simplex keratitis: Thygeson Lecture 1996, presented at the Ocular Microbiology and Immunology Group meeting, October 26th, 1997. Cornea 16:503-506. [PubMed] [Google Scholar]
- 24.Hendricks, R. L. 1999. Immunopathogenesis of viral ocular infections. Chem. Immunol. 73:120-136. [DOI] [PubMed] [Google Scholar]
- 25.Herold, B. C., D. WuDunn, N. Soltys, and P. G. Spear. 1991. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 65:1090-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill, J. M., B. M. Gebhardt, R. Wen, A. M. Bouterie, H. W. Thompson, R. J. O'Callaghan, W. P. Halford, and H. E. Kaufman. 1996. Quantitation of herpes simplex virus type 1 DNA and latency-associated transcripts in rabbit trigeminal ganglia demonstrates a stable reservoir of viral nucleic acids during latency. J. Virol. 70:3137-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu, H., I. Solovyev, A. Colombero, R. Elliott, M. Kelley, and W. J. Boyle. 1997. ATAR, a novel tumor necrosis factor receptor family member, signals through TRAF2 and TRAF5. J. Biol. Chem. 272:13471-13474. [DOI] [PubMed] [Google Scholar]
- 28.Kaye, S. B., K. Baker, R. Bonshek, H. Maseruka, E. Grinfeld, A. Tullo, D. L. Easty, and C. A. Hart. 2000. Human herpesviruses in the cornea. Br. J. Ophthalmol. 84:563-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krummenacher, C., A. V. Nicola, J. C. Whitbeck, H. Lou, W. Hou, J. D. Lambris, R. J. Geraghty, P. G. Spear, G. H. Cohen, and R. J. Eisenberg. 1998. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J. Virol. 72:7064-7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krummenacher, C., A. H. Rux, J. C. Whitbeck, M. Ponce-de-Leon, H. Lou, I. Baribaud, W. Hou, C. Zou, R. J. Geraghty, P. G. Spear, R. J. Eisenberg, and G. H. Cohen. 1999. The first immunoglobulin-like domain of HveC is sufficient to bind herpes simplex virus gD with full affinity while the third domain is involved in oligomerization of HveC. J. Virol. 73:8127-8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krummenacher, C., I. Baribaud, M. Ponce de Leon, J. C. Whitbeck, H. Lou, G. H. Cohen, and R. J. Eisenberg. 2000. Localization of a binding site for herpes simplex virus glycoprotein D on herpesvirus entry mediator C by using antireceptor monoclonal antibodies. J. Virol. 74:10863-10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwon, B. S., K. B. Tan, J. Ni, K. O. Oh, Z. H. Lee, Y. J. Kim, S. Wang, R. Gentz, G. L. Yu, J. Harrop, S. D. Lyn, C. Silverman, T. G. Porter, A. Truneh, and P. R. Young. 1997. A newly identified member of the tumor necrosis factor receptor superfamily with a wide tissue distribution and involvement in lymphocyte activation. J. Biol. Chem. 272:14272-14276. [DOI] [PubMed] [Google Scholar]
- 33.Liesegang, T. J., L. J. Melton, P. J. Daly, and D. M. Ilstrup. 1989. Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn., 1950 through 1982. Arch. Ophthalmol. 107:1155-1159. [DOI] [PubMed] [Google Scholar]
- 34.Liesegang, T. J. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1-13. [DOI] [PubMed] [Google Scholar]
- 35.Liu, J., Z. Shriver, P. Blaiklock, K. Yoshida, R. Sasisekharan, and R. D. Rosenberg. 1999. Heparan sulfate d-glucosaminyl 3-O-sulfotransferase-3A sulfates N-unsubstituted glucosamine residues. J. Biol. Chem. 274:38155-38162. [DOI] [PubMed] [Google Scholar]
- 36.Lopez, M., F. Cocchi, L. Menotti, E. Avitabile, P. Dubreuil, and G. Campadelli-Fiume. 2000. Nectin2α (PRR2α or HveB) and nectin2δ are low-efficiency mediators for entry of herpes simplex virus mutants carrying the Leu25Pro substitution in glycoprotein D. J. Virol. 74:1267-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marsters, S. A., T. M. Ayres, M. Skubatch, C. L. Gary, M. Rothe, and A. Ashkenazi. 1997. Herpesvirus entry mediator, a member of the tumor necrosis factor receptor (TNFR) family, interacts with members of the TNFR-associated factor family and activates the transcription factors NF-κB and AP-1. J. Biol. Chem. 272:14029-14032. [DOI] [PubMed] [Google Scholar]
- 38.Martinez, W. M., and P. G. Spear. 2002. Amino acid substitution in the V domain of nectin-1 (HveC) that impair entry activity for herpes simplex virus type 1 and 2 but not for pseudorabies virus or bovine herpesvirus 1. J. Virol. 76:7255-7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milne, R. S., S. A. Connolly, C. Krummenacher, R. J. Eisenberg, and G. H. Cohen. 2001. Porcine HveC, a member of the highly conserved HveC/nectin 1 family, is a functional alphaherpesvirus receptor. Virology 281:315-328. [DOI] [PubMed] [Google Scholar]
- 40.Montgomery, R. I., M. S. Warner, B. J. Lum, and P. G. Spear. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427-436. [DOI] [PubMed] [Google Scholar]
- 41.O'Donnell, C., V. Tiwari, M. Jin-Oh, T. Valyi-Nagy, and D. Shukla. 2005. A role for 3-O-sulfotransferase isoform-2 in assisting HSV-1 entry and spread. Virology 346:452-459. [DOI] [PubMed] [Google Scholar]
- 42.Panoutsakopoulou, V., M. E. Sanchirico, K. M. Huster, M. Jansson, F. Granucci, D. J. Shim, K. W. Wucherpfennig, and H. Cantor. 2001. Analysis of the relationship between viral infection and autoimmune disease. Immunity 15:137-147. [DOI] [PubMed] [Google Scholar]
- 43.Parry, C., S. Bell, T. Minson, and H. Browne. 2005. Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J. Gen. Virol. 86:7-10. [DOI] [PubMed] [Google Scholar]
- 44.Patel, A. H., and J. B. Maclean. 1995. The product of the UL6 gene of herpes simplex virus type 1 is associated with virus capsids. Virology 206:465-478. [DOI] [PubMed] [Google Scholar]
- 45.Perez-Romero, P., A. Perez, A. Capul, R. Montgomery, and A. O. Fuller. 2005. Herpes simplex virus entry mediator associates in infected cells in a complex with viral proteins gD and at least gH. J. Virol. 79:4540-4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pertel, P. 2002. Human herpesvirus 8 glycoprotein B (gB), gH, and gL can mediate cell fusion. J. Virol. 76:4390-4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Polcicova, K., P. S. Biswas, K. Banerjee, T. W. Wisner, B. T. Rouse, and D. C. Johnson. 2005. Herpes keratitis in the absence of anterograde transport of virus from sensory ganglia to the cornea. Proc. Natl. Acad. Sci. USA 102:11462-11467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richart, S. M., S. A. Simpson, C. Krummenacher, J. C. Whitbeck, L. I. Pizer, G. H. Cohen, R. J. Eisenberg, and C. L. Wilcox. 2003. Entry of herpes simplex virus type 1 into primary sensory neurons in vitro is mediated by nectin-1/HveC. J. Virol. 77:3307-3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ritterband, D. C., and D. N. Friedberg. 1998. Virus infection of the eye. Rev. Med. Virol. 8:187-201. [DOI] [PubMed] [Google Scholar]
- 50.Scanlan, P. M., V. Tiwari, S. Bommireddy, and D. Shukla. 2003. Cellular expression of gH confers resistance to herpes simplex virus type-1 entry. Virology 312:14-24. [DOI] [PubMed] [Google Scholar]
- 51.Scanlan, P., V. Tiwari, S. Bommireddy, and D. Shukla. 2005. Spinoculation of heparan sulfate deficient CHO 745 cells enhances herpes simplex type-1 entry, but does not abolish the need for essential glycoproteins involved in the virus fusion mechanism. J. Virol. Methods 128:104-112. [DOI] [PubMed] [Google Scholar]
- 52.Schneider-Schaulies, J. 2000. Cellular receptors for viruses: links to tropism and pathogenesis. J. Gen. Virol. 81:1413-1429. [DOI] [PubMed] [Google Scholar]
- 53.Shieh, M. T., D. WuDunn, R. I. Montgomery, J. D. Esko, and P. G. Spear. 1992. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J. Cell Biol. 116:1273-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shukla, D., C. L. Rowe, Y. Dong, V. R. Racaniello, and P. G. Spear. 1999. The murine homolog (Mph) of human herpesvirus entry protein B (HveB) mediates entry of pseudorabies virus but not herpes simplex virus types 1 and 2. J. Virol. 73:4493-4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shukla, D., and P. G. Spear. 2001. Herpesviruses and heparan sulfate: an intimate relationship in aid of viral entry. J. Clin. Investig. 108:503-510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shukla, D., M. C. Dal Canto, C. L. Rowe, and P. G. Spear. 2000. Striking similarity of murine nectin-1α to human nectin-1α (HveC) in sequence and activity as a glycoprotein D receptor for alphaherpesvirus entry. J. Virol. 74:11773-11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shukla, D., J. Liu, P. Blaiklock, N. W. Shworak, X. Bai, J. D. Esko, G. H. Cohen, R. J. Eisenberg, R. D. Rosenberg, and P. G. Spear. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13-22. [DOI] [PubMed] [Google Scholar]
- 58.Spear, P. G. 1993. Entry of alphaherpesviruses into cells. Semin. Virol. 4:167-180. [Google Scholar]
- 59.Spear, P. G., R. Eisenberg, and G. Cohen. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1-8. [DOI] [PubMed] [Google Scholar]
- 60.Spear, P. G., and R. Longnecker. 2003. Herpesvirus entry: an update. J. Virol. 77:10179-10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tiwari, V., C. Clement, M. B. Duncan, J. Chen, J. Liu, and D. Shukla. 2004. A role for 3-O-sulfated heparan sulfate in cell fusion induced by herpes simplex virus type 1. J. Gen. Virol. 85:805-809. [DOI] [PubMed] [Google Scholar]
- 62.Tiwari, V., C. Clement, P. M. Scanlan, B. Y. J. T. Yue, and D. Shukla. 2005. A role for herpesvirus entry mediator as the receptor for herpes simplex virus 1 entry into primary human trabecular meshwork cells. J. Virol. 79:13173-13179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tiwari, V., C. O'Donnell, M. Jin-Oh, T. Valyi-Nagy, and D. Shukla. 2005. A role for 3-O-sulfotransferase isoform-4 in assisting HSV-1 entry and spread. Biochem. Biophys. Res. Commun. 338:930-937. [DOI] [PubMed] [Google Scholar]
- 64.Valyi-Nagy, T., V. Sheth, C. Clement, V. Tiwari, P. Scanlan, J. H. Kavouras, L. Leach, G. Guzman-Hartman, T. S. Dermody, and D. Shukla. 2004. Herpes simplex virus entry receptor nectin-1 is widely expressed in the murine eye. Curr. Eye Res. 29:303-309. [DOI] [PubMed] [Google Scholar]
- 65.Warner, M. S., R. J. Geraghty, W. M. Martinez, R. I. Montgomery, J. C. Whitbeck, R. Xu, R. J. Eisenberg, G. H. Cohen, and P. G. Spear. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246:179-189. [DOI] [PubMed] [Google Scholar]
- 66.Whitbeck, J. C., C. Peng, H. Lou, R. Xu, S. H. Willis, M. Ponce de Leon, T. Peng, A. V. Nicola, R. I. Montgomery, M. S. Warner, A. M. Soulika, L. A. Spruce, W. T. Moore, J. D. Lambris, P. G. Spear, G. H. Cohen, and R. J. Eisenberg. 1997. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the TNFR superfamily and a mediator of HSV entry. J. Virol. 71:6083-6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.WuDunn, D., and P. G. Spear. 1989. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 63:52-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xia, G., J. Chen, V. Tiwari, W. Ju, J. P. Li, A. Malmstrom, D. Shukla, and J. Liu. 2002. Heparan sulfate 3-O-sulfotransferase isoform 5 generates both an antithrombin-binding site and an entry receptor for herpes simplex virus, type 1. J. Biol. Chem. 277:37912-37919. [DOI] [PubMed] [Google Scholar]
- 69.Xu, D., V. Tiwari, G. Xia, C. Clement, D. Shukla, and J. Liu. 2005. Characterization of heparan sulphate 3-O-sulphotransferase isoform 6 and its role in assisting the entry of herpes simplex virus type 1. Biochem. J. 385:451-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yue, B. Y., E. J. Higginbotham, and I. L. Chang. 1990. Ascorbic acid modulates the production of fibronectin and laminin by cells from an eye tissue-trabecular meshwork. Exp. Cell Res. 187:65-68. [DOI] [PubMed] [Google Scholar]
- 71.Zhao, Z. S., F. Granucci, L. Yeh, P. A. Schaffer, and H. Cantor. 1998. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science 279:1344-1347. [DOI] [PubMed] [Google Scholar]