

Abstract
Electrochemical biosensors have revolutionized glucose monitoring but have not yet fulfilled their promise of a low cost, direct detection replacement for genetic amplification tests such as PCR [K. Kerman, M. Kobayashi, E. Tamiya, Recent trends in electrochemical DNA biosensor technology, Meas. Sci. Technol. 15 (2004) R1–R11; A. Chaubey, B.D. Malhotra, Mediated biosensors. Biosens. Bioelectron. 17 (6–7) (2002) 441–456]. It has been anticipated that the integration of nanoscale chemical structures such as self-assembled monolayers with electrochemical biosensors would increase sensitivity by decreasing inherent system noise. We have designed a novel biosensing approach incorporating such integration and achieved rapid, ultra-low concentration sensitivities without target amplification. Raw samples are mixed with lysis buffer to allow hybridization of nucleic acid targets with anchor and signal probes before immobilizing a signaling enzyme proximate to the biosensor surface. A bias potential is subsequently applied and the secondary byproduct of a cyclic peroxidase reaction measured. Further studies have demonstrated the application of our approach in protein, clinical chemistry, and ionic assays.
Keywords: Electrochemical detection, Cyclic enzymatic reaction, Bionanotechnology, Genetic assay, Immunoassay, Simultaneous multi-channel detection
1. Introduction
Biosensors combine biochemical recognition with signal transduction for the detection of specific molecules. The recognition component, such as a nucleic acid sequence, an antibody, an enzyme, or other biomolecule, specifically binds to or catalyzes a reaction with the target of interest. The transducer component transforms this recognition event into a measurable signal. Together, these elements enable the specific detection of targets in complex samples.
These analytical tools have been utilized in a variety of industrial applications ranging from clinical diagnostics and environmental monitoring to biothreat detection and forensics. Their broad applicability comes from their promise of overcoming the limitations of traditional measurement techniques, which can be expensive, complicated, and slow.
Biosensors can be categorized by the reactant that they measure after the recognition event. Specifically, biosensor types include optical (light), bioluminescent (photons), thermal (heat), mass (resonance frequency changes), and electrochemical (electron transfer) [3]. While each of these types has inherent strengths and weaknesses, optical and electrochemical biosensors have become the most widely used.
Optical biosensors have been adopted throughout clinical diagnostics and life science research due to their speed and sensitivity compared with other techniques. However, most optical biosensors still require the use of target amplification such that the signal is enhanced to a measurable level. Of these amplification methods, real-time PCR has emerged as the most widely adopted and is now considered the gold standard for the detection of nucleic acids from a variety of origins [4]. Nevertheless, real-time PCR has several limitations and its avoidance remains a holy grail for the industry. Of primary importance are its time requirement for sample processing and amplification, false positives from sample or reagent contamination, and high instrumentation and labor costs.
Electrochemical biosensors promise superior speed and selectivity at a low cost [1], [2], [5]. Historically, however, electrochemical biosensors lacked sufficient sensitivity for use beyond glucose monitoring and clinical chemistry analysis. Recently, however, companies such as GeneFluidics have developed platforms that address this sensitivity question and point to electrochemical biosensors’ widespread use in the near future [11].
2. Novel electrochemical detector array
2.1. Overview
This platform consists of a novel electrochemical detection system embodied in disposable, multi-assay sensor chips and a user-friendly potentiostat reader instrument (Fig. 1 A). The reader system can acquire pico ampere current signal from the sensor array chip through a chip mount interface (Fig. 1B). Raw samples such as blood or urine can be directly applied onto the sensor chip (Fig. 1C). Unlike previously reported electrochemical DNA sensors that used graphite or carbon electrodes, we use a single layer of gold for all three electrodes, i.e., working, auxiliary, and reference electrodes. Typically, Ag/AgCl or a saturated calomel electrode (SCE) is used as the reference electrode so that reversible oxidation/reduction occurs at a fixed potential at the reference electrode. In contrast, we use Au as the reference electrode because its properties of malleability and durability simplify fabrication and allow use of extremely thin electrodes. In our particular application, where the reduction of a mediator is monitored, Au can be successfully used as the reference electrode because a low voltage difference is maintained for short periods of time. The Au/Au/Au electrode system is characterized by cyclic voltammetry and targets are detected by amperometry.
Fig. 1.

(A) Multi-channel potentiostat reader, (B) sensor chip mount, and (C) well structure on sensor array chip.
2.2. Optical-grade sensor surface
We have developed a robust and proprietary manufacturing process that enables the direct deposition of gold onto silicon, glass, or plastic. It is this process that provides the optical-grade flat sensors upon which extremely uniform, stable, and “pinhole-free” self-assembled monolayers (SAMs, described below), can be constructed. This optical-grade smooth surface is essential to manufacture a non-conductive SAM with dense packing density free of deposition defects. SAM integrity is essential because any leakage current due to structural defects would result in high background signal. As shown in Fig. 2 , a comparison of the X and Y units with those of the Z-axis over the 20 μM × 60 μm area elucidates the exceptional smoothness of our 50 nm thick gold sensor.
Fig. 2.
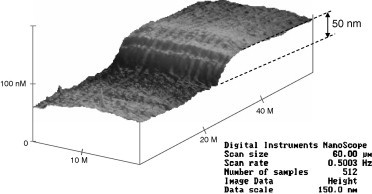
Optical-grade sensor surface with 50 nm Au electrode.
2.3. Nanoscale surface treatment
The interface between engineering transducers and biological assays plays an important role in the sensitivity and specificity of a biosensor. Currently, interfaces consisting of SAMs are of great interest because of their potential applications in electronic surface treatment, protective passivation, adhesion, and nanotechnology [6], [7], [8], [9]. SAMs of organosulfurs on gold films have been shown to form highly organized and ordered systems, which are suitable for membrane mimetic studies such as molecular recognition, catalysis, and cellular interactions. The difference between SAMs and other chemical binding techniques is that SAMs can provide specific and uniform molecular bonding.
SAMs are widely used to chemically modify surfaces by forming a thin layer of uniform molecules with or without exposed functional terminal groups. Alkanethiols can tether organic compounds or be terminated with alcohol (ethylene glycol) groups to form SAMs that resist the adsorption of proteins or nucleic acids (so-called ‘inert surfaces’). These alkanethiols, when used in mixed SAMs, prevent biomolecular interactions of interest from producing non-specific effects.
Fig. 3A shows the dense molecular stems on the surface of our electrochemical sensor with the optimal number of recognition binding sites. We uniformly build our SAM across two dimensions with an optimized ratio between the spacing stem and the recognition stem. The spacing stem provides electrical shielding from conductive raw specimens such as blood and urine, while the recognition stem immobilizes the target/probe hybrid onto the sensor surface. This provides the advantages of liquid phase hybridization and solid phase detection. Note that the high density of our SAM also makes it an electrical insulator (Fig. 3B), a unique property that facilitates the quality control of each sensor chip batch and ensures that leakage current is minimal.
Fig. 3.
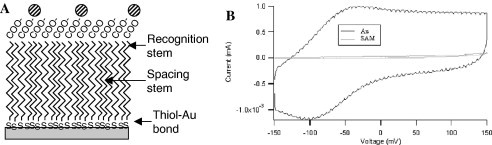
(A) Structural detail of the self-assembled monolayer (SAM), (B) comparison of cyclic voltammetry of a bare Au and SAM-treated sensor using potassium ferric cyanide.
A second aspect to our nanoscale surface treatment is our simultaneous use of three chemical blockers. In our “pre-cap” protocol, in which we immobilize our anchor probe onto the sensor surface, we add a slightly negatively charged material such as polyethylene glycol (PEG) on top of the pre-cap layer. We have shown that this layer reduces the non-specific binding of oligonucleotide probes, which are also slightly negatively charged. A second blocker is bovine serum albumin (BSA), which is added to the probe solution and mixed with the sample prior to introduction onto the sensor surface. BSA coats the SAM, reducing background by decreasing non-specific binding to the sensor surface. The third blocker is casein, which is included with our enzyme solution. Casein works much like BSA, but primarily blocks the enzyme from binding to the SAM.
2.4. Conjugation flexibility
Our electrochemical sensor array chip can be constructed with various binding chemistries. The most commonly used chip is built with a uniform layer of streptavidin binding sites on the working electrode surface. This chip can be used for either genetic or immunologic assays when functionalized with biotinylated oligonucleotide probes or biotinylated monoclonal antibodies, respectively, as target-anchoring agents.
Other immobilization chemistries such as thiol:gold, FITC:anti-FITC Ab, carboxyl:amine and direct bonding provide the user with a variety of potential methods for coupling anchor molecules to the sensor surface. This enables the user to maintain previously studied binding kinetics while adapting the sensor platform to new detection strategies. All chips are fabricated by our manufacturing partner, U-tech, in a clean room facility and meet the highest standards for chemical stability, reproducibility, and low cumulative variance.
3. Electrochemistry
3.1. Analytical electrochemistry
A major difference between our genetic detection array and the DNA microarrays currently available is the detection mechanism applied. Our on-chip detector is based on electrochemical detection, while others primarily rely on off-chip optical detection with image processing. To derive a quantitative correlation between the current readout and analyte concentration, a series of electroanalytical validations are performed. Cyclic voltammetry is used for quality control of electrochemical detector chips and to monitor for organic contamination. Amperometry is used for detection due to its ability to distinguish minute differences in measured signal.
3.2. Cyclic voltammetry
Cyclic voltammetry (CV) is one of the most versatile analytic techniques used in the study of electroactive species and the characterization of biosensors. It is widely used as both an industrial and academic research tool in the fundamental characterization of electrochemical systems. In cyclic voltammetry (Fig. 4 ), the potential is ramped from an initial potential (E 0) to a maximum potential (E m) at a fixed sweep rate (V/s). Repeated cycles of reduction and oxidation of the analyte generate alternating anodic and cathodic currents in and out of the working electrode. Since the solution is not stirred, diffusion effects are observed at different analyte concentrations and scan rates.
Fig. 4.
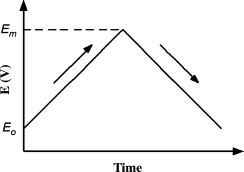
Cyclic voltammetry scans with potential over time.
Separation of the anodic (i pa) and cathodic (i pc) current peaks can be used to predict the number of electrons involved in the redox reaction. The peak current is directly proportional to the analyte concentration, C, and scan rate, v. Experimental results are typically plotted as current versus potential, as shown in Fig. 5 . In this CV scan, the potential is graphed along the X-axis with more positive (or oxidizing) potentials plotted to the right, and more negative (or reducing) potentials to the left. The current is plotted on the Y-axis with cathodic (i.e., reducing) currents plotted in the positive direction and anodic (i.e., oxidizing) currents plotted in the negative direction.
Fig. 5.
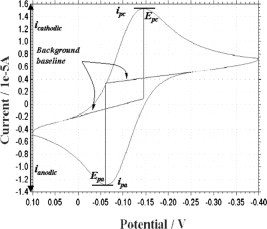
Typical cyclic voltammetry measurement (one cycle) taken by GeneFluidics sensor on K3Fe(CN)6.
3.2.1. Randles–Sevcik equation
The important parameters of a cyclic voltammogram are the magnitudes of the cathodic and anodic peak currents (i pc and i pa, respectively) and the potentials at which these currents are observed (E pc and E pa, respectively). Using these parameters, it is possible to calculate the formal reduction potential (E 0—which is centered between E pa and E pc) and the number of electrons (n) transferred in the charge transfer reaction.
The peak current (i pa or i pc) can be expressed by the Randles–Sevcik equation as shown in Eq. (1):
| (1) |
where n is the number of electrons appearing in half-reaction for the redox couple, F the Faraday’s constant (96,485 C/mol), A the electrode area (cm2), v the rate at which the potential is swept (V/s), D the analyte’s diffusion coefficient (cm2/s), R the universal gas constant (8.314 J/mol K), and T is the absolute temperature (K).
At 25 °C, the Randles–Sevcik equation can be reduced to the following Eq. (2):
| (2) |
where the constant has units (i.e., 2.687 × 105 C mol−1 V −1/2).
The Randles–Sevcik equation predicts that the peak current should be proportional to the square root of the sweep rate when voltammograms are taken at different scan rates. As shown in Fig. 6 , the plot of peak current versus the square root of sweep rate yields a straight line. The Randles–Sevcik equation can be modified to give an expression for the slope of this straight line as follows
| (3) |
The scan rate dependence of the peak potentials and peak currents is used to evaluate the number of electrons participating in the redox reaction as well as to provide a qualitative account of the degree of reversibility in the overall reaction. The number of electrons transferred in the electrode reaction (n) for a reversible redox couple is determined by the separation between the peak potentials (ΔEp = E pa − E pc). For a simple, reversible (fast) redox couple, the ratio of the anodic and cathodic peak currents should be equal to one. Our results deviate only slightly from unity, indicating that our sensors closely approximate an idealized system. Large deviations would indicate interfering chemical reactions coupled to the electrode processes.
Fig. 6.
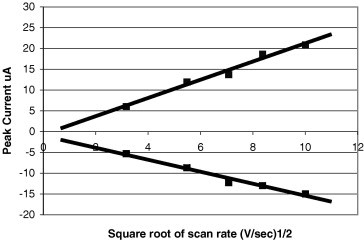
Scan rate dependence of peak current (taken by GeneFluidics sensor on K3Fe(CN)6).
3.3. Chronoamperometry
Chronoamperometry involves stepping the bias potential at the working electrode from an initial potential to a final potential and holding that potential while the current is recorded at the electrode. These potentials are chosen so that they bracket the formal potential, E 0, of the analyte. At the initial potential, no significant current flows. Once the potential is stepped to the final potential, the analyte is consumed at the electrode surface via oxidation or reduction (depending on the direction of the step). This depletes the concentration of the analyte near the electrode. The current response is thus a rapid increase followed by decay as the analyte is depleted and equilibrium is reached. The analysis of chronoamperometry or amperometry data is based on the Cottrell equation, which defines the current–time dependence for linear diffusion control. The final bias potential is determined such that the baseline current generated from electrolyte oxidation or reduction is minimized. This yields a better signal-to-noise ratio since the majority of background noise is from the electrolyte baseline.
If the point in time when the potential is stepped is taken as time zero, then the Cottrell equation in Eq. (4) describes how the current, i (t) decays as a function of time, t
| (4) |
where n is the number of electrons appearing in half-reaction for the redox couple, F the Faraday’s constant (96,485 C/mol), A the electrode area (cm2), C the concentration of analyte (mole/L), D the analyte’s diffusion coefficient (cm2/s), π = 3.14159, and t is the time the current was measured (s).
The current decays as the reciprocal of the square root of time. This dependence on the square root of time reflects the fact that physical diffusion is responsible for transport of the analyte to the electrode surface. The experimental data are usually plotted as i (t) vs. t −1/2, yielding a straight line graph called a Cottrell plot. In addition to being used to determine concentration, the Cottrell plot is also commonly used to determine the working electrode area or an analyte’s diffusion coefficient.
4. Cyclic enzymatic reaction
Horseradish peroxidase (HRP) is one of the most widely used enzymes for analytical purposes because its high kinetic rate maximizes enzymatic signal amplification [10]. By converting HRP-catalyzed electron transfer to an amperometric signal, our electrochemical sensor effectively measures the number of HRP molecules immobilized on the sensor surface. Therefore, the output current is proportional to the number of molecular targets in the sample.
4.1. Mediated electron transfer
The generalized reaction of peroxidases is an irreversible ping-pong mechanism that can be described by three sequential steps:
| (i) |
| (ii) |
| (iii) |
In this mechanism, the resting state ferric enzyme FeIII (porphyrin) (the original form of HRP) is oxidized by 1 equiv. of H2O2 to yield an active intermediate (compound I), in which the iron is oxidized to a ferryl (FeIV O−II) species. Compound I is a porphyrin-cation radical containing FeIV, with one oxidizing equivalent (• +) stored as an organic radical. This is a two-electron oxidation/reduction reaction where H2O2 is reduced to water and the enzyme is oxidized. One oxidizing equivalent resides on iron, giving rise to the oxyferryl (FeIV O−II) intermediate.
The oxidized form of HRP is reduced back to its native form in two steps. In each step, one organic mediator (AH2) is oxidized. In our protocol, TMB (3,3′,5,5′tetramethylbenzidine—C16H20N2) is used as the organic substrate to compound I. First, compound I oxidizes a TMB molecule at its active site and as a result, a free radical (AH•) is produced. In the process, compound I then undergoes a one-electron oxidation reaction with one molecule of TMB yielding compound II, which contains an oxyferryl center coordinated to a normal (dianionic) porphyrin ligand. Compound II is finally reduced back to its native ferric state with concomitant one-electron substrate oxidation. The mediator (AH2) is oxidized to a free radical product (AH•). The overall charge on the resting state of compound I is +1, while compound II is neutral.
5. Sensor and reagent preparation
Although pre-modified sensor chips such as biotin and streptavidin with a variety of modifications are available from GeneFluidics, specialized chips with specialized functionalities can also be easily constructed by the researcher from the GeneFluidics SAM chip. The preparation protocols described in this section are designed for use in general life science laboratories, not for mass production. The distance between the sensors on the GeneFluidics sensor array chip is designed for use with a standard multi-channel pipetter.
5.1. Streptavidin chip preparation
Start with a SAM chip. Obtain stock EDC and NHS from the freezer then thaw the contents. Vortex the vials after thawing. Mix equal volume (e.g., 75 μL each) of NHS and EDC into one vial, vortex, and apply 4 μL to each sensor to cover each working electrode. After dispensing NHS/EDC onto each working electrode, allow the mixture to incubate at room temperature (RT) for approximately 10 min. Carefully remove the sensor chip, take it to the sink, and rinse with deionized (DI) water and dry lightly with N2. Place the sensor chip in a covered glass Petri dish with a small piece of damp filter paper to prevent evaporation. Pipette approximately 4 μL biotin solution to cover each working electrode. Incubate the sensor chip at RT for approximately 15 min. Rinse with DI water and dry lightly with N2. Pipette approximately 20 μL of 1 M ethanolamine to cover the working electrode or all three electrodes. Incubate at RT for approximately 10 min. Rinse with DI water and dry lightly with N2. Pipette approximately 4 μL streptavidin onto each working electrode. Incubate at RT for approximately 25 min. Rinse with DI water and dry lightly with N2. Store in a dry condition in an atmosphere of N2 at RT (see Table 1 ).
Table 1.
Reagents used in streptavidin chip preparation
| NHS | 100 mM N-hydroxysuccinimide in water | Sigma E9884 |
| EDC | 400 mM ethylenediaminetetraacetic acid in water | Aldrich 130672 |
| Biotin | 5 mg/mL biotin in 50 mM sodium acetate buffer | Pierce 0021346 |
| Ethanolamine | 1 M ethanolamine in DEPC water | Aldrich 110167 |
| Streptavidin | 0.5 mg/mL streptavidin in PBS buffer | Sigma S4762 |
| PBS | 100 mM phosphate-buffered saline, pH 7.4 | Sigma P4417 |
5.2. Pre-cap chip preparation for genetic assay
Start with a streptavidin chip prepared as described in Section 5.1. Pipette approximately 4 μL of 1 μM biotinylated anchor probe to cover each working electrode. Incubate at RT for approximately 30 min. Rinse with DI water and dry lightly with N2. Pipette approximately 10 μL PEG 3350 to cover each working electrode or all three electrodes. Incubate at RT for approximately 10 min. Rinse with DI water and dry lightly with N2. Store in a dry condition in an atmosphere of N2 at RT (see Table 2 ).
Table 2.
Reagents used in pre-cap chip preparation
| PEG | 0.05% polyethylene glycol 3350 in PBS | Sigma P3640 |
5.3. Pre-cap chip preparation for immunoassay
Start with a SAM chip. Pipette an equal volume (e.g., 70 μL) of NHS and EDC into a new vial. Vortex and immediately apply 4 μL of the NHS/EDC mixture to cover each working electrode. Allow the mixture to incubate at RT for 10–15 min in a covered, humidified glass Petri dish. During the incubation period, make dilutions of the monoclonal anchor antibody in PBS to obtain concentrations ranging from 0.01 to 1 mg/mL (depending on the purity and affinity of the antibody). At the completion of the NHS/EDC incubation period, carefully rinse the sensor chip with DI water. Dry lightly with N2. Place the sensor chip back in the Petri dish. Pipette 4 μL of the anchor antibody to cover each working electrode. Cover the Petri dish and incubate at RT for 10–90 min (depends on the antibody concentration). At the end of anchor antibody incubation, carefully take the sensor chip to the sink and rinse with DI water. Dry lightly with N2. Place the sensor chip back in the Petri dish. Pipette 20 μL of 1 M ethanolamine to cover the working electrode or all three electrodes. Cover the Petri dish. Incubate at RT for 15 min. At the end of ethanolamine incubation, carefully take the sensor chip to the sink and rinse with DI water. Dry lightly with N2. Place the sensor chip back in the Petri dish. Store in a dry condition in an atmosphere of N2 at RT.
5.4. Preparation of the oligonucleotide probe solution
Reconstitute oligonucleotides with DEPC water according to the specifications listed on the synthesis report that accompanies the order. The amount of DEPC water to be added is specified by Volume for 100 pmol/μL in the synthesis report. This figure indicates how much water is necessary to create a 100 μM concentration of the oligonucleotide. Carefully remove the cap, add the DEPC water, re-cap, and briefly vortex the tube to make a homogeneous oligonucleotide stock solution. Make a 20 μM probe solution and aliquot into different vials. Keep the 100 and 20 μM stock solutions in −20 °C freezer.
Prepare the appropriate concentrations of signal probe in diluent. For high target concentration samples (e.g., target >1 nM), use a 1 μM signal probe solution. For lower target concentration samples (e.g., target <100 pM), use a 0.25 μM signal probe solution (see Table 3 ).
Table 3.
Reagents used in probe solution preparation
| DEPC water | Diethyl pyrocarbonate treated water | Ambion 9920 |
6. Sensor quality control
6.1. Sensor characterization
Before testing or releasing any sensor chips, GeneFluidics sensors are carefully examined under a microscope, paying particular attention to the electrode area to identify any areas of uneven Au coating, unknown particulates, or scratches. However, potential nano-meter scale defects may only be detected electrochemically. Organic contamination on bare gold sensors will affect the impedance of the sensor while defects of SAM-coated chips will increase the leakage current. Both defects cannot be easily detected optically, but can be monitored electrochemically.
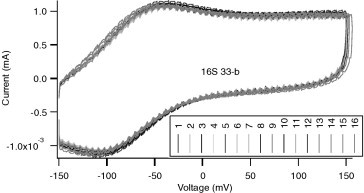
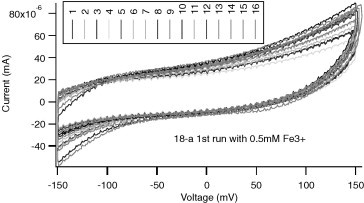
Sensors are characterized by amperometric (potentiostatic) measurement and cyclic voltammetry in a QC mixture solution (see Section 6.2). Amperometric measurement of bare gold and SAM-coated sensors is performed at −150 mV (Fig. 7 ). The signal variance of all sensors on one chip should be less than 5% and the difference between minimum and maximum readings should be less than 10%. Cyclic voltammetry of bare gold sensors is performed with voltage ranging from −150 to 150 mV at 50 mV/s (Fig. 8 ). On the CV curves of bare gold sensors, the redox peaks should be around −110 mV for the reduction peak and −55 mV for the oxidation peak. SAM-coated sensors are characterized by conducting cyclic voltammetry with voltage ranging from ±150 mV (Fig. 9 ). The redox peak position of all sensors should match the rest of the sensors on the chip (see Table 4 ).
Fig. 7.
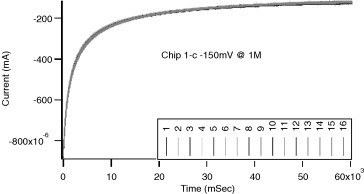
Amperometric QC measurement of a 16-sensor bare gold chip with signal from all channels with tight variance.
Fig. 8.
Cyclic voltammetry QC measurement of a 16-sensor bare gold chip with clear oxidation and reduction peak.
Fig. 9.
Cyclic voltammetry QC measurement of a 16-sensor SAM chip with no REDOX peak and much smaller current.
Table 4.
Reagents used in sensor characterization
| 0.1 mM 3+ QC solution | 0.1 mM K3Fe(CN)6 potassium ferric cyanide prepared using 0.1 M KCl potassium chloride as diluents | Sigma P8131, Sigma P9541 |
| 0.5 mM mixture solution | Mixture of 0.5 mM K3Fe(CN)6 potassium ferric cyanide and K4Fe(CN)6 potassium ferric cyanide prepared using 0.1 M KCl potassium chloride as diluents | Sigma P8131, Sigma P3289, Sigma P9541 |
6.2. Materials and reagents
K3Fe(CN)6 should be placed in an amber bottle and the measurement should be conducted in a yellow light room or similar environment. Use 0.1 mM K3Fe(CN)6 solution for bare gold sensor characterization. Use 0.5 mM mixture of the K3Fe(CN)6 and K4Fe(CN)6 for SAM modified sensor characterization.
7. Assay protocols
7.1. Cell lysing for different sample types
Cell lysis is required only when target molecules are intracellular. For the immunoassay, electrolyte and ionic detection, one can typically assay the raw sample directly without a lysis step. Cell lysis is defined as disruption of cells by physical, chemical, mechanical or enzymatic means to obtain intracellular materials, such as DNA and RNA. Detergents, solvents, and antibiotics are commonly used for the chemical method and these protocols are applicable to both bacteria and eukaryotic cell types. Different lysis approaches are listed in Table 5 .
Table 5.
Reagents used in cellular lysis
| Blood, plasma sample, and saliva | Directly add alkaline lysis solution at 1:9 ratio to break the cell membrane. Blood lysates are very viscous so the probe solution has to be added immediately after lysis | 0.4 M NaOH (Sigma S0899) |
| Sputum | Add PBS buffer at 1:1 ratio then alkaline lysis solution at 1:9 ratio to buffered sputum | 0.4 M NaOH (Sigma S0899) |
| Urine and cell culture | Universal lysis buffer containing 1% Triton X-100, 0.1 M KH2PO4, 2 mM EDTA, and 1 mg/mL lysozyme | Triton X-100 (Sigma T9284), KH2PO4 (Sigma P0662), EDTA (Sigma E-5134), lysozyme (Sigma, L-6876) |
7.2. Genetic assay using a mixture of anchor and signal probes
Prepare specimen or nucleic acids samples in an appropriate diluent. Pipette a 1:1 ratio of target sample and probe mixture solutions (anchor and signal probe) into reaction vials. Cap and gently vortex the vials. Leave the vials at room temperature or place in oven for hybridization. Allow sufficient time (e.g., 15 min) for target/probe hybridization.
Pipette 4 μL of hybridized solution onto each working electrode. Incubate at RT for approximately 15 min. Rinse sensor chip with DI water and dry lightly with N2. Pipette 4 μL of HRP conjugate onto each working electrode. Incubate at RT for approximately 15 min. Approximately 5 min before the HRP conjugate step is complete, remove the TMB substrate bottle from the refrigerator and place sufficient volume into a clean trough covered with aluminum foil. After the HRP incubation, rinse the chip with DI water and dry lightly with N2. Apply a clean multi-sensor well structure (Fig. 1C) onto the sensor chip. Align the well structure so that all three electrodes are exposed within the well. Press firmly to seal the well structure onto the sensor chip. Place the sensor chip onto the chip mount. Add 80 μL of TMB substrate to each sensor well using a multi-channel pipetter and click “START” on the potentiostat reader to initiate electrochemical measurement. At the completion of amperometric measurement, conduct an optional cyclic voltammetry measurement to confirm sensor array integrity. This last step can be performed immediately at the completion of the amperometric measurement or a few minutes later. Table 6 lists commonly used assay reagents.
Table 6.
Commonly used assay reagents
| HRP conjugate | Prepare 0.5 U/mL anti-fluorescein-POD from 150 U/mL stock solution using 0.5% casein/PBS buffer | Roche 1426346 |
| TMB substrate | TMB = 3,3′,5,5′-tetramethylbenzidine; Neogen K-blue low activity substrate | Neogen 306176 |
| Casein/PBS buffer | Dissolve phosphate-buffered saline (PBS) tablet and casein powder with water to make PBS solution | Sigma C7078, Sigma P4417 |
7.3. Pre-cap genetic assay
Prepare specimen or nucleic acid samples in an appropriate diluent. Pipette a 1:1 ratio of target sample and signal probe solutions into reaction vials. Cap and gently vortex the vials. Leave the vials at room temperature or place in a hybridization oven. Allow sufficient time (e.g., 15 min) for target/probe hybridization.
Pipette 4 μL samples to the pre-cap sensors. Incubate for approximately 15 min at 65 °C for RNA targets or at room temperature for DNA targets. Rinse with DI water and dry lightly with N2. Pipette 4 μL of HRP conjugate onto each working electrode. Incubate at RT for approximately 15 min. During enzyme incubation, ready the potentiostat reader for measurement. Approximately 5 min before the enzyme conjugate step is complete, remove the TMB substrate from the refrigerator and place a sufficient volume into a clean trough covered with aluminum foil. Rinse the chip with DI water and dry lightly with N2. Apply a clean multi-sensor well structure onto the sensor chip. Place the sensor chip onto the chip mount. Add 80 μL of TMB substrate to each sensor well using a multi-channel pipetter and click “START” on the potentiostat reader to initiate electrochemical measurement. At the completion of amperometric measurement, conduct an optional cyclic voltammetry measurement to confirm sensor array integrity. This last step can be performed immediately at the completion of the amperometric measurement or a few minutes later.
7.4. Immunoassay with antibody-coated sensor chip
Pipette 4 μL samples to cover each working electrode. Place a lightly damp filter paper in the Petri dish under the sensor chip to prevent the sample from drying out. Cover the Petri dish. Allow the sample to incubate for 5–30 min (depends on the target concentration, 30 min for pg/mL and 5 min for ng/mL and more). At the end of sample incubation, carefully rinse the sensor chip with DI water. Dry lightly with N2. Place the sensor chip back in the Petri dish. Apply 4 μL of the biotin-conjugated monoclonal signal antibody onto each sensor to cover the working electrode only. Incubate for 5–30 min (depends on the target concentration). Rinse with DI water. Dry lightly with N2. Apply 4 μL of the streptavidin–HRP conjugate onto each sensor to cover the working electrode only. Incubate at RT for 30 min. During enzyme incubation, ready the potentiostat reader for measurement. Approximately 5 min before the enzyme conjugate step is complete, remove the TMB substrate from the refrigerator and place a sufficient volume into a clean trough covered with aluminum foil. Rinse the chip with DI water and dry lightly with N2. Before the HRP conjugate incubation step is complete, obtain a clean multi-channel plastic well, a clean trough, and multi-channel pipetter. Calculate the TMB substrate needed (use 80 μL on each sensor) and place the required volume into the clean trough. Cover the trough with aluminum foil. Allow the TMB substrate to equilibrate to room temperature for 5 min before use. Dry lightly with N2. Place the sensor chip back in the Petri dish. Apply a clean multi-sensor well structure onto the sensor chip. Place the sensor chip onto the chip mount. Add 80 μL of TMB substrate to each sensor well using a multi-channel pipetter and click “START” on the potentiostat reader to initiate electrochemical measurement. At the completion of amperometric measurement, conduct an optional cyclic voltammetry measurement to confirm sensor array integrity. This last step can be performed immediately at the completion of the amperometric measurement or a few minutes later.
8. Results
This detection platform has demonstrated both extraordinary sensitivity and specificity with multiple target and sample types. Fig. 10, Fig. 11, Fig. 12, Fig. 13, Fig. 14 present experimental results from five experiments. The Y-axis of each chart depicts current measured by our reader instrument at the sensor surface. This current correlates with the number of enzymes and therefore targets in the sample. Standard deviations are shown by the error bars and P values listed where appropriate.
Fig. 10.
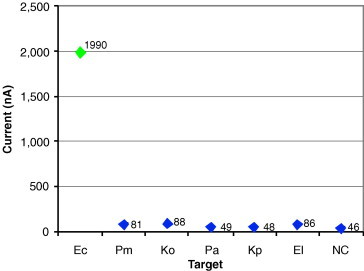
Specificity of the E. coli capture and detector probe pair.
Fig. 11.
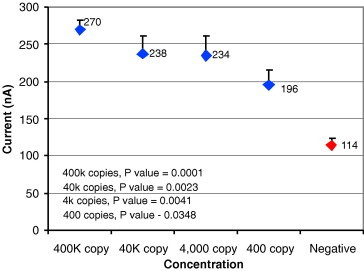
Sensitivity for SARS viral copy from infected monkey cells.
Fig. 12.
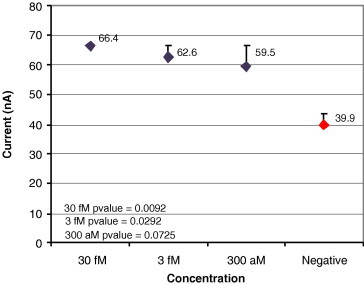
Sensitivity for TREC plasmid spiked in serum sample.
Fig. 13.
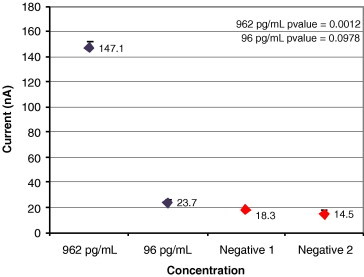
Sensitivity for IL-8 protein in clinical saliva sample.
Fig. 14.
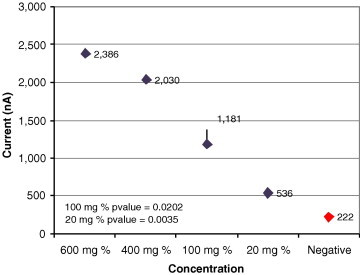
Sensitivity for glucose in BSA.
Fig. 10 depicts results from a uropathogen study conducted at UCLA. In this study, an Escherichia coli-specific capture and detector probe pair were created and tested against E. coli (Ec), Proteus mirabilis (Pm), Klebsiells oxytoca (Ko), Pseudomonas aeruginosa (Pa), Klebsiella pneumoniae (Kp), and Enterobacter cloacae (El). Negative control (NC) readings were taken from a sample without bacteria. As shown, readings for E. coli were significantly greater than those for the five other species of uropathogenic bacteria tested (P < 0.0001).
Fig. 11, Fig. 12, Fig. 13, Fig. 14 show results from four sensitivity experiments. In the study shown in Fig. 11, we assayed a SARS coronavirus probe set against a serial dilution of SARS virus, identifying the signal from 400 viruses above that of the negative control. In the study shown in Fig. 12, we detected the TREC single nucleotide polymorphism in serum at 300 aM levels. Fig. 13’s study compared IL-8 levels in a cancer patient saliva sample plus a 10-fold dilution of that sample against two negative controls. In the study shown in Fig. 14, we tested its sensor against four typical glucose concentrations, showing strong signal separation even at the lowest concentration.
9. Concluding remarks
This review is intended to provide a foundation for future studies investigating target-amplification-free, low concentration molecular detection. The principles underlying our electrochemical detection platform are summarized as is the methodology for its use in rapid molecular analysis. Enabling nanoscale sensor surface treatments are presented along with electrochemical measurement techniques. Generic assay protocols are described that can be adapted for detection of the protein or nucleic acid target(s) of interest. When combined with microfluidic-based automated sample preparation techniques under development at GeneFluidics, this electrochemical sensor platform is a promising tool for a wide variety of commercial and research applications.
Acknowledgments
The uropathogen study is supported by Bioengineering Research Partnership Grant EB00127 from the National Institute of Biomedical Imaging and Bioengineering. The SARS study is supported by Innovative Technology Transfer program from National Science Council, Taiwan. The CA125 investigation is supported by the Early Detection Research Network (EDRN) at the National Cancer Institute. We thank Dr. David Wong and Dr. Ren Sun from University of California, Los Angeles, for providing the oral cancer saliva specimens and SARS infected monkey cells. Special thanks to the R/D group at U-tech media for sensor chip production.
References
- 1.Kerman K., Kobayashi M., Tamiya E. Meas. Sci. Technol. 2004;15:R1–R11. [Google Scholar]
- 2.Chaubey A., Malhotra B.D. Biosens. Bioelectron. 2002;17(6–7):441–456. doi: 10.1016/s0956-5663(01)00313-x. [DOI] [PubMed] [Google Scholar]
- 3.Mehrvar M., Abdi M. Anal. Sci. 2004;20(August) doi: 10.2116/analsci.20.1113. [DOI] [PubMed] [Google Scholar]
- 4.Mackay I.M., Arden K.E., Nitsche A. Nucleic Acids Res. 2002;30(6) doi: 10.1093/nar/30.6.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drummond G., Hill M.G., Barton J.K. Nat. Biotechnol. 2003;21(10) doi: 10.1038/nbt873. [DOI] [PubMed] [Google Scholar]
- 6.Revell D.J., Knight J.R., Blyth D.J., Haines A.H., Russell D.A. Langmuir. 1998;14(16):4517–4524. [Google Scholar]
- 7.Motesharei K., Myles D.C. J. Am. Chem. Soc. 1998;120(29):7328–7336. [Google Scholar]
- 8.Xia Y., Whitesides G.M. Angew Chem. Int. Ed. 1998;37:550–575. doi: 10.1002/(SICI)1521-3773(19980316)37:5<550::AID-ANIE550>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 9.Lahiri J., Ostuni E., Whitesides G.M. Langmuir. 1999;15(6):2055–2060. [Google Scholar]
- 10.Ruzgas T., Cs regi E., Emneéus J., Gorton L., Marko-Varga G. Anal. Chim. Acta. 1996;330:123–138. [Google Scholar]
- 11.Sun C.-P., Liao J.C., Zhang Y.H., Gau V., Mastali M., Babbitt J., Grundfest W., Churchill B.M., McCabe E.R.B., Haake D.A. Mol. Gen. Metab. 2005;84(1):90–99. doi: 10.1016/j.ymgme.2004.11.006. [DOI] [PubMed] [Google Scholar]