

Abstract
We have found that binding sites for interleukin-13 (IL-13) are overexpressed in a vast majority of high-grade astrocytomas (HGAs). These binding sites for IL-13 are distinct from the physiological receptor in that it does not bind IL-4. We also demonstrated that IL-13 receptor alpha 2 protein chain (IL-13Rα2), an IL-4-independent receptor for IL-13, is abundant among HGAs, but not in normal organs. To examine if IL-13Rα2 is the tumor-associated site for IL-13, we stably transfected normal Chinese hamster ovary (CHO) cells and glioma G-26 cells to express either human (h) or murine (m) IL-13Rα2. CHO-hIL-13Rα2(+) cells and G-26-h/mIL-13Rα2(+) cells, and not CHO and G-26 parental or mock-transfected cells, specifically bound IL-13 in an IL-4-independent manner. The IL-13Rα2(+) cells also became highly susceptible to the killing by an IL-13-based cytotoxic fusion protein. In loss of function studies, a HGA cell line, SNB-19, was transfected with antisense (as) hIL-13Rα2. as-SNB-19-hIL-13Rα2(+) cells lost their natural affinity towards IL-13 and became resistant to IL-13-based cytotoxins. The fact, that IL-13Rα2-positive cells bind IL-13 independent of IL-4, become susceptible to IL-13 cytotoxins, and cells deprived of IL-13Rα2 receptor lose these features, demonstrates that IL-13Rα2 is the brain tumor-associated receptor for IL-13.
Keywords: brain tumors, gliomas, cytotoxin, receptor, interleukin-13, cytokine
Introduction
High-grade astrocytomas (HGAs) are rapidly progressive heterogeneous brain tumors of glial origin that are extremely resistant to current therapies. Recently, we demonstrated that the vast majority of HGAs bind interleukin-13 (IL-13) and are highly susceptible to the cytotoxicity of IL-13-based cytotoxins [1–4].
IL-13 is an immune regulatory cytokine that shares a variety of functions with IL-4 [5–7]. IL-13 is secreted by B and T cells and binds to a plethora of different normal hemopoietic and somatic cells [5–7]. Its physiological functions are mediated through the shared IL-13/4 receptor [8–11]. The IL-13/4 receptor is a heterodimer that first binds IL-13 through the IL-13 binding component, IL-13 receptor alpha 1 (IL-13Rα1) (42 kDa), at a low affinity and subsequently recruits the IL-4 receptor component, IL-4Rα (140 kDa) [11]. The complex of these two proteins results in a high-affinity binding site for IL-13, which is competed for by IL-4 [11,12].
We demonstrated that a vast majority of HGAs bind IL-13. Unexpectedly, their affinity for IL-13 remained even in the presence of IL-4 [1,3,13–15]. This indicated that there might exist a more restricted HGA-associated receptor for IL-13 that is distinct from the physiological IL-13/4 receptor [4]. Additionally, mutants of IL-13 that showed a diminished capacity to signal retained the ability to bind HGAs [14–16]. We demonstrated that a prototype mutant of IL-13, IL-13.E13K, loses the ability to proliferate TF-1 cells, a downstream effect of IL-13 signaling [15]. Furthermore, we showed that IL-13.E13K competes for the HGA IL-13 binding site approximately 50 times better than wild-type IL-13 [14]. These mutants were fused to derivatives of Pseudomonas exotoxin (PE), such as PE38QQR, PE1E, and PE4E, and showed ability to very potently kill glioma cells [1,14,17]. Because these derivatives of PE lack a fully functional α2 macroglobulin receptor binding domain of PE, the only means the IL-13-based fusion protein can enter a cell is through IL-13 binding to cell surface receptors and subsequent internalization [12,13].
Additional experimental evidence for a second IL-13 receptor was provided by the work of Caput et al. [18]. These investigators isolated a protein termed IL-13 receptor alpha 2 (IL-13Rα2), which binds IL-13 in an IL-4-independent manner, a feature resembling a glioma-restricted receptor for IL-13. However, the link between IL-13Rα2 and the IL-13 binding to the restricted sites on HGAs or the susceptibility of HGA to IL-13-based cytotoxins did not appear to be clear-cut. Several cell lines from which either the gene was cloned or had tested positive for IL-13Rα2 gene expression did not demonstrate the anticipated response to IL-13-PE cytotoxins (not shown). This and similar other initial lack of correlation between IL-13Rα2 expression and, e.g., susceptibility to IL-13-based cytotoxins manifested the need to further investigate whether or not IL-13Rα2 and the restricted binding sites for IL-13 on HGA are in fact the same IL-13 binding site.
Materials and Methods
Materials
DNA ligase and restriction endonucleases were purchased from New England Biolaboratories (Beverly, MA). Pfu turbo DNA polymerase was purchased from Stratagene (La Jolla, CA). Oligonucleotide primers were synthesized in the Macromolecular Core Laboratory of the Pennsylvania State University College of Medicine. All polymerase chain reaction (PCR) purifications, DNA extraction from agarose gels, and plasmid minipreps were done using respective Qiagen kits (Qiagen, Valencia, CA). Tissue culture equipment was from Corning Glass (Corning, NY). RNA blotted membranes were purchased from Clontech (Palo Alto, CA). Cell lines, such as U-87 MG, SW-1088, H-4, A-431, U-373 MG, SNB-19, and CHO-K1, were purchased from American Type Culture Collection (Manassas, VA) or were primary HGA cultures isolated in this laboratory (G-48a). The ATCC CHO-K1 cell line was derived as a subclone from the parental Chinese hamster ovary (CHO) cell line initiated from a biopsy of an ovary of an adult Chinese hamster. G-26 mouse glioma cells were the gift of Dr. Marzenna Wiranowska.
Production of Recombinant Proteins
Plasmids and recombinant proteins for IL-13.E13K, IL-13, IL-4, and IL-13.E13K-PE38QQR were prepared as described in Debinski et al. [14] and Thompson and Debinski [15]. The procedure used for protein production, isolation, refolding, and purification of IL-13 and IL-13-based cytotoxins was described previously [12].
RNA Isolation and Northern Blots
Total RNA from CHO, G-26, and SNB-19 cells were obtained using an acid-guanidium isothiocyanate-phenol-chloroform method [19]. Procedures for poly(A) + isolation and Northern blots were described previously [20]. Blots were probed with random-primed 32P cDNA for hIL-13Rα2 or mIL-13Rα2. 32P riboprobes were also constructed as described previously and used to probe for sense-strand hIL-13Rα2. Additionally, blots were probed for actin to demonstrate the integrity of the RNA.
Isolation of hIL-13Rα2 and mIL-13Rα2
hIL-13Rα2 was isolated by screening a cDNA library derived from renal cell carcinoma (RCC) cells. A custom cDNA library was prepared by Stratagene using poly(A)+ RNA isolated from the RCC cell line. The library was screened using the protocol described by the manufacturer. cDNA probes for hIL-13Rα2 were identical to the ones described above.
mIL-13Rα2 was isolated from murine brain mRNA obtained from Clontech. Superscript II Moloney murine leukemia virus (MMLV) with a poly(T) primer was used to synthesize first-strand cDNA. The resulting murine brain cDNA was purified using a Qiagen PCR purification kit. For PCR amplification of mIL-13Rα2, forward primer, 5′-CAGACTAGGGGTACCATGGCTTTTGTGCAT-3′, and reverse primer, 5′-TGATGCTAGTCTAGATTAACAGAGGGTATCTTC-3′, were used. Pfu turbo DNA polymerase was used for high-fidelity amplification of mIL-13Rα2. KpnI and XbaI restriction endonucleases were used to create compatible ends and the resulting fragments were ligated into pcDNA 3.1 vector (Invitrogen, Carlsbad, CA). Constructs were sequenced in the Pennsylvania State University College of Medicine Core Macromolecular Facility using an ABI 377 automated sequencer (PE, Norwalk, CT).
Transfection of CHO and G-26 Cells
The gene for hIL-13Rα2 that was transfected into CHO cells was kindly provided by Dr. Pascual Ferrara of Sanofi BioRecherche, Labège Innopole (France) and the gene for hIL-13Rα2 that was transfected into G-26 cells was isolated by us. The primers used to PCR-amplify hIL-13Rα2 were forward primer, 5′-AAGATTTGGGCTAGCATGGCTTTCGTTTGC-3′, and reverse primer, 5′-TCCCTCGAAGCTTCATGTATCACAGAAAAA-3′. For antisense constructs of hIL-13Rα2, forward primer, 5′-AAGATTTGGAAGCTTATGGCTTTCGTTTGC-3′, and reverse primer, 5′-TCCCTCGAAGCTTCATGTATCACAGAAAAA-3′, were used and the purified product was confirmed by sequencing. For the hIL-13Rα2 forward construct, NheI and HindIII restriction endonucleases were used to create compatible ends, and for the antisense hIL-13Rα2 construct, HindIII restriction endonuclease was used. Fragments were ligated into pcDNA 3.1 vector.
DNA was transfected into CHO and G-26 cells using lipofectin reagent as indicated by the manufacturer (GibcoBRL, Rockville, MD). After 48 hours, cells were passaged 1:5 and grown in selection medium (800 µg/ml geneticin; GibcoBRL) until individual clones were isolated. Cells were clonally expanded and maintained in 200 µg/ml geneticin.
Transfection of SNB-19 Cells with Antisense hIL-13Rα2 (as-hIL-13Rα2)
The construction of pcDNA3.1-as-hIL-13Rα2, which contains the full-length gene of IL-13Rα2 in an antisense orientation under a CMV promoter, is described above. DNA was transfected into SNB-19 cells using lipofectin reagent. In addition to geneticin, cells were selected in media containing 10 µg/ml IL-13-based cytotoxin, IL-13.E13K-PE38QQR. This procedure efficiently helped isolate clones expressing low amounts of hIL-13Rα2. IL-13.E13K-PE38QQR was added to the SNB-19 cells transfected with as-IL-13Rα2 48 hours after transfection and surviving cells were maintained in media containing the cytotoxin for five passages. Additionally, mock-transfected cells were treated with IL-13.E13K-PE38QQR in the same manner as the transfected cells and no surviving clones were observed.
Reverse Transcription (RT) PCR Analysis of IL-13Rα2 Gene Expression
RNA was isolated from CHO-IL-13Rα2(+), G-26-IL-13Rα2(+) cells, and controls as described above and cDNA was produced using Superscript reverse transcriptase from GibcoBRL and Taq DNA polymerase. The sequence of the primers used to specifically amplify 970 bp of cDNA encoding hIL-13Rα2 was as follows: forward primer, 5′-AGCTTGACCGCATATGTCATCTTCAGACACCGA-3′, and reverse primer, 5′-TGGTACGTGAGGTACCACGTAGCAAAGTTTTCT-3′. To amplify a 900-bp fragment of murine actin, forward primer, 5′-CAGAGCAAGAGGGGTATCCTGA-3′, and reverse primer, 5′-TGATCCACATCTGCTGGAAGGT-3′, were used. The primers used to amplify mIL-13Rα2 were identical to the ones used to isolate the gene.
Autoradiography
Recombinant IL-13 and IL-13.E13K were labeled with 125I by using Iodo-Gen reagent (Pierce Chemical, Rockford, IL) as described [1]. 5x104 cells were dotted onto sterile slides and allowed to adhere overnight. Cells were then fixed using ice-cold ethanol. Procedure used for autoradiography was previously described in detail [1].
All films from autoradiography were scanned on a transparency scanner at a pixel size of 88x88 µm(Molecular Dynamics, Sunnyvale, CA) and the densities were plotted using Quantity 1 software (Bio-Rad, Hercules, CA). The images were compiled in Paint Shop Pro V 5.0 (Jasc Software, Eden Prairie, MN).
Cell Proliferation Assay
Cytotoxicity assays were performed as described previously [14]. 3x103 cells were plated in each well of a 96-well plate in 150 µl of media and allowed to adhere overnight. The following day, 25 µl of blocker (1 µg/ml final concentration) of IL-13.E13K, or IL-4 in 0.1% BSA/PBS (phosphate-buffered saline) buffer, or buffer alone, was added. After an hour of incubation at 37°C, varying concentrations of IL-13.E13K-PE38QQR were added in triplicate and incubated at 37°C in 5% CO2/95% O2 for 48 hours. After 48 hours, MTS/PMS cell proliferation assay was performed as instructed by the manufacturer (Promega, Madison, WI). Ten microliters of dye was added to each well and incubated for 4 hours. Plates were then read by a microplate reader (Cambridge Technology, Watertown, MA) at absorbance of 490 nm. Cyclohexamide-treated, cell-containing wells served as the background for the assay. Background was subtracted from each data point and was divided by the value from the wells that were not treated with cytotoxin to obtain the fraction of cells remaining. The fraction of cells remaining was multiplied by 100 to obtain the percent of control.
Production of Anti-hIL-13Rα2 Serum
A monoclonal antibody against IL-13Rα2, clone B-D13, was purchased from Cell Sciences (Norwood, MA). This antibody was found to be inadequate in our hands for immunohistochemistry (IH) and adequate for flow cytometry in the presence of signal's amplification. We therefore commenced the production of anti-IL-13Rα2 serum in house. One hundred micrograms of pcDNA3.1-hIL-13Rα2 DNA was injected intrasplenically into 10-week-old BALBc mice and boosted 2 weeks later intramuscularly again with 100 µg of the plasmid. Four weeks later, mice were reboosted every other week for a total of three boosts with 500 µg of affinity-purified 6-Histidine-IL-13Rα2 extracellular domain (6his-hIL-13Rα2ex) recombinant protein produced in Escherichia coli. One week after the last injection, serum was obtained.
IH
IH was performed on 1x104 ethanol-fixed GBM cells or snap-frozen sections of HGA tumors fixed to glass slides. Samples were exposed to a 1:50 dilution of anti-hIL-13Rα2 serum or serum from an nonimmunized mouse. The procedure used for immunostaining was described previously [21].
Cell ELISA Assay
1x106 cells were plated in each well of a 96-well plate and allowed to adhere overnight. Media were removed and nonspecific binding sites were blocked using 2.5% dry milk dissolved in media. After 20 minutes of blocking, 1:100 dilution of anti-hIL-13Rα2 serum or nonimmunized mouse serum was added to each well for 30 minutes and shaken gently at room temperature (RT). Cells were washed three times with PBS and 1:1000 dilution of secondary antibody (anti-mouse polyclonal alkaline phosphatase) (Roche, Indianapolis, IN) was added for 30 minutes at RT. Cells were washed three times with PBS and p-nitrophenyl phosphate substrate was added (1 mg/1 ml) in alkaline phosphatase buffer (100 mM NaCl, 5 mM MgCl2, 100 mM Tris-HCl; pH 9.5) (Invitrogen). Plates were then read using a microplate reader (Cambridge Technology) at absorbance of 405 nm. Values were obtained by subtracting the background immunoreactivity observed in wells treated with nonimmunized mouse serum. Each experiment was performed in quadruplicates and repeated at least twice.
Flow Cytometry
Cells were detached from flask using 1xPBS/4 mm EDTA and counted using a hemocytometer. 1x106 cells in a volume of 100 µl were stained for enhanced signal using Flow-Amp enzymic enhancement kit (Flow-Amp Systems, Cleveland, OH) according to the manufacturer's instructions. Cells were incubated with anti-hIL-13Rα2 monoclonal antibody (clone B-D13; Cell Sciences). Ten thousand events (gated on viable cells) were measured for each sample and controls using a Becton Dickinson FACS scan flow cytometer and analyzed with CellQuest software (Becton Dickinson Biosciences, Franklin Lakes, NJ). Results are expressed in mean fluorescence intensity (MFI). Each experiment was repeated three separate times.
Results
HGA Tumor Specimens and Glioma Cell Lines are Immunoreactive for hIL-13Rα2
Previously, we demonstrated that HGAs expressed the transcript for hIL-13Rα2 [20]. Additionally, we demonstrated that a vast majority of HGAs bind IL-13 independent of IL-4 [1,3,13–15]. Here we demonstrate the presence of immunoreactive hIL-13Rα2 on HGAs directly using anti-hIL-13Rα2 mouse serum. Only the anti-hIL-13Rα2 serum, but not serum from nonimmunized mice, immunoreacted with the membranes of the HGA tumor section (Figure 1A).
Figure 1.
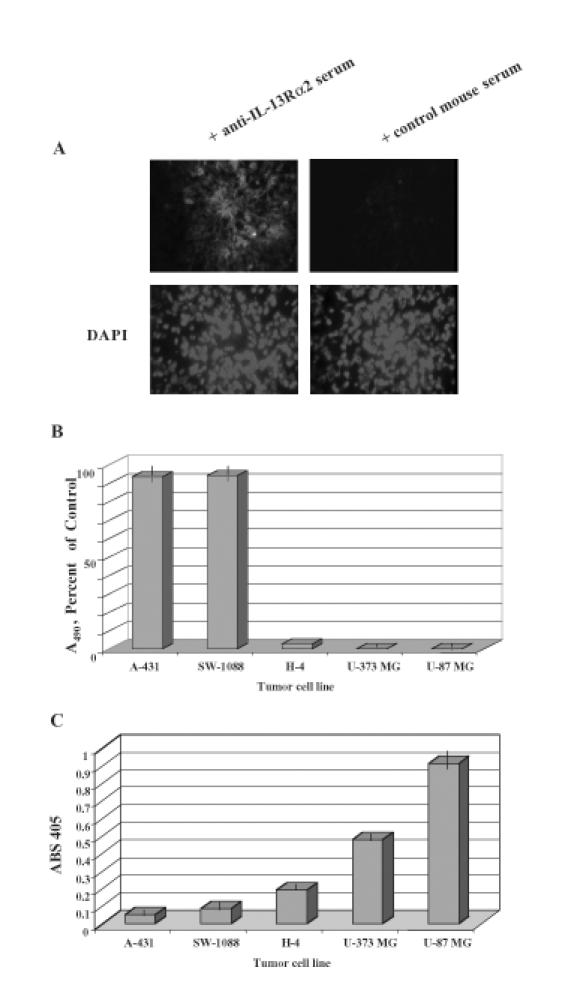
(A) hIL-13Rα2 immunoreactivity in a GBM tumor specimen. Nonimmunized serum was used on a contiguous section of the same tumor as a negative control. Tissues were also stained for nuclear presence using DAPI. (B) Cytotoxicity of high-grade astrocytoma cells (SW-1088, U-373 MG, U-87 MG), low-grade astrocytoma cells (H-4), and epidermoid tumor cells (A-431) using 100 ng/ml IL-13.E13K-PE38QQR determined in a colorimetric cell proliferation assay. Vertical lines represent SD. (C) Reactivity of high-grade astrocytoma cells (U-87 MG, U-373 MG, SW-1088), low-grade astrocytoma cells (H-4), and epidermoid tumor cell (A-431) to anti-hIL-13Rα2 serum in a cell ELISA assay. Vertical lines represent SD.
IL-13Rα2 Immunoreactivity is Present in Brain Tumor Cells
To further document the presence of immunoreactive hIL-13Rα2 in HGAs, various tumor cell lines were tested using a cell ELISA assay. Because whole cells were used in this assay, the only proteins that are exposed to the anti-hIL-13Rα2 antibodies are plasma membrane proteins. Two HGA cell lines, U-87 MG, U-373 MG, and one low grade glioma cell line, H-4, all which are potently killed by IL-13-based cytotoxins (Figure 1B), demonstrated prominent reactivity towards anti-hIL-13Rα2 serum (Figure 1C). In contrast, a HGA cell line, SW-1088, and an epidermoid carcinoma cell line, A-431, both which are resistant to IL-13-based cytotoxins (Figure 1B), did not react with the anti-hIL-13Rα2 serum (Figure 1C). These results demonstrate a link between the presence of IL-13Rα2 immunoreactive protein and the susceptibility of tumor cells to IL-13-based cytotoxins.
Gain of Function Studies: Transfecting CHO and G-26 Cells with hIL-13Rα2
In order to directly demonstrate that hIL-13Rα2 is the glioma-restricted receptor for IL-13 and is responsible for the restrictive IL-13 binding properties seen in HGAs, we transfected normal CHO cells and murine glioma-derived G-26 cells with hIL-13Rα2 in a CMV promoter-containing vector.
Total RNA was isolated from clones that showed highest transgene expression in the initial screening: CHO-hIL-13Rα2(+) clone 5 and G-26-hIL-13Rα2(+) clone 2. Then, Northern blot analysis was performed using 32P-labeled hIL-13Rα2 cDNA. CHO-hIL-13Rα2(+) clone 5 (Figure 2A, lane 3) and G-26-hIL-13Rα2(+) clone 2 (Figure 2A, lane 6) demonstrated the presence of hIL-13Rα2 transcript. Of importance, CHO and G-26 parental clones (Figure 2A, lanes 1 and 4) and control clones containing empty vector (Figure 2A, lanes 2 and 5) failed to show any hybridization to 32P-hIL-13Rα2. Blots were also probed with human 32P actin, which hybridizes to RNA encoding murine actin as well (Figure 2A). Additionally, RT-PCR was performed to confirm the presence of the transgene message using primers to specifically amplify hIL-13Rα2. Only CHO-hIL-13Rα2(+) clone 5 (Figure 2B, lane 3) and G-26-hIL-13Rα2(+) clone 2 (Figure 2B, lane 6) and not the controls, such as G-26 and CHO parental cell lines (Figure 2B, lanes 1 and 4) or G-26 and CHO transfected with empty vector (Figure 2B, lanes 2 and 5), showed amplification of hIL-13Rα2 transcript. Murine actin was amplified to show the integrity of the RNA in cells of murine origin (Figure 2B, lanes 4 to 7).
Figure 2.
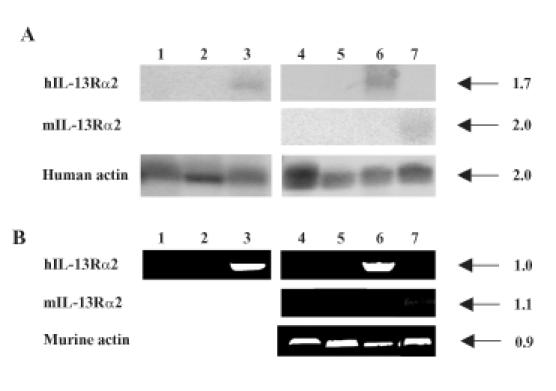
(A) Northern blot of CHO and G-26 cells transfected with hIL-13Rα2 or mIL-13Rα2 and controls. Parental CHO cells (lane 1), mock-transfected CHO (lane 2), CHO-hIL-13Rα2(+) clone 5 (lane 3), parental G-26 cells (lane 4), mock-transfected G-26 (lane 5), G-26-hIL-13Rα2(+) clone 2 (lane 6), and G-26-mIL-13Rα2(+) clone 17 (lane 7) are shown. The size of transcripts is given in kilobases (kb). (B) RT-PCR of hIL-13α2 or mIL-13Rα2 in G-26 and CHO controls and transfected cells. The order of analyzed cells is the same as in (A). The size of PCR-amplified fragments is given in kilobases (kb).
Transfecting mIL-13Rα2 into G-26 Murine Glioma Cells
In order to further analyze IL-13Rα2 in gliomas, a murine glioma model that expresses IL-13Rα2 was needed. Of the several rodent glioma cell lines that we tested, all failed to demonstrate detectable levels of IL-13Rα2 (unpublished). We therefore transfected mIL-13Rα2 into the murine G-26 glioma cell line in order to generate an immunocompetent syngeneic murine glioma model that reflects the expression of IL-13Rα2 found in human HGAs. mIL-13Rα2-transfected clones were analyzed using Northern blot analysis and RT-PCR. Northern blot analysis of total RNA from mIL-13Rα2(+) clone 17 and controls was performed using 32P-labeled mIL-13Rα2. G-26-mIL-13Rα2(+) clone 17 showed hybridization to 32P mIL-13Rα2 cDNA (Figure 2A, lane 7). In addition, RTPCR was performed on total RNA isolated from G-26-mIL-13Rα2(+) clone 17. The resulting fragment of full-length mIL-13Rα2 was demonstrated only in G-26-mIL-13Rα2(+) clone 17 (Figure 2B, lane 7) and not in parental G-26 (Figure 2B, lane 4) or G-26 cells containing vector alone (Figure 2B, lane 5).
CHO-hIL-13α2(+) and G-26-hIL-13Rα2(+) Cells Specifically Bind IL-13 or its Mutant in an IL-4-Independent Manner
IL-13.E13K is a mutant of IL-13 that has been shown to have altered interactions with the physiological shared IL-13/4 receptor, while preserving or even strengthening its affinity towards the HGA-restricted IL-13 receptor [14–16]. To examine the actual hIL-13Rα2 protein expression and whether or not transgene-expressed hIL-13Rα2 is capable of binding IL-13.E13K, iodo-labeled IL-13.E13K was applied to ethanol-fixed cells and autoradiography was performed. CHO and G-26 parental or mock-transfected cells were not bound by the labeled cytokine (Figure 3, column 1, 2, 4 and 5). However, in the absence of blocker, CHO-hIL-13Rα2(+) clone 5 (Figure 3, column 3) and G-26-hIL-13Rα2(+) clone 2 (Figure 3, column 6) showed high retention of labeled IL-13.E13K. To ascertain that this binding was IL-13 receptor-specific, an excess of unlabeled IL-13.E13K was applied prior to 125I IL-13.E13K in a blocking assay. Efficient, more than 75% blocking of labeled IL-13.E13K binding was observed in IL-13Rα2-expressing clones (Figure 3, columns 3 and 6), indicating that the binding of IL-13.E13K is an IL-13 receptor-specific phenomenon. In addition, CHO-hIL-13Rα2-transfected cells and controls were exposed to wild-type 125I IL-13 in the presence and absence of excess IL-13. Only CHO-IL-13Rα2(+) clones and not parental or mock-transfected CHO cells demonstrated retention of labeled wild-type IL-13, which was competed for by excess IL-13 (not shown).
Figure 3.
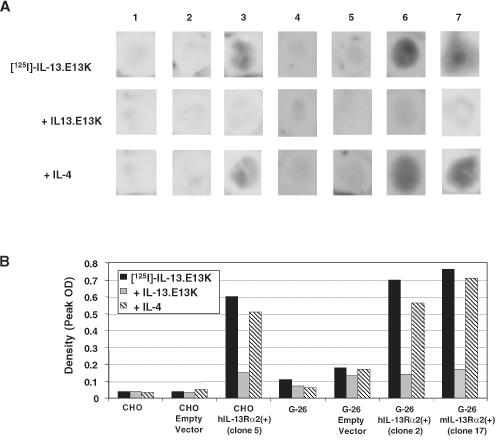
(A) Autoradiography of CHO-hIL-13Rα2(+) clone 5, G-26-hIL-13Rα2(+) clone 2, G-26-mIL-13Rα2(+) clone 17, and control cells using labeled IL-13.E13K. Parental CHO cells (column 1), mock-transfected CHO (column 2), CHO-hIL-13Rα2(+) clone 5 (column 3), parental G-26 cells (column 4), mock-transfected G-26 (column 5), G-26-hIL-13Rα2(+) clone 2 (column 6), and G-26-mIL-13Rα2(+) clone 17 (column 7). The assay was performed in the absence of blocker or in the presence of an excess of unlabeled IL-13.E13K or IL-4. Each experiment was repeated at least four times and the results shown are a representative sample of one of the experiments. (B) Histogram of peak densities of 125I IL-13.E13K binding in the absence or presence of IL-13.E13K and IL-4 in CHO-hIL-13Rα2(+) clone 5, G-26-hIL-13Rα2(+) clone 2, G-26-mIL-13Rα2(+) clone 17, and control cells. Column designations are the same as in (A). Measurements were taken from scanned X-rays shown in (A).
Unlike the physiological IL-13/4 receptor, the glioma-restricted IL-13 receptor has the distinct characteristic of not being competed for by IL-4 [1]. To determine whether hIL-13Rα2 exhibits this property in our CHO-hIL-13Rα2(+) and G-26-hIL-13Rα2(+) transfectants, we performed a competition assay, applying unlabeled IL-4 prior to labeled IL-13.E13K. As it was found on the majority of HGAs, no significant blocking was observed in CHO-hIL-13Rα2(+) clone 5 (Figure 3, column 3) and G-26-hIL-13Rα2(+) clone 2 (Figure 3, column 6).
G-26-mIL-13Rα2(+) Cells Specifically Bind IL-13.E13k in an IL-4-Independent Manner
To examine if the functional characteristics of the IL-13 binding sites resulting from the presence of mIL-13Rα2 inG-26 cells are similar to those of hIL-13Rα2, autoradiography was performed on G-26-mIL-13Rα2(+) clone 17. In the absence of blocker, G-26-mIL-13Rα2(+) clone 17 avidly bound labeled IL-13.E13K (Figure 3, A and B, column 7). The addition of unlabeled IL-13.E13K competed for the IL-13 binding sites on mIL-13Rα2(+) cells, indicating IL-13-specific binding (Figure 3, A and B, column 7). Furthermore, cold IL-4 did not compete for the IL-13 binding sites on G-26-mIL-13Rα2(+) cells (Figure 3, A and B, column 7), indicating that mIL-13Rα2 binds IL-13 independently of IL-4, a characteristic of hIL-13Rα2.
CHO-hIL-13Rα2(+) and G-26-hIL-13Rα2(+) Clones Become Highly Susceptible to Killing by IL-13-Based Cytotoxins
HGAs demonstrate a high sensitivity toward IL-13-based PE derivative-containing cytotoxins [11,14]. To examine if CHO and G-26 cells, which are otherwise resistant to IL-13-based cytotoxins (Figure 4, A and B), gain sensitivity towards IL-13-based cytotoxins when transfected with hIL-13Rα2, we performed a colorimetric cell proliferation assay. CHO-hIL-13Rα2(+) cells, G-26-hIL-13Rα2(+) cells, and controls were exposed to varying concentrations of IL-13.E13K-PE38QQR. The hIL-13Rα2(+) clones, CHO clone 5, and G-26 clone 2 became responsive to the cytotoxin, with IC50 values of 3 and 0.1 ng/ml, respectively, whereas parental and mock-transfected CHO and G-26 cells remained resistant even in the presence of up to 100 ng/ml cytotoxin (Figure 4, A and B). In addition, the susceptibility to IL-13-based cytotoxins varied between hIL-13Rα2(+) clones (Figure 4A for CHO-hIL-13Rα2 clones 2 and 5, and Figure 4B) for G-26-hIL-13Rα2 clones 2 and 5), suggesting that the cell killing is in proportion to the amounts of hIL-13Rα2 transcript and protein present. This was supported by Northern blot analysis of CHO-hIL-13Rα2(+) clone 2 that demonstrated lesser amounts of hIL-13Rα2 mRNA than hIL-13Rα2(+) clone 5 (not shown).
Figure 4.
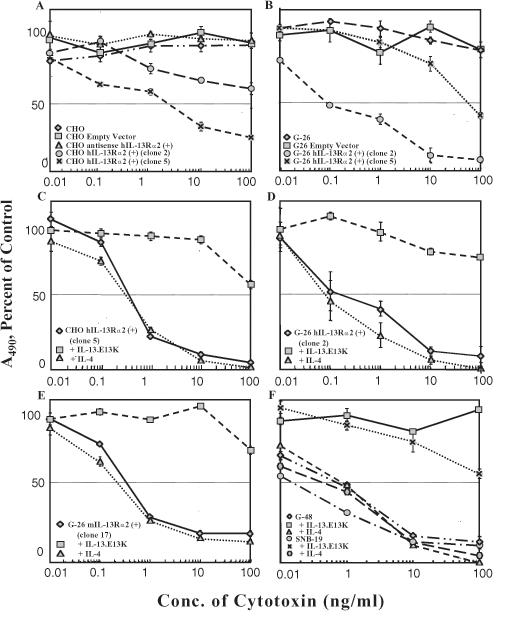
Cytotoxicity using IL-13.E13K-PE38QQR determined in a colorimetric cell proliferation assay. Vertical bars represent SD. (A) CHO parental cells, CHO mock-transfected, and CHO-hIL-13Rα2(+) clones 2 and 5. (B) G-26 parental cells, G-26 mock-transfected, and G-26-hIL-13Rα2(+) clones 2 and 5. (C-E) Neutralization assays in which the cell killing by IL-13.E13K-PE38QQR in IL-13Rα2(+) clones is blocked in the presence of an excess of IL-13.E13K, but not IL-4, in CHO and G-26 cells. (F) G-48a and SNB-19 HGA cells and neutralization of IL-13.E13K-PE38QQR cytotoxicity in the presence of an excess of IL-13.E13K, but not IL-4.
To determine if the susceptibility of hIL-13Rα2 expressing clones to cytotoxin is IL-4-independent and IL-13 receptor-mediated, a neutralization assay was performed using an excess of IL-13.E13K. In the CHO-hIL-13Rα2(+) and G-26-hIL-13Rα2(+) clones tested, the killing by the cytotoxin was completely inhibited in the presence of excess IL-13.E13K but not IL-4 (Figure 4, C and D).
mIL-13Rα2 Transgene Turns G-26 Murine Glioma Cells into Responders to the hIL-13 Cytotoxin
In order to examine if a mIL-13Rα2 transgene changes the G-26 cells' responsiveness to IL-13-based cytotoxin, the G-26-mIL-13Rα2(+) clone 17 cells and controls were treated with IL-13.E13K-PE38QQR. G-26-mIL-13Rα2(+) clone 17 and not the controls were killed by IL-13-based cytotoxin at an IC50 of 0.1 to 1 ng/ml (Figure 4E). Excess IL-13.E13K neutralized the cell killing by IL-13 cytotoxin in mIL-13Rα2-expressing cells, whereas excess IL-4 had no effect on this phenomenon (Figure 4E).
IL-13Rα2(+) Clones are Highly Susceptible to IL-13-Based Cytotoxins Similar to HGA Cells
We demonstrated above that IL-13Rα2-transfected cells became highly responsive to the killing by IL-13-based cytotoxins. In order to juxtapose the susceptibility to IL-13Rα2-transfected cells with human HGAs, HGA cell lines SNB-19 and G-48a were treated with IL-13 cytotoxin. The two HGA cell lines were potently killed by IL-13.E13K-PE38QQR at IC50 values of 0.01 to 0.1 ng/ml (Figure 4F), which is in a close range to what was seen in IL-13Rα2-transfected cells. In addition, neutralization assays were performed using an excess of mutated IL-13, IL-13.E13K, but not IL-4, which blocked the cytotoxicity of IL-13.E13K-PE38QQR in HGAs (Figure 4F).
Loss of Function Studies: Transfecting SNB-19 Cells with Antisense hIL-13Rα2
We demonstrated in gain of function studies that the IL-13Rα2 transgene mimics the IL-13 binding properties of HGA in that it 1) binds IL-13 independent of IL-4; 2) binds a mutant of IL-13, IL-13.E13K; and 3) is able to mediate the cytotoxicity of IL-13.E13K-based cytotoxins. In order to demonstrate that IL-13Rα2, and not other proteins, mediates these IL-13 binding properties in HGAs, loss of function studies were performed. Antisense hIL-13Rα2 was stably transfected into SNB-19 cells in a CMV-containing vector. Clones containing low amounts of hIL-13Rα2 were obtained by a previously described method of selection in which IL-2-based cytotoxins had been used to identify cell lines that were unable to express high-affinity IL-2 receptors [22]. In our experiment, an IL-13-based cytotoxin was used to select for cells that had significantly reduced their expression level of IL-13Rα2. Only as-SNB-19-hIL-13Rα2 transfectants, but not the mock-transfected SNB-19 cells, survived this selection. Total RNA was isolated from mock-transfected SNB-19 cells, as-SNB-19-hIL-13Rα2(+) clone 1, and as-SNB-19-hIL-13Rα2(+) clone 6, and Northern blot analysis was performed. Mock-transfected SNB-19 cells and not as-SNB-19-hIL-13Rα2(+) clones showed strong signal for the presence of hIL-13Rα2 gene expression (Figure 5A).
Figure 5.
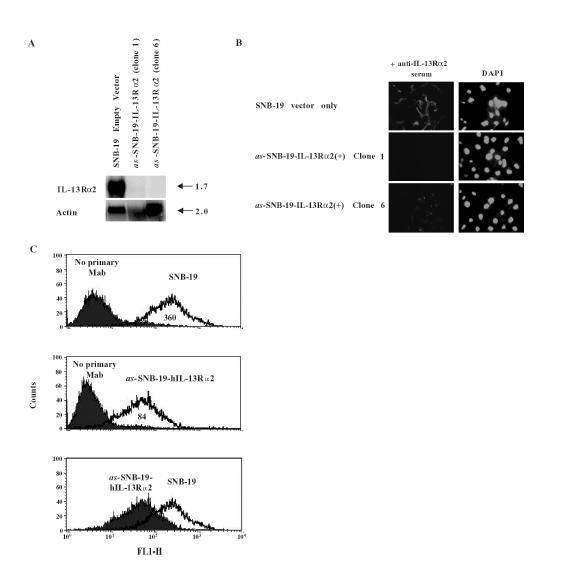
(A) Northern blot of IL-13Rα2 and actin in SNB-19 mock-transfected, and as-SNB-19-hIL-13Rα2(+) clones 1 and 6. (B) IL-13Rα2 immunoreactivity in SNB-19 cells transfected with as-IL-13Rα2 (clones 1 and 6) and with vector only. DAPI nuclear staining was also performed. (C) Flow cytometry performed on SNB-19 parental cells and as-SNB-19-IL-13Rα2(+) clone 6 cells. The bottom graph juxtaposes the distribution of the two experiments. Mean values of each distribution are listed in the area under the peaks. Experiment was repeated three times.
hIL-13Rα2 Protein Expression is Drastically Reduced in as-SNB-19-hIL-13Rα2(+)-Transfected Cells
In order to demonstrate actual reduction in immunoreactive IL-13Rα2 protein expression in SNB-19 cells transfected with as-IL-13Rα2, IH was performed using anti-hIL-13Rα2 serum. Mock-transfected SNB-19 cells demonstrated intense immunoreactivity towards the anti-hIL-13Rα2 serum (Figure 5B). In contrast, such immunoreactivity was not seen in either of the as-SNB-19-hIL-13Rα2(+) clones. Furthermore, flow cytometry performed on as-SNB-19-hIL-13Rα2(+) clone 6 using an anti-hIL-13Rα2 monoclonal antibody demonstrated a decreased amount of hIL-13Rα2 expression compared to parental SNB-19 cells (Figure 5C).
as-SNB-19-hIL-13Rα2(+) Clones no Longer Possess the Restrictive IL-4-Independent IL-13 Binding Site
To examine if the IL-4-independent IL-13 binding sites that are a characteristic of HGAs are still present in SNB-19 cells transfected with antisense hIL-13Rα2, autoradiography was performed. 125I-labeled IL-13.E13K was applied to as-SNB-19-hIL-13Rα2(+) clone 1, clone 6, and their controls. Only parental and mock-transfected SNB-19 cells, but not as-SNB-19-hIL-13Rα2(+) clones, bound 125I-IL-13.E13K (Figure 6A).
Figure 6.
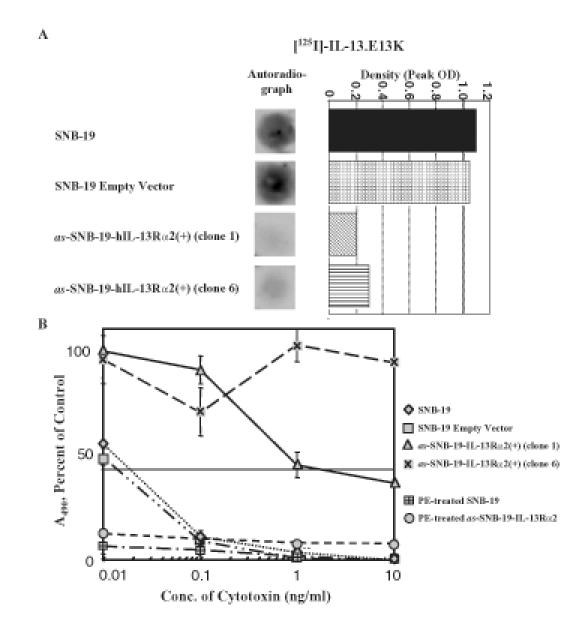
(A) Autoradiography and histogram of peak densities of IL-13 binding sites in SNB-19 parental cells, SNB-19 mock-transfected, and in as-SNB-19-hIL-13Rα2(+) clones treated with labeled IL-13.E13K. Each experiment was repeated at least four times and the results shown are a representative sample of the experiments. (B) Cytotoxicity assay using IL-13.E13K-PE38QQR cytotoxin or wild-type PE in SNB-19 parental cells, SNB-19 mock-transfected, and in as-SNB-19-hIL-13Rα2(+) clones 1 and 6. Vertical bars represent SD.
as-SNB-19-hIL-13Rα2(+) Clones Become Resistant to the Killing by IL-13-Based Cytotoxins
We demonstrated that CHO and G-26 cells transfected with IL-13Rα2 are susceptible to IL-13-based cytotoxins similar to HGA cells. To examine if this susceptibility is truly mediated by hIL-13Rα2 in human HGA cells, as-SNB-19-hIL-13Rα2(+) cells were treated with IL-13-based cytotoxins. Parental and mock-transfected SNB-19 cells were characteristically killed by IL-13.E13K-PE38QQR, an IL-13-based cytotoxin, at an IC50 of 0.01 to 0.1 ng/ml (Figure 6B). Conversely, as-SNB-19-hIL-13Rα2(+) clones 1 and 6 were no longer killed by IL-13.E13K-PE38QQR at similarly low concentrations (Figure 6B). Of importance, as-SNB-19-hIL-13Rα2(+) clone 6 failed to show susceptibility to IL-13.E13K-PE38QQR at concentrations exceeding 1000 times the IC50 of parental and mock-transfected SNB-19 cells (Figure 6B). To examine if this loss of susceptibility to IL-13.E13K-PE38QQR was due to an induced resistance of as-SNB-19-hIL-13Rα2(+) cells to PE, as-SNB-19-hIL-13Rα2(+) clones and controls were exposed to varying concentrations of wild-type toxin. Because PE contains an α2 macroglobulin binding region, its cytotoxicity is IL-13 receptor-independent. as-SNB-19-hIL-13Rα2(+) clones and controls were completely killed by wild-type PE at all concentrations tested (Figure 6B). This result convincingly demonstrated that the resistance of as-SNB-19-hIL-13Rα2(+) clones to IL-13.E13K-PE38QQR is due to the loss of IL-13 binding sites and not to an inadvertently introduced resistance to PE.
Discussion
This study shows, for the first time, a direct link between IL-13Rα2 and the IL-13 binding site properties seen in the vast majority of HGAs. The gain of function studies using cells transfected with hIL-13Rα2 demonstrate that hIL-13Rα2 binds IL-13 in an IL-4-independent manner, a characteristic not found in nondiseased IL-13 binding cells. Moreover, we demonstrate that a mutant of IL-13, which has altered interaction with the shared IL-13/4 receptor, is bound by hIL-13Rα2-expressing cells and internalized, a characteristic feature of HGA-associated IL-13 binding. Importantly, CHO cells and tumor G-26 cells became extremely sensitive to IL-13.E13K-PE38QQR cytotoxin when transfected with IL-13Rα2. IL-13.E13K-PE38QQR cytotoxic fusion protein has been shown, in vitro and in vivo, to be able to safely and efficiently treat HGA tumors [14]. Moreover, we showed for the first time that HGA cells transfected with antisense hIL-13Rα2 1) lose an immunoreactive IL-13Rα2; 2) lose the restricted IL-4-independent IL-13 binding site; and 3) become resistant to IL-13-based cytotoxins. Thus, both gain of function and loss of function studies provide direct evidence for the link between IL-13Rα2 and the restricted IL-13 binding site seen in the vast majority of HGAs.
Furthermore, we demonstrated that murine IL-13Rα2 can mimic the restrictive IL-13 binding properties of human IL-13Rα2 and can be used to generate a syngeneic, fully immunocompetent murine HGA model that reflects the presence and function of hIL-13Rα2 (Mintz et al., manuscript in preparation). mIL-13Rα2 transfected into G-26 murine glioma cells caused the cells to bind IL-13.E13K in an IL-4-independent manner and to become highly susceptible to IL-13-based cytotoxins, characteristics of cells containing hIL-13Rα2. This indicates a conserved function between hIL-13Rα2 and mIL-13Rα2 and confirms the absence of IL-13 species specificity between humans and mice.
Recently, we demonstrated that the gene for hIL-13Rα2 is expressed in HGAs, but little, if at all, in normal tissue, with the exception of the testes [20]. We therefore classified hIL-13Rα2 as a member of the cancer/testis antigen (CTA) family. This finding, together with the direct evidence provided in the current work that identifies IL-13Rα2 as a glioma-restricted receptor for IL-13, demonstrates the potential of efficient and specific targeting of HGAs.
It is tempting to speculate that the expression of IL-13Rα2 may be just one part of a more widespread phenomenon in malignancy. Recently, a number of genes located on chromosome X were found to be abnormally expressed in cancer [23]. CTA are a group of antigens located on the X chromosome and almost exclusively expressed in cancer and the normal testes. It is plausible that epigenetic pathomechanisms, such as abnormal methylation or aneuploidy, are responsible for the abundance of CTAs in malignancy [23]. Of interest, IL-13Rα2 is only sporadically present in low-grade astrocytomas and becomes abundant only with the progression to higher-grade malignancy [24,25]. Thus, its expression contributes and/or reflects an important step in astrocytoma oncogenesis.
Our previous and present studies, suggest that molecular targeting of IL-13Rα2 is an attractive strategy for molecular detection and treatment of HGAs (Figure 7). Targeted gene therapy, targeted radiation, and targeted chemotherapy all have the potential of being applied to patients with HGAs. The evidence presented above suggests that IL-13Rα2 is a unique molecular target to accomplish this goal.
Figure 7.
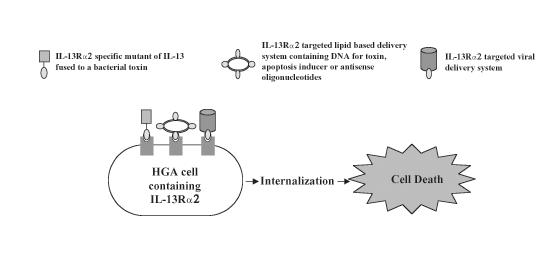
Schemata of possible therapeutic interventions by using IL-13Rα2 as a molecularly defined target.
Abbreviations
- as
antisense
- CHO cells
Chinese hamster ovary cells
- CTA
cancer/testis antigen
- IH
immunohistochemistry
- h
human
- HGA
high-grade astrocytoma
- IL-4
interleukin-4
- IL-13
interleukin-13
- m
murine
- PE
Pseudomonas exotoxin
Footnotes
This work was supported by the National Institutes of Health grant R01 CA74145.
References
- 1.Debinski W, Gibo DM, Hulet SW, Connor JR, Gillespie GY. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin Cancer Res. 1999;5:985–990. [PubMed] [Google Scholar]
- 2.Debinski W, Gibo DM, Slagle B, Powers SK, Gillespie GY. Receptor for interleukin 13 is abundantly and specifically over-expressed in patients with glioblastoma multiforme. Int J Oncol. 1999;15:481–486. doi: 10.3892/ijo.15.3.481. [DOI] [PubMed] [Google Scholar]
- 3.Debinski W, Obiri NI, Powers SK, Pastan I, Puri RK. Human glioma cells over-express receptor for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and Pseudomonas exotoxin. Clin Cancer Res. 1995;1:1253–1258. [PubMed] [Google Scholar]
- 4.Debinski W. An immune regulatory cytokine and high-grade gliomas: an unexpected link. Crit Rev Oncog. 1998;9:256–268. [PubMed] [Google Scholar]
- 5.Minty A, Chalon P, Derocq JM, Dumont X, Guillemot JC, Kaghad M, Labit C, Leplatois P, Liauzun P, Miloux B, Minty C, Casellas P, Loison G, Lupker J, Shire D, Ferrara P, Caput D. Interleukin-13 is a new human lymphokine regulating inflammatory and immune responses. Nature. 1993;36:248–251. doi: 10.1038/362248a0. [DOI] [PubMed] [Google Scholar]
- 6.McKenzie ANJ, Culpepper JR, De Waal Malefyt R, Briere F, Punnonen J, Aversa G, Sato A, Dang W, Cocks BG, Menon S, De Vries JE, Banchereau J, Zurawski G. Interleukin 13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proc Natl Acad Sci USA. 1993;90:3735–3739. doi: 10.1073/pnas.90.8.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zurawski G, de Vries JI. Interleukin 13 elicits a subset of the activities of its close relative interleukin 4. Stem Cells. 1993;12:169–174. doi: 10.1002/stem.5530120204. [DOI] [PubMed] [Google Scholar]
- 8.Zurawski SM, Vega F, Jr, Huyghe B, Zurawski G. Receptors for interleukin 13 and interleukin-4 are complex and share a novel component that functions in signal transduction. EMBO J. 1993;12:26663–26670. doi: 10.1002/j.1460-2075.1993.tb05927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hilton DJ, Zhang J-G, Metcalf D, Alexander WS, Nicola NA, Wilson TA. Cloning and characterization of a binding subunit of the interleukin 13 receptor that is also a component of the interleukin 4 receptor. Proc Natl Acad Sci USA. 1996;93:497–501. doi: 10.1073/pnas.93.1.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aman MJ, Tayebi N, Obiri NI, Puri PK, Modi WS, Leonard WJ. cDNA cloning and characterization of the human interleukin-13 receptor α chain. J Biol Chem. 1996;19:29265–29270. doi: 10.1074/jbc.271.46.29265. [DOI] [PubMed] [Google Scholar]
- 11.Miloux B, Laurent P, Bonnin O, Lupker J, Caput D, Vita N, Ferrara P. Cloning of the human IL-13R alpha1 chain and reconstitution with the IL4R alpha of a functional IL-4/IL-13 receptor complex. FEBS Lett. 1997;401:163–166. doi: 10.1016/s0014-5793(96)01462-7. [DOI] [PubMed] [Google Scholar]
- 12.Debinski W, Obiri NI, Pastan I, Puri RK. A novel chimeric protein composed of interleukin 13 and Pseudomonas exotoxin is highly cytotoxic to human carcinoma cells expressing receptors for interleukin 13 and interleukin 4. J Biol Chem. 1995;270:16775–16780. doi: 10.1074/jbc.270.28.16775. [DOI] [PubMed] [Google Scholar]
- 13.Debinski W, Miner R, Leland P, Obiri NI, Puri RK. Receptor for IL 13 does not interact with IL4 but receptor for IL4 interacts with IL13 on human glioma cells. J Biol Chem. 1996;271:22428–22433. doi: 10.1074/jbc.271.37.22428. [DOI] [PubMed] [Google Scholar]
- 14.Debinski W, Gibo D, Obiri NI, Kealiher A, Puri RK. Novel anti-brain tumor cytotoxins specific for cancer cells. Nat Biotechnol. 1998;16:449–453. doi: 10.1038/nbt0598-449. [DOI] [PubMed] [Google Scholar]
- 15.Thompson JP, Debinski W. Mutants of interleukin 13 with altered reactivity toward interleukin 13 receptors. J Biol Chem. 1999;274:29944–29950. doi: 10.1074/jbc.274.42.29944. [DOI] [PubMed] [Google Scholar]
- 16.Debinski W, Thompson JP. Retargeting interleukin 13 for radioimmunodetection and radioimmunotherapy of human high-grade gliomas. Clin Cancer Res. 1997;5:3143s–3147s. [PubMed] [Google Scholar]
- 17.Nash KT, Thompson JP, Debinski W. Molecular targeting of malignant gliomas with novel multiply-mutated interleukin 13-based cytotoxins. Crit Rev Oncol/Hemato. 2001;l39:87–98. doi: 10.1016/s1040-8428(01)00124-x. [DOI] [PubMed] [Google Scholar]
- 18.Caput D, Laurent P, Kaghad M, Lelias J-M, Lefort S, Vita N, Ferrara P. Cloning and characterization of a specific interleukin (IL-13) binding protein structurally related to the IL-5 receptor α chain. J Biol Chem. 1996;271:16921–16926. doi: 10.1074/jbc.271.28.16921. [DOI] [PubMed] [Google Scholar]
- 19.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 20.Debinski W, Gibo DM. Molecular expression analysis of restrictive receptor for interleukin 13, a brain tumor associated cancer/testis antigen. Mol Med. 2000;6:440–449. [PMC free article] [PubMed] [Google Scholar]
- 21.Debinski W, Slagle-Webb B, Achen MG, Stacker SA, Tulchinsky E, Gillespie GY, Gibo DM. VEGF-D is an X-linked/AP-1 regulated putative onco-angiogen in human glioblastoma multiforme. Mol Med. 2001;7:598–608. [PMC free article] [PubMed] [Google Scholar]
- 22.Furse RK, Malek TR. Selection of internalization-deficient cells by interleukin-2-Pseudomonas exotoxin chimeric protein: the cytoplasmic domain of the interleukin-2 receptor β chain does not contribute to internalization of interleukin-2. J Immunol. 1993;23:3181–3188. doi: 10.1002/eji.1830231221. [DOI] [PubMed] [Google Scholar]
- 23.Mintz A, Debinski W. Cancer genetics/epigenetics and the X chromosome: possible new links for malignant glioma pathogenesis and immune-based therapies. Crit Rev Oncog. 2000;11:77–95. [PubMed] [Google Scholar]
- 24.Debinski W, Slagle B, Gibo DM, Powers SK, Gillespie GY. Expression of a restrictive receptor for interleukin 13 is associated with glial transformation. J Neuro-Oncol. 2000;48:103–111. doi: 10.1023/a:1006446426611. [DOI] [PubMed] [Google Scholar]
- 25.Liu H, Jacobs BS, Liu J, Prayson RA, Estes ML, Barnett GH, Barna BP. Interleukin-13 sensitivity and receptor phenotypes of human glial cell lines: non-neoplastic glia and low-grade astrocytoma differ from malignant glioma. Cancer Immunol Immunother. 2000;49:319–324. doi: 10.1007/s002620000110. [DOI] [PMC free article] [PubMed] [Google Scholar]