

Abstract
Herpes simplex virus (HSV) normally undergoes productive cytocidal infection in culture and is thought of as relatively resistant to innate immune responses such as interferon. We previously described an unusual pattern of infection in culture in MDBK cells, which after initial productive infection, surprisingly resulted in progressive suppression of replication and cell recovery. The dominance of the refractory state was due to the inability to suppress interferon production and subsequent paracrine signaling. Here, using a wild-type HSV-1 strain expressing green fluorescent protein (GFP)-VP16, we analyze aspects of long-term HSV persistence resulting from this oscillating refractory state. We show that the gradual suppression of GFP-VP16 expression correlated with a biphasic pattern of accumulation of viral DNA and extracellular virus titers. We quantify virus maintenance in a minor subpopulation of cells during subculture, show the reemergence of virus by infectious center assay, and demonstrate that this required intracellular events over a 24- to 48-h time course. We also demonstrate that conditioned medium (cMed) from infected cells induced a profound shutoff of HSV gene expression at the transcriptional level. Finally, we demonstrate that this suppression was extremely rapid, requiring only 1 h of treatment to essentially abolish HSV immediate-early expression, and surprisingly persisted for almost 2 days after removal of the cMed. These combined effects underpin the oscillating effect both in plaque progression, where infection spreads but is overwhelmed by the accumulation of inhibitory components, enabling cell recovery, and virus maintenance in a subpopulation of cells. These results may be relevant to consider in studies of HSV latency in different animal models.
In cell culture, herpes simplex virus (HSV) normally undergoes productive cycles of replication resulting in cell destruction and virus production (7). Replication is studied not only in human cells but in a wide range of cells, including those of primate, murine, canine, or bovine origin. In vivo, in its natural host, after an acute stage of productive infection in epithelial cells at the site of primary infection, the virus is transported to neuronal cells innervating the primary sites, where it undergoes a nonproductive infection resulting in latency. Periodic reactivation occurs whereby, after undergoing productive rounds of infection within reactivating cells, HSV is transported back to surface sites where it may undergo further rounds of replication (12, 42). Various systems have been established in culture to recapitulate this cyclical pathway of productive infection, repression, and reactivation. These systems usually involve the use of virus mutants defective in several functions and/or the use of inhibitors to suppress virus replication (10, 30, 31, 33, 36, 38, 41, 43).
Cells have evolved mechanisms of innate and acquired antiviral responses (7). One of the most important innate response mechanisms is the production and secretion of interferon (IFN) and the subsequent paracrine activation of signaling via IFN receptors (8). IFN-α and IFN-β are secreted by most cells in response to infection, while IFN-γ production is largely restricted to T cells and NK cells. IFN-α/β activate a common single receptor, while IFN-γ recognizes a separate receptor (37). IFN binding results in the activation of the Jak/Stat pathway and the ensuing induction of expression of antiviral components, such as double-stranded RNA-dependent protein kinase R (PKR), 2′,5′-oligoadenylate synthase (OAS), and RNase L (17, 34, 35, 37). By analysis of the IFN pathway after infection with HSV in the presence of protein synthesis inhibitors (26, 29) or infection with UV-inactivated or defective viruses (6, 21), virus binding or the entry of virion components appears to be sufficient to trigger expression of IFN-stimulated genes. In turn, HSV-1 products, synthesized de novo during normal infections with wild-type viruses, are very efficient blockers of such triggering (6, 9, 19, 21, 26, 29). For example, the HSV protein ICP34.5 recruits a cellular phosphatase to dephosphorylate the eukaryotic initiation factor 2α (eIF2α) and thus counteracts the activation of PKR (11). The HSV immediate-early protein ICP0 is also required and sufficient to repress the induction of IFN-stimulated genes (6, 9, 18), and mutant viruses lacking functional ICP0 were shown to be hypersensitive to IFN in culture (22, 23). These countermeasures are normally very effective, with the result that, e.g., the induction of ISG54, one of the most responsive genes observed during infection in the presence of cycloheximide, is undetectable during wild-type infection (26). HSV also promotes additional countermeasures, both ICP0 dependent and independent, to block the IRF3 pathway for production of IFN itself (18, 29). These effective countermeasures and others—e.g., suppression of Jak-Stat phosphorylation (44, 45) or reduction in Jak1 (5)—account for the general observation that wild-type HSV replication is comparatively resistant to IFN signaling and IFN-mediated responses in culture (6, 22, 26, 32). This notwithstanding, interferons, and in particular IFN-α/β, play a key role in vivo in controlling the early acute phase of HSV-1 infection (9, 15, 39).
In the present study, we further characterize HSV infection in a culture system in which infection does not block the production of IFN and where such IFN production results in a paracrine suppression of virus replication. With time, the result of the progressive resistance to infection is the apparent clearance of active replication and cell recovery (4). Here we show that virus is maintained in a minor subpopulation of cells in a type of “persistent” state which can be periodically reactivated. Those cells harboring reactivatable virus require 24 to 48 h of growth before infectious centers could be detected. This is consistent with additional results examining the potency and longevity of suppression. First, we show that conditioned medium (cMed) from a primary infection potently and rapidly suppresses gene expression even from a high-multiplicity of infection (MOI), with inhibition seen within 1 h after application to uninfected cells. Second, in reversal experiments, where cMed was applied and removed for various intervals prior to subsequent infection, we show that the inhibitory effect persists in cells for up to 2 days after reversal of the cMed.
The inability to repress IFN production in a cell-type-specific fashion and the consequent effect of virus suppression, together with the rapidity and longevity of the suppressed state, may have important implications for general studies of HSV-1 replication in culture and in animal models.
MATERIALS AND METHODS
Cells and viruses.
MDBK, MDBK/V (4), and Vero cells were grown in Dulbecco's modified minimal essential medium (DMEM), supplemented with 10% newborn calf serum (NBCS). Virus stocks used were HSV-1 [17] and HSV-1 [v44], which is an otherwise wild-type derivative of HSV-1 [17] expressing green fluorescent protein (GFP)-VP16 (14). Encephalomyocarditis virus (EMCV), was used in bioassays to estimate IFN activity in cMed.
Virus infection and plaque assays.
Cells were seeded in six-well cluster dishes at a density of 2 × 106 cells per dish such that monolayers were confluent the following day. Monolayers were infected with serial dilutions of virus in serum-free DMEM for 1 h, the inoculum was removed, and the cells were then incubated in medium containing 2% NBCS with or without 1% neutralizing human serum (hS) as indicated.
Analysis of individual plaque progression using HSV-1 expressing GFP-VP16.
MDBK cells were infected with a standard dose (2,000 PFU per well) of HSV-1 [v44]. GFP-VP16 expression was then monitored in live cells by confocal microscopy with a Zeiss LSM 410 confocal microscope using a ×20 magnification objective lens. The fate of single plaques was monitored by moving into position a marker objective (which contains an inked endpiece in place of a lens). The marker objective was raised to contact the dish, ringing an area which could then be relocated and repeatedly imaged. The same plaque was thus monitored every 24 h thereafter.
Virus titration during HSV-1 infection of MDBK cells.
MDBK monolayers were infected for 1 h with a standard dose (2,000 PFU) of HSV-1 [v44] and rinsed with 0.1 M glycine (pH 3.5) to inactivate the remaining inoculum, and DMEM containing 2% NBCS was then added. Aliquots of infected cell medium were then collected at time points during infection and stored at −70°C for subsequent virus titration in Vero cells. In parallel experiments, infected cell medium was replaced with fresh medium at 12 days post infection (dpi) and samples of infected medium were harvested thereafter and assayed in Vero cells.
PCR analysis of the viral genome.
Total genomic DNA was extracted from MDBK cells at different stages during infection using the DNeasy kit (QIAGEN), according to the manufacturer's instructions. DNA preparation included RNase treatment on the column and quantification performed using a NanoDrop ND-1000 spectrophotometer (Labtech). A standard dose (150 ng) of total DNA per sample was used for PCR analysis. PCR was performed with the Taq polymerase kit (QIAGEN) according to the manufacturer's instructions. To examine the viral genome, the following specific DNA primers for HSV-1 ICP0 were used: forward, 5′-GGGAAGATCTGAGGACGGGGGGAGCGAC-3′; and reverse, 5′-GGGAAGATCTGCTGGGCGTCACGCCCAC-3′.
Production of cMed.
cMed was produced as before (4) by infecting MDBK cells at an MOI of 1, harvesting medium 24 h later, and removing virus by filtration. The medium was then stored at −70°C, during which time IFN levels, as measured by virus inhibition or the ability to induce Stat1 phosphorylation, remained stable.
EMCV bioassay to analyze IFN activity in cMed.
MDBK cells, plated in triplicate in 96-well cluster dishes, were treated for 24 h with control medium or serial dilutions of cMed. Cells were then infected with 100 50% tissue culture infective doses (TCID50) of EMCV in DMEM containing 2% NBCS. Infection was stopped at 4 dpi, and monolayers were fixed with formaldehyde and stained with crystal violet. Cells were scored as infected (with disruption and clear background) or uninfected (with no or very partial disruption and stained background).
Subculture and propagation of infected MDBK cells.
MDBK cells were infected with a standard dose (2,000 PFU) of HSV-1 [v44], as described above and monitored daily by live-cell confocal microscopy. At 12 days, when GFP expression was virtually undetected and the monolayer appeared healthy, cells were trypsinized, split (1:20 ratio), and replated in fresh six-well cluster dishes in DMEM containing 10% NBCS and 1% hS. Subcultured cells were then daily monitored for GFP reappearance.
Quantification of infectious centers in repressed infected cultures.
MDBK cells infected for 12 days, as described above, with a standard dose (2,000 PFU) of HSV-1 [v44] were rinsed with warm phosphate-buffered saline (PBS), trypsinized, and counted. Cell dilutions were then plated immediately onto monolayers of Vero cells in the presence of hS, and infectious centers were counted 3 days later. Alternatively, samples of the trypsinized MDBK cells were diluted (1:20) and plated onto fresh culture dishes for various times, before retrypsinization and plating onto Vero monolayers for the infectious center assay (ICA).
Analysis of protein synthesis by pulse-labeling with [35S]methionine.
Vero, MDBK, and MDBK/V cells were plated as described above and treated or mock treated for 16 h with universal IFN (uIFN) (IFN αA/D; Sigma product no. 14401) at a standard dose of 1 U/μl in all experiments unless otherwise stated or with cMed obtained from a previous infection of MDBK cells. Cells were then rinsed several times with PBS and infected or mock infected with 10 MOI of HSV-1 [17]. At different times during infection, the medium was removed and cells were incubated for 1 h with methionine-free DMEM containing 10 μCi/ml of [35S]methionine (Amersham). Monolayers were then washed in cold PBS and lysed in sodium dodecyl sulfate (SDS) lysis buffer. Proteins were subsequently separated by SDS-polyacrylamide gel electrophoresis (PAGE) and analyzed by autoradiography.
Kinetics of repression of HSV protein synthesis by uIFN or cMed.
Two different aspects were analyzed in relation to the effects that IFN and cMed have on infected cell protein synthesis. First, cells were pretreated (either with uIFN or cMed), for different times, from 16 h to 1 h, prior to infection with HSV at an MOI of 10. The aim here was to address the length of time required to induce total selective shutoff of viral protein synthesis. In a second type of experiment, cells were treated for a standard time (16 h) and then washed several times and incubated in normal medium for different lengths of time prior to infection with HSV (MOI of 10). This was aimed at assessing the duration of the refractory state (induced by uIFN or cMed treatment) and the time taken to reverse this state allowing HSV gene expression.
Western blot analysis.
Cell lysates (produced during the cell metabolic labeling experiments) were separated by SDS-PAGE, and proteins were then transferred on nitrocellulose membranes that were then blocked with PBS containing 0.1% Tween 20 and 5% milk powder. After blocking, the membranes were incubated overnight at 4°C with the anti-ICP0 monoclonal antibody (11060; 1:1,000). The membranes were washed and processed using a horseradish peroxidase-conjugated secondary antibody, and proteins were detected by standard methods.
RNA isolation and RT-PCR analysis.
MDBK cells were treated for 16 h with a standard dose of uIFN or mock treated and then infected with HSV-1 [17] at an MOI of 10. At different time points after infection, cells were rinsed, trypsinized, and pelleted and the pellets were resuspended in 50 mM Tris-HCl, pH 8, 140 mM NaCl, 1.5 mM MgCl2 and 0.5% NP-40. Cytoplasmic RNA was extracted and purified using the RNeasy mini kit (QIAGEN). A DNase step was performed on the column according to the manufacturer's instructions. Reverse transcription-PCR (RT-PCR) was performed using the OneStep RT-PCR kit (QIAGEN), with a starting RNA concentration of 500 ng. The primers used for this step were described above.
RESULTS
Suppression and reactivation of HSV-1 infection in MDBK cells.
We previously reported that HSV infection in MDBK cells triggers production of a soluble mediator, including but not necessarily limited to interferon, and that this mediator promoted a progressive refractory state (4). To pursue this system further and in particular to examine the nature of repression and possible maintenance of virus in the recovered population, we made use of a derivative of HSV-1 [17] that expresses the GFP-VP16 protein in an otherwise wild-type background (HSV-1 [v44]) (14). In parallel, with analysis of plaque progression, we wished to examine the titer of infectious virus being produced in the infected monolayers and obtain a measure of the relative abundance of virus DNA. Cells were infected at an MOI of 0.02, and infection in the same plaque was monitored by confocal microscopy. A typical example is shown in Fig. 1. Plaques expressing GFP-VP16 were visible by 2 dpi (panel 1) and increased in size by 5 dpi, maintaining GFP-VP16 expression (panel 2). However, plaque progression was comparatively slow and thereafter actually regressed, with the result that by 12 dpi there was almost complete suppression of GFP-VP16 expression (panel 3), and by phase analysis, the monolayer appeared relatively healthy. Consistent with our previous results, replacement of the medium at this time (panel 4) caused the delayed reactivation of GFP-VP16 expression, which could be observed in the monolayer, initiating as small clusters and progressing by 5 days to encompass a renewed plaque expressing GFP-VP16 (panel 5).
FIG. 1.

HSV-1 suppression in MDBK cells. MDBK cells were infected with an MOI of 0.02 of HSV-1 [v44]. (a) Plaque progression was monitored by confocal microscopy on live cells. Images represent 2 (panels 1), 5 (panels 2), and 12 (panels 3) dpi. At 12 dpi, the medium was replaced with fresh medium in a population of cells, which were then imaged at 1 (panels 4) and 7 (panels 5) days post-medium replacement. (b) Samples of infected cell medium were harvested at the times indicated (x axis of the histogram), and their titers were determined on Vero cells, counting plaques after 3 days. Virus yield is expressed in PFU/ml (y axis). (c) Total (cellular/viral) DNA was extracted from cells (at the time points indicated) and quantified, and PCR was performed to amplify the ICP0 gene. As a negative control, DNA from uninfected cells (Un) was amplified for the absence of the ICP0 band (lane 1).
Titers of extracellular virus were examined under the same conditions. MDBK cells were infected as described above, and samples were harvested at different time points and subsequently titrated altogether on Vero cells (Fig. 1b). No measurable extracellular virus could be detected at 8 h postinfection (hpi), while significant titers were readily measurable by 1 dpi, reaching a peak by 5 dpi but decreasing quite dramatically thereafter. Interestingly, upon medium reversal at this time, while no extracellular virus was detected initially, low but significant levels of infectious virus were observed, gradually increasing at least up to 7 days after medium replacement.
We next used PCR analysis to obtain a more relative measure of the viral DNA content, from the initial productive infection through the establishment of the suppressed state. Infections were carried out as described above, and total DNA was extracted after RNase treatment. A standard dose per sample (150 ng) was then amplified using virus-specific primers: in this case for the ICP0 gene (Fig. 1c). DNA analysis followed a similar profile, although slightly in advance of the virus titer. DNA was first readily detected by 1 dpi (lane 3), reaching a peak at 2 dpi (lane 4) and decreasing thereafter (lanes 5 and 6). DNA did not completely disappear and was still detectable at 12 dpi (lane 7), at which time no GFP-VP16 expression was detected and the virus titer had reached very low levels. Upon medium reversal, DNA levels were initially similar to the levels at 12 dpi and, consistent with the results from assaying infectious virus titers, increased slowly thereafter (lanes 9 and 10).
Maintenance and propagation of virus in cell culture.
We next wished to examine whether during subcultivation and propagation of the infected cell population, virus was maintained in a form that could be reactivated. Cells were infected as before with HSV-1 [v44], and the progression of infection was monitored by confocal microscopy for 12 days. When the suppressed state of infection was established (Fig. 2, panel 1), the cells were trypsinized, split (1:20 ratio), and replated in fresh dishes. The cells were then cultured, and daily thereafter cell growth and any potential virus reactivation were monitored by phase and confocal microscopy. After 1 day (panel 2), the replated cells appeared flat and healthy and no GFP-VP16 expression could be detected. Similarly, by 2 days (panel 3) cells appeared normal and healthy, growing to confluence by about 3 days after plating (panel 4). GFP-VP16 expression was still not generally detectable at this stage. However, by this time, we could also observe, by scanning across the monolayer, individual plaques exhibiting significant GFP-VP16 (panel 5). Scanning the entire monolayer indicated that by this stage there were approximately 20 to 50 plaques expressing GFP-VP16 (see also below). Interestingly, if incubation of this monolayer was maintained, infection initially progressed with moderate increase in plaque size. But, as with the original infection, this progression was again suppressed, with inhibition of GFP-VP16 expression, contraction of plaque size, and cell recovery (data not shown).
FIG. 2.
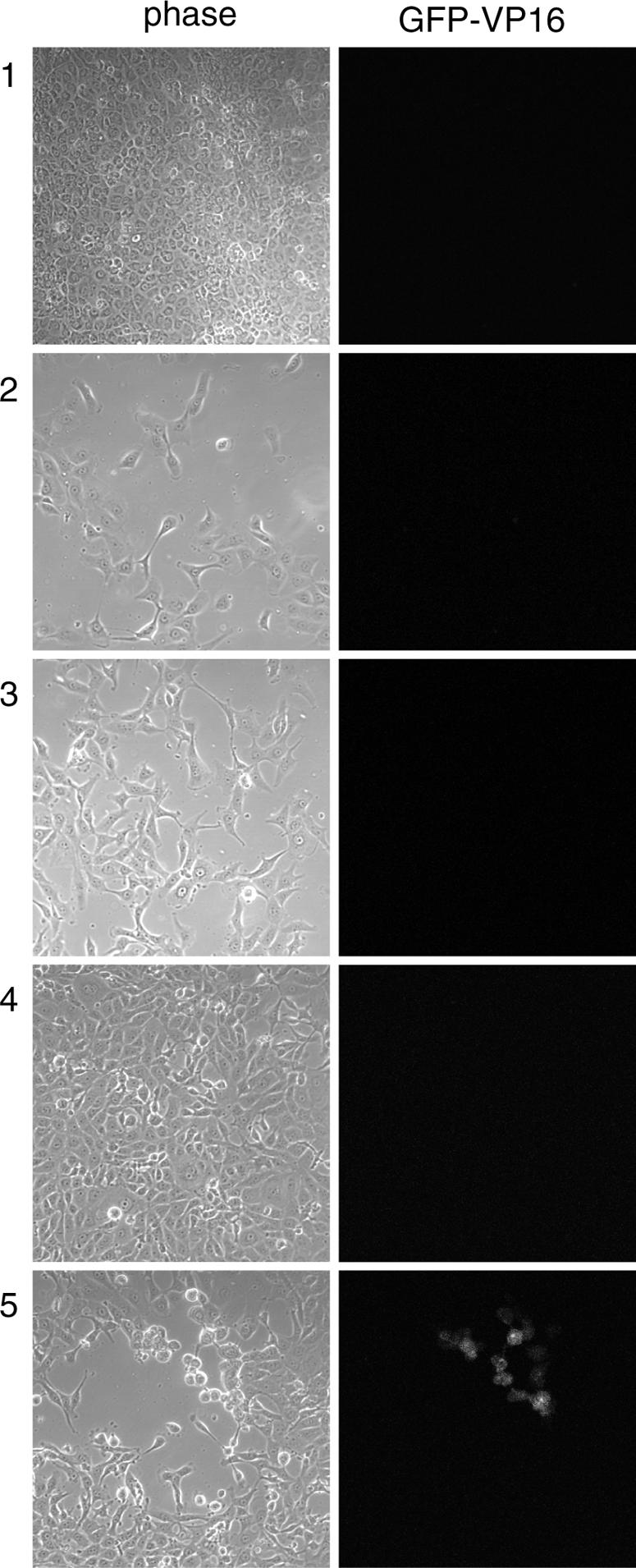
Propagation of an MDBK monolayer in a repressed state of infection. MDBK cells infected for 12 days with 2,000 PFU of HSV-1 [v44] were trypsinized, split (1:20 ratio), and replated in six-well cluster dishes. Cell progression was monitored thereafter by confocal microscopy. Images were taken at 12 dpi (panels 1) and 1 (panels 2), 2 (panels 3), and 3 (panels 4 and 5) days posttrypsinization.
Consistent with previous data, this analysis indicated absence of GFP-VP16 expression immediately after trypsinization and a lag phase to allow virus reappearance. We note that while we did not detect GFP-VP16 expression initially after subcultivation, it was formally difficult to rule out the presence of very small numbers of single cells expressing GFP-VP16. To analyze this further, we wished to examine the repressed infected cell monolayers by ICA. We wished to quantify what subpopulation of cells in the repressed state harbored virus which was able to be derepressed or “reactivated.”
MDBK monolayers were infected with HSV-1 [v44] at an MOI of 0.001. Plaques progressed as before regressing and establishing a repressed state of infection (12 dpi). Cells were trypsinized, counted, and immediately used to perform ICA by titration on Vero monolayers. In parallel, samples of the cell suspension (1:20) were plated in separate fresh dishes and these dishes were then incubated for 1, 2, 3, 4, 5, 6, and 10 days. At each subsequent time point, the replated cells were again trypsinized, counted, and immediately used for an ICA. This scheme and the results are summarized in Fig. 3. When we assayed for infectious centers in the original repressed state, without any cultivation period prior to assay, surprisingly no infectious centers were observed. However, if cells were cultivated for some time before trypsinization and ICA, infectious centers were now detectable. The number of infectious centers increased after 1 day in culture, to a maximum of between 30 and 40 after 4 days in culture. However, upon longer cultivation times, the number of infectious centers again declined and infectious centers completely disappeared after 10 days of cultivation.
FIG. 3.

ICA of MDBK cells in a repressed state of infection. An MDBK monolayer in a repressed state of infection, corresponding to 12 dpi, was trypsinized and split (1:20 ratio). Cells in the 1:20 split were counted, and one aliquot was immediately used to perform an ICA on fresh Vero cells, while other aliquots were plated in six-well cluster dishes, incubated for different lengths of time (t = 1 day, 2 days, etc), and then used to perform ICA on Vero cells. Vero monolayers were analyzed when cytopathic effects were visible, and plaques were counted. Results of the ICA are summarized above.
These results are consistent with the results from the earlier analyses (shown above). Together the data indicate that the repressed monolayer contained a small subpopulation of cells that harbored viral DNA in a “silent” state, unable to immediately propagate infection when assayed. They indicate that some type of lag phase is required, during which some event or events are needed to allow the appearance of infectious virus and initial replication, resulting in an increase in viral DNA content or increase in infectious centers. They also show the recapitulation of a progressive repression as gene expression declines and infectious centers disappear.
Potent suppression of infection by priming with cMed.
We have previously shown that pretreatment of fresh naïve MDBK cells with cMed causes a pronounced inhibition of plaque formation (4). Repressively infected MDBK cells are in the presence of IFN initially produced upon infection (4), and the delayed reactivation of infection observed after medium reversal is presumably due to delay in reversible events maintained by IFN signaling. We next wished to examine aspects of the kinetics of the establishment of repression and its longevity decided during pretreatment regimens and to compare the effects of cMed with IFN alone.
First, we wished to analyze whether after pretreatment of cells with cMed, primed cells would be less susceptible to the first events that characterize virus infection rather than, e.g., infection initiating with equal efficiency but not proceeding with production and cell-to-cell spread. Cells were treated with cMed or mock treated and 24 h later were infected with HSV-1 [v44] at an MOI of 0.1. Initial GFP-VP16-expressing cells were visualized at 14 hpi (Fig. 4a), and a clear decrease in the number of expressing cells was noticed in the treated cells (GFP-VP16 panels).
FIG. 4.

Ability of cMed to inhibit a new infection. (a) MDBK cells were treated (cMed) or mock treated (mock) for 24 h with cMed. Monolayers were then infected with HSV-1 [v44] at an MOI of 0.1. Initial numbers of infected cells were visualized and imaged at 14 hpi, using a confocal microscope. (b) MDBK cells were treated for 16 h with serial dilutions of cMed or uIFN (1 U/μl) as indicated (lanes 1 to 5). Monolayers were then infected with 100 TCID50 of EMCV and fixed and stained when evident cytopathic effects were visible. As a negative control, cells were pretreated with mock medium and then infected with 100 TCID50 of EMCV (lanes 7 to 12). Undil, undiluted.
We wished to verify whether the effect of cMed specifically inhibited HSV-1 infection or whether, as expected, cMed would function as a broad-range cytokine (such as IFN), also limiting other viruses. To verify this, we used an antiviral bioassay based on the reduction of cytopathic effects during EMCV infection. MDBK cells were plated in triplicate and treated with serial dilutions of either cMed or IFN as indicated. After 16 h, MDBK monolayers were infected with 100 TCID50 of EMCV and incubated until cytopathic effects were visible in the control cultures (Fig. 4b). Controls for EMCV infectivity of MDBK cells without prior treatment with cMed or uIFN (lanes 6 and 12), show pronounced cytopathic effect at the doses used. Both cMed and uIFN inhibited EMCV infection, with the results indicating the equivalent of approximately 0.5 to 1 U of IFN/μl in undiluted cMed.
cMed treatment selectively inhibits virus protein synthesis and transcription.
We next analyzed the effect of pretreatment with cMed on infected cell protein synthesis. In these experiments, we compared Vero cells, MDBK cells, and a derivative MDBK cell line in which IFN signaling has been effectively suppressed by the expression of the V protein of simian virus 5, which promotes Stat1 degradation (2, 4). Cells were treated or mock treated for 16 h with uIFN (1 U/μl) or cMed and then infected or mock infected with HSV-1 [17] at a high MOI (MOI of 10). At different time after infection, cells were pulse-labeled for 1 h with [35S]methionine and profiles of protein synthesis were analyzed by SDS-PAGE and autoradiography (Fig. 5a). Major viral products were readily observed in extracts from mock-treated cultures of each of the cell types (lanes 2, 3, and 4). Virus protein expression was only partially reduced in Vero cells pretreated with either uIFN or cMed, with, if anything, less effect of cMed (Vero panel, lanes 6 to 8 and 10 to 12). In marked contrast, in MDBK cells, the pattern of viral protein synthesis changed dramatically in response to pretreatment with either uIFN or cMed. There was virtually a complete shutoff of viral protein synthesis which persisted for the duration of the experiment up to 12 hpi (MDBK panel, lanes 6 to 8 and 10 to 12). This shutoff was selective to viral products and cellular protein levels were maintained in the pretreated cells (MDBK panel, lanes 8 and 12). This repression was clearly due to IFN signaling, since in cells (MDBK/V, bottom panel) in which the pathway was suppressed (by knockdown of Stat1), pretreatment with cMed or uIFN did not abrogate HSV protein expression. We note, though, there was a difference between cMed and uIFN pretreatment, in that in MDBK/V, cMed still reduced virus protein synthesis compared to that in untreated cells (cf. lanes 2 to 4 with 10 to 12), while the effect of IFN was completely abrogated (cf. lanes 2 to 4 with 6 to 8). This result may indicate that the effect of cMed may be more potent or may operate by additional pathways to IFN (see below). We did note that cMed had a minor effect on uninfected protein synthesis (e.g., cf lanes 1 and 9) the reasons for which are unknown, though it could be a general nonspecific effect of labeling in preincubated medium. However, the main conclusion of this analysis is that even with infection with 10 PFU/cell of wild-type HSV, unlike, e.g., Vero cells, pretreatment with uIFN and more so with cMed profoundly and selectively inhibited virus protein synthesis by mechanisms which HSV was unable to counteract.
FIG. 5.

Effects of IFN and cMed on protein synthesis. (a) Vero, MDBK, and MDBK/V cells were untreated (mock, from lane 1 to lane 4) or treated for 16 h with uIFN-α (from lane 5 to lane 8) or cMed (from lane 9 to lane 12) and then infected with HSV-1 [17] at an MOI of 10. At the time points indicated (hpi), cells were labeled and lysed and lysates were run on SDS-PAGE gels for analysis of total protein synthesis. As a control, cells were pretreated as described but left uninfected (lanes 1, 5, and 9, respectively). (b) Cell lysates corresponding to MDBK cells, mock or uIFN pretreated, from the previous experiment were separated by SDS-PAGE and probed by Western blotting for ICP0. Un, uninfected. (c) MDBK cells mock or uIFN-α pretreated for 16 h were infected as described above, and at the time points indicated, cytoplasmic RNA was extracted. RT-PCR was used to specifically amplify for ICP0 mRNA.
It seemed unlikely that the effect of cMed or uIFN pretreament was due to blocking early stages in adsorption or penetration, and we ruled this out by demonstrating no effect of either treatment on infectious center formation (data not shown). To examine whether the effect on protein expression was due to a posttranscriptional effect on translation of virus RNA or due to inhibition of virus transcription, we analyzed levels of protein and RNA for a specific immediate-early product, in this case ICP0 (Fig. 5b and c). ICP0 protein and RNA (Fig. 5b and c, lanes 2) were readily detectable at 2 to 4 hpi in infected control cells, increasing thereafter (lanes 3 and 4). In contrast, IFN pretreatment completely suppressed ICP0 expression at both protein and RNA levels at 4 and 8 hpi (lanes 5 and 6), with only very minor amounts detected at 12 hpi (lanes 7).
These data provide robust evidence that uIFN or cMed treatments caused virtually a total and selective shutoff of viral proteins and that this shutoff is likely to cause a profound inhibition of virus transcription.
Establishment and longevity of repressive effects of cMed.
In the experiments shown above, we routinely used a pretreatment time of 16 h. We next wished to determine the minimal length of treatment required to inhibit viral protein synthesis and in particular whether relatively short times were sufficient. Thus, MDBK cells were treated for different lengths of time (from 0 to 16 h) with uIFN (1 U/μl) or cMed. After treatment, cells were washed, infected, and labeled for 1 h as described above. Equivalent cell lysates were then processed by SDS-PAGE and autoradiography. For uIFN treatment (Fig. 6a), a decrease in viral protein synthesis was observed after approximately 4 h (lane 5). With longer times of IFN pretreatment, virus protein synthesis was further repressed and from about 6 to 8 h onwards of uIFN treatment, similar profiles, exhibiting residual virus protein synthesis, were observed (lanes 6 to 11). Treatment with cMed had a striking effect (Fig. 6b), causing a dramatic inhibition of viral protein synthesis after just 1 h (lane 15). With longer exposure with cMed (up to 6 to 8 h, lanes 16 to 19), there was a further reduction in virus protein synthesis, until with treatments of 10 h or longer (lanes 20 to 23), total shutoff of viral protein synthesis was observed. The rapidity of the effect of cMed on inhibition of virus protein synthesis was noteworthy, as it demonstrated the virtually complete inhibition of virus protein synthesis, whereas reduced, but residual levels of protein synthesis were observed with uIFN treatment, even after longer periods of treatment (see below and Discussion).
FIG. 6.

Differences between uIFN-α and cMed treatment on MDBK cells. Schematics depicting experimental timing are shown above the relevant data sets. MDBK cells treated with uIFN-α (a) or cMed (b) for the time indicated (h of treatment) were infected for 6 h with HSV-1 [17] at an MOI of 10 and then processed as described for Fig. 5a. Lanes 1 and 13 are uninfected cells, used as a control. MDBK cells treated for 16 h with uIFN-α (c) or cMed (d) were reversed in normal medium for the time indicated (h of reversal) and then infected and processed as previously described. As control, prior to infection, cells were incubated for 16 h without being treated with uIFN-α or cMed and then reversed in normal medium for 48 h (lanes 2 and 10) or 24 h (lanes 3 and 11).
We next examined the duration of the inhibitory effects of uIFN and cMed in treatment/reversal experiments. In these experiments, cells were treated for a standard duration (16 h) with a standard dose of uIFN or cMed. The cells were then washed several times and incubated in normal medium for different lengths of time, from 2 h to 48 h. After reversal, the cells were infected and labeled as before (labeling from 6 to 7 h postinfection) and lysates were analyzed by SDS-PAGE and autoradiography gel. For uIFN treatment (Fig. 6c), partial recovery of viral protein synthesis could be observed after 2 h of reversal (cf., Fig. 6c, lane 8, with Fig. 6a, lane 11). With 8 h of reversal (Fig. 6c, lane 7), increased levels of virus protein synthesis were detected, representing almost the maximal recovery observed. Increased durations of reversal resulted in only modest further increases in virus protein synthesis (lanes 7 to 4). The time course of the reversal of cMed treatment appeared qualitatively different (Fig. 6d). Thus, very low levels of viral products were observed with reversal times from 2 up to 24 h (lanes 16 to 13)—even after 2 days of reversal (lane 12), virus protein synthesis exhibited only marginal recovery.
Taken together these experiments indicate several points. First, it appears likely that the repressive effects of cMed, while certainly mediated through the IFN and and the Stat pathway (4), may involve additional inhibitory cytokines whose combinations make a more powerful repressive effect than uIFN alone. Second, whatever the precise identity of these components or combinations, these results demonstrate a very powerful, rapid, and long-lasting effect such that 1 h of treatment has the ability to substantially suppress gene expression even from a high-multiplicity infection with wild-type HSV. Moreover, this repressive effect is surprisingly long-lasting such that even after 1 to 2 days of reversal, a significantly repressed environment pertains within cells. As discussed below, these combined effects are likely to underpin the oscillating effect both in plaque progression, where infection initiates and spreads but is overwhelmed by the accumulation of inhibitory components enabling cell recovery, and the maintenance of virus in a population of cells during cell passage. These results may also be relevant to consider in studies of HSV latency in different animal models.
DISCUSSION
In studies with tissue culture, herpes simplex virus normally undergoes a productive cycle of replication resulting in cell destruction and virus production. In vivo, after acute-stage productive infection, HSV undergoes a nonproductive infection in neurons resulting in latency. Periodic reactivation occurs, resulting in a productive infection and virus transport back to surface sites where further rounds of productive replication ensue (7). Various systems have been pursued to recapitulate aspects of the cyclical pathway of productive infection, repression, and reactivation, usually involving the use of virus mutants and/or the use of inhibitors, e.g., acycloguanosine and cycloheximide (10, 30, 31, 36, 38, 41, 43). Here we expand on the characterization of an unusual pattern of infection with wild-type HSV-1 in MDBK cells, which results in the progressive suppression of replication, cell recovery and regrowth of infected monolayers, and maintenance of virus in a cyclical manner by virtue of a paracrine signaling involving the IFN pathway (4). In these cells, HSV-1 triggers and is unable to counteract the IFN pathway. In turn, IFN and possibly other cytokines are responsible for the establishment of a refractory state that extinguishes virus replication and allows the maintenance of HSV-1 in a subpopulation of cells from which infectious virus can reemerge. We show that the biphasic nature of initial cell destruction and then monolayer recovery is paralled by a biphasic production of infectious virus and DNA abundance. By 12 to 14 days after infection, DNA had peaked and decreased but was clearly still present. We have demonstrated that IFN is continually present in these cultures, maintaining suppression. Upon reversal, we observed increased DNA content and renewed virus production, underlining the maintenance of potentially infectious virus in a suppressed state.
Interestingly, resumed DNA synthesis and virus production took place after a significant lag of several days. We wished to examine whether this was mainly a quantitative effect, in that only very low numbers of cells harbored virus, or whether IFN (and any additional cytokines) induced a long-lasting suppressive effect that was maintained long after removal. The results indicated that both explanations likely contributed to the delayed time course of virus emergence. Analysis by infectious center assay indicated that only a small population of cells (<1:104) harbored infectious virus. However, consistent with a lag phase upon medium reversal in the primary population, when the population was trypsinized and assayed immediately, no infectious centers were detected. If the cells were plated for 1 to 2 days prior to assay, then infectious centers were readily detected. This apparent delay could be due to a quantitative effect, with infectious centers initially being below the level of detection and plating serving to allow some limited amplification of virus, which then registers as infectious centers. However, the results examining the population at the single-cell level, using virus expressing GFP-VP16, indicated that individual cells expressing the protein were observed only several days after removal of the cMed. Therefore, there is also a qualitative delay, with some event(s) required within the harboring cells to convert them to infectious centers. This is consistent with the results discussed below.
To further explore the dynamics of this virus-host interaction, we examined the time required for IFN to exert its repressive effect on the one hand and the duration of the effect on the other. Prior treatment of MDBK cells with uIFN or conditioned medium had a very dramatic inhibitory effect on HSV protein synthesis even after a high-MOI infection, and we show that this was likely due to inhibition at the transcriptional level. In contrast, parallel treatment of Vero cells (which while unable to produce IFN are IFN responsive) had a relatively modest effect on HSV protein synthesis. These results are consistent with the interpretation that HSV both triggers a potent IFN response and is unable to overcome that response in primed cells. By varying the duration of uIFN or cMed treatment, we make two conclusions. First, cMed appeared to have a more potent inhibitory effect than uIFN alone, which as discussed previously may indicate that while IFN and Stat signaling are clearly involved, other cytokines could contribute to the suppressive effects of cMed. This is consistent with previous results, for example, showing synergistic effects of combined IFN-α/β treatment (9, 32), but other cytokines could presumably also enhance IFN effects. Second, while pretreatment with IFNs has been shown previously to inhibit HSV replication and gene expression (1, 20, 25, 27), this inhibition is relatively modest in comparison to the effects on, e.g., RNA viruses, and for HSV even the combined effects if IFN-α/β and -γ are not potent at high multiplicities of infection (32). However, in the current system under study, and examining high-multiplicity infection, even within 1 h of treatment, cMed had induced a very significant inhibitory effect, which was virtually complete by 6 h. For uIFN alone, a delay in the onset of the inhibitory effect was observed. Surprisingly, but consistent with a difference between uIFN alone and cMed, in experiments where infection was delayed for progressively longer periods after reversal, cMed had a more sustained effect than uIFN. Thus, even 2 days after cMed removal, the MDBK cells retained a suppressed state and after a high-MOI infection only limited virus protein synthesis ensued.
Our results also raise several intriguing questions on the mechanism of inhibition. As indicated above, several reports have demonstrated the ability of HSV to block interferon production (19), to block the induction of interferon-stimulated genes (6, 18, 25), and to block interferon-mediated signaling (5). Furthermore, although HSV immediate-early gene expression has been shown to be inhibited by IFN pretreatment (1, 25, 27, 40), this inhibition is relatively weak. For example, in human fetal lung cells, HSV transcription was inhibited by three- to fivefold after treatment with IFN-α for 24 h, and this required the presence of cycloheximide to block virus protein synthesis (25). We show that, within 60 min of treatment with cMed, a potent inhibition of virus protein synthesis was observed, and this was due to transcription, rather than inhibition of protein synthesis per se, although the latter may well operate in addition. This inhibition ensued in the face of a high-MOI infection with wild/type virus. Currently there is no information on the mechanism by which IFN pretreatment inhibits HSV gene expression, although we know that inhibition is not likely to be at the level of adsorption or entry, since similar numbers of infectious centers are established with or without cMed pretreatment. Interestingly, in addition to candidates such as OAS and PKR, several recent publications have highlighted the importance of the bovine Mx gene products. It has been shown that these proteins, which are constitutively expressed at low levels in MDBK cells, are also upregulated upon IFN-α/β treatment (13, 24), and compared to the human Mx proteins, they exhibit a broader ability to block different viruses when expressed in Vero cells (3, 16). Whether or not mechanisms may be involved similar to those operating in other situations (i.e., in cells of other animal species which are used to study replication and pathogenesis), this system may, by virtue of its potency, allow insight into the mechanism which suppresses HSV gene expression and by which the refractory state is maintained even in the absence of the exogenous agent.
Together, these results also raise interesting questions regarding the dynamics of cell-virus interactions, suppression, persistence, and reemergence. We have shown that the suppressive effect of cMed (and uIFN) is extremely stable. A single dose of uIFN applied to cells and incubated at 37°C remains active and able to signal for at least 2 weeks. Furthermore, even when the external component(s) is removed (i.e., production ceases or, if production has already ceased, removing cMed from the site), cells can remain refractory to new infection for days. Thus, it could readily be envisaged that this situation could modulate dynamics of virus-host interaction. The extracellular in situ longevity of the components themselves and secondarily the duration of their intracellular effect could modulate the rate of production, suppression, and “reemergence” of infectious virus. Due to its stability, IFN could remain in situ long after any stimulus promoting its expression had disappeared. Furthermore, even when removed, the suppressive effect on recipient cells could remain in effect, until in the attempt to reemerge, a new round of virus stimuli induces further expression of the cytokine, restoring the suppressive effect. The extent to which this might operate in vivo is unclear, but IFN and IFN signaling are known to play a role in vivo in different animal model systems (9, 15, 28, 39). While paracrine suppressive mechanisms may not account for other models of refractory virus-host cell interactions in culture, such as those employing inhibitors or virus deletion variants, further studies with this system should yield useful insight into the nature of this state, how it is maintained for a long duration, the nature of the suppressed state, and key steps in its reversal.
Acknowledgments
This work is supported by Marie Cure Cancer Care.
REFERENCES
- 1.Altinkilic, B., and G. Brandner. 1988. Interferon inhibits herpes simplex virus-specific translation: a reinvestigation. J. Gen. Virol. 69:3107-3112. [DOI] [PubMed] [Google Scholar]
- 2.Andrejeva, J., D. F. Young, S. Goodbourn, and R. E. Randall. 2002. Degradation of STAT1 and STAT2 by the V proteins of simian virus 5 and human parainfluenza virus type 2, respectively: consequences for virus replication in the presence of alpha/beta and gamma interferons. J. Virol. 76:2159-2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baise, E., G. Pire, M. Leroy, J. Gerardin, N. Goris, K. De Clercq, P. Kerkhofs, and D. Desmecht. 2004. Conditional expression of type I interferon-induced bovine Mx1 GTPase in a stable transgenic Vero cell line interferes with replication of vesicular stomatitis virus. J. Interferon Cytokine Res. 24:513-521. [DOI] [PubMed] [Google Scholar]
- 4.Barreca, C., and P. O'Hare. 2004. Suppression of herpes simplex virus 1 in MDBK cells via the interferon pathway. J. Virol. 78:8641-8653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chee, A. V., and B. Roizman. 2004. Herpes simplex virus 1 gene products occlude the interferon signaling pathway at multiple sites. J. Virol. 78:4185-4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eidson, K. M., W. E. Hobbs, B. J. Manning, P. Carlson, and N. A. DeLuca. 2002. Expression of herpes simplex virus ICP0 inhibits the induction of interferon-stimulated genes by viral infection. J. Virol. 76:2180-2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flint, S. J., L. W. Enquist, R. M. Krug, V. R. Racaniello, and A. M. Skalka. 2000. Principles of virology. ASM Press, Washington, D.C.
- 8.Goodbourn, S., L. Didcock, and R. E. Randall. 2000. Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol. 81:2341-2364. [DOI] [PubMed] [Google Scholar]
- 9.Harle, P., B. Sainz, Jr., D. J. Carr, and W. P. Halford. 2002. The immediate-early protein, ICP0, is essential for the resistance of herpes simplex virus to interferon-alpha/beta. Virology 293:295-304. [DOI] [PubMed] [Google Scholar]
- 10.Harris, R. A., and C. M. Preston. 1991. Establishment of latency in vitro by the herpes simplex virus type 1 mutant in1814. J. Gen. Virol. 72:907-913. [DOI] [PubMed] [Google Scholar]
- 11.He, B., M. Gross, and B. Roizman. 1997. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1 alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiator factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 94:843-848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hill, T. 1985. Herpes simplex virus latency, p. 175-240. In B. Roizman (ed.), The herpesviruses, vol. 3. Plenum Press, New York, N.Y. [Google Scholar]
- 13.Horisberger, M. A., and M. C. Gunst. 1991. Interferon-induced proteins: identification of Mx proteins in various mammalian species. Virology 180:185-190. [DOI] [PubMed] [Google Scholar]
- 14.La Boissière, S., A. Izeta, S. Malcomber, and P. O'Hare. 2004. Compartmentalization of VP16 in cells infected with recombinant herpes simplex virus expressing VP16-green fluorescent protein fusion proteins. J. Virol. 78:8002-8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leib, D. A., T. E. Harrison, K. M. Laslo, M. A. Machalek, N. J. Moorman, and H. W. Virgin. 1999. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med. 189:663-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leroy, M., G. Pire, E. Baise, and D. Desmecht. 2006. Expression of the interferon-alpha/beta-inducible bovine Mx1 dynamin interferes with replication of rabies virus. Neurobiol. Dis. 21:515-521. (First published 3 October 2005; doi: 10.1016/j.nbd.2005.08.015.) [DOI] [PubMed] [Google Scholar]
- 17.Levy, D. E., and A. Garcia-Sastre. 2001. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 12:143-156. [DOI] [PubMed] [Google Scholar]
- 18.Lin, R., R. S. Noyce, S. E. Collins, R. D. Everett, and K. L. Mossman. 2004. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 78:1675-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melroe, G. T., N. A. DeLuca, and D. M. Knipe. 2004. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol. 78:8411-8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mittnacht, S., P. Straub, H. Kirchner, and H. Jacobsen. 1988. Interferon treatment inhibits onset of herpes simplex virus immediate-early transcription. Virology 164:201-210. [DOI] [PubMed] [Google Scholar]
- 21.Mossman, K. L., P. F. Macgregor, J. J. Rozmus, A. B. Goryachev, A. M. Edwards, and J. R. Smiley. 2001. Herpes simplex virus triggers and then disarms a host antiviral response. J. Virol. 75:750-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mossman, K. L., H. A. Saffran, and J. R. Smiley. 2000. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 74:2052-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mossman, K. L., and J. R. Smiley. 2002. Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon-induced barriers to virus replication. J. Virol. 76:1995-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Müller-Doblies, D., M. Ackermann, and A. Metzler. 2002. In vitro and in vivo detection of Mx gene products in bovine cells following stimulation with alpha/beta interferon and viruses. Clin. Diagn. Lab. Immunol. 9:1192-1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nicholl, M. J., and C. M. Preston. 1996. Inhibition of herpes simplex virus type 1 immediate-early gene expression by alpha interferon is not VP16 specific. J. Virol. 70:6336-6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicholl, M. J., L. H. Robinson, and C. M. Preston. 2000. Activation of cellular interferon-responsive genes after infection of human cells with herpes simplex virus type 1. J. Gen. Virol. 81:2215-2218. [DOI] [PubMed] [Google Scholar]
- 27.Oberman, F., and A. Panet. 1988. Inhibition of transcription of herpes simplex virus immediate early genes in interferon-treated human cells. J. Gen. Virol. 69:1167-1177. [DOI] [PubMed] [Google Scholar]
- 28.Openshaw, H., J. I. McNeill, X. H. Lin, J. Niland, and E. M. Cantin. 1995. Herpes simplex virus DNA in normal corneas: persistence without viral shedding from ganglia. J. Med. Virol. 46:75-80. [DOI] [PubMed] [Google Scholar]
- 29.Preston, C. M., A. N. Harman, and M. J. Nicholl. 2001. Activation of interferon response factor-3 in human cells infected with herpes simplex virus type 1 or human cytomegalovirus. J. Virol. 75:8909-8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Preston, C. M., and J. Russell. 1991. Retention of nonlinear viral DNA during herpes simplex virus latency in vitro. Intervirology 32:69-75. [DOI] [PubMed] [Google Scholar]
- 31.Russell, J., N. D. Stow, E. C. Stow, and C. M. Preston. 1987. Herpes simplex virus genes involved in latency in vitro. J. Gen. Virol. 68:3009-3018. [DOI] [PubMed] [Google Scholar]
- 32.Sainz, B., Jr., and W. P. Halford. 2002. Alpha/beta interferon and gamma interferon synergize to inhibit the replication of herpes simplex virus type 1. J. Virol. 76:11541-11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samaniego, L. A., L. Neiderhiser, and N. A. DeLuca. 1998. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72:3307-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Samuel, C. E. 2001. Antiviral actions of interferons. Clin. Microbiol. Rev. 14:778-809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sen, G. C. 2001. Viruses and interferons. Annu. Rev. Microbiol. 55:255-281. [DOI] [PubMed] [Google Scholar]
- 36.Shiraki, K., and F. Rapp. 1989. Protein analysis of herpes simplex virus latency in vitro established with cycloheximide. Virology 172:346-349. [DOI] [PubMed] [Google Scholar]
- 37.Stark, G. R., I. M. Kerr, B. R. Williams, R. H. Silverman, and R. D. Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227-264. [DOI] [PubMed] [Google Scholar]
- 38.Stuart-Jamieson, D. R., L. H. Robinson, J. I. Daksis, M. Nicholl, and C. M. Preston. 1995. Quiescent viral genomes in human fibroblasts after infection with herpes simplex virus type 1 Vmw65 mutants. J. Gen. Virol. 76:1417-1431. [DOI] [PubMed] [Google Scholar]
- 39.Su, Y.-H., J. E. Oakes, and R. N. Lausch. 1990. Ocular avirulence of a herpes simplex virus type 1 strain is associated with heightened sensitivity to alpha/beta interferon. J. Virol. 64:2187-2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thiele, K., S. Mittnacht, and H. Kirchner. 1989. Persistent replication of herpes simplex virus type 1 in JOK-1 cells. J. Gen. Virol. 70:1907-1911. [DOI] [PubMed] [Google Scholar]
- 41.Wigdahl, B., A. C. Scheck, R. J. Ziegler, E. De Clerq, and F. Rapp. 1984. Analysis of the herpes simplex virus genome during in vitro latency in human diploid fibroblasts and rat sensory neurons. J. Virol. 49:205-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wildy, P., H. J. Field, and A. A. Nash. 1982. Classical herpes latency revisited, p. 133-167. In B. W. J. Mahy, A. C. Minson, and G. K. Darby (ed.), Symposium 33, Society for General Microbiology. Cambridge University Press, Cambridge, United Kingdom.
- 43.Wu, N., S. C. Watkins, P. A. Schaffer, and N. A. DeLuca. 1996. Prolonged gene expression and cell survival after infection by a herpes simplex virus mutant defective in the immediate-early genes encoding ICP4, ICP27, and ICP22. J. Virol. 70:6358-6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yokota, S., N. Yokosawa, T. Kubota, T. Suzutani, I. Yoshida, S. Miura, K. Jimbow, and N. Fujii. 2001. Herpes simplex virus type 1 suppresses the interferon signaling pathway by inhibiting phosphorylation of STATs and janus kinases during an early infection stage. Virology 286:119-124. [DOI] [PubMed] [Google Scholar]
- 45.Yokota, S.-I., N. Yokosawa, T. Okabayashi, T. Suzutani, S. Miura, K. Jimbow, and N. Fujii. 2004. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 78:6282-6286. [DOI] [PMC free article] [PubMed] [Google Scholar]