

Introduction
Androgens acting via androgen receptor (AR) play essential roles in the prostate development, growth and pathogenesis of benign prostate hyperplasia (BPH) and prostate cancer. Over the last three decades, intensive studies have been carried out to elucidate the molecular basis of androgen action in the prostate. It has been realized that there are two natural potent androgens in the mammal including humans. Although testosterone is the major androgen secreted from the testes, dihydrotestosterone (DHT) is the main androgen in the prostate to mediate the androgen action via the AR. Only one AR has been identified up to date, a member of the steroid/nuclear receptor superfamily, which is a ligand-dependent nuclear transcription factor. When androgens bind to the AR, this results in a conformational change within the AR, leading to the recruitment of co-regulators and transcription factors which mediate androgen-target gene expression. Androgen actions in the prostate can also be modulated by other hormones such as estrogens via estrogen receptors (ER). There are two known isoforms of the ER, ERα and ERβ, which are both co-expressed with AR in the prostate and prostate tumor cells, which provides an anatomical basis for a direct interplay between AR and ER. Although it is well known that androgens are important for prostate development and for the pathogenesis of BPH and prostate cancer, the precise mechanisms as to how androgens control these processes are not yet fully understood. Furthermore, evidence for the direct modulation of androgenAR actions by other hormones within the prostate or prostate tumor cells is emerging, and the clinical implications of these hormones is being explored. In this article, we will assess the importance of androgens, and especially DHT, in prostate physiology and in the pathogenesis of BPH and prostate cancer. We will also review the molecular nature of the interactions between AR and ER and their respective ligands in prostate cells, as well their potential clinical implications.
Molecular Biology And Structure Of the Androgen Receptor
The androgen receptor, a member of the nuclear steroid receptor superfamily, and an associated ligand-dependent nuclear transcription factor were cloned in 1988 (1,2). A polypeptide product of around 910-919 amino acids is encoded by the AR gene, which is located in Xq11-12 (see Figure 1). The AR gene itself is a single-copy gene that spans approximately 90 kilobases of genomic DNA within the long arm of the X chromosome. The encoding region of the AR gene is comprised of 8 exons separated by 7 introns. Like other steroid receptors, the AR is a single polypeptide comprised of relatively distinct domains (Figure 1, 3): an amino-terminal A/B domain, a DNA binding domain (DBD, domain C), a hinge region (domain D), and a ligand-binding domain (LBD, domain E/F).
Figure 1.
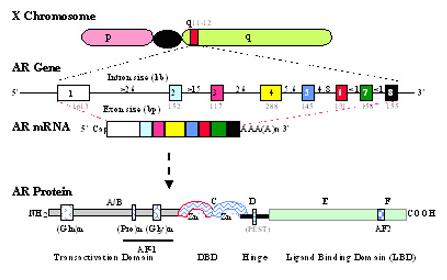
Schematic illustration of the location, exon structure and protein domain structure of the AR gene. (Top) The location of AR gene at the q11-12 of X chromosome. (Middle) The AR gene and its mRNA. The AR gene consists of 8 exons (boxes) and 7 introns (line), and the size of each exon and introns is indicated in kbases. (Bottom) The AR protein. The domains of AR are indicated. Relative positions of glutamine (Gln), proline (Pro) and glycine (Gly) repeats within the N-terminal domain are shown by the indicated boxes. The transactivation function domains, AF-1 and AF-2 are located within the N-terminal domain and ligand-binding domain, respectively. Two zinc fingers in the DNA binding domain and a PEST sequence in hinge region are indicated.
The large amino-terminal domain that comprises nearly half of the AR molecule is encoded by exon 1. It is involved in the transcriptional activation of target genes and contains a transactivation domain, known as Activation Function 1 (AF-1). This domain plays an integral role in AR functions via intramolecular and/or intermolecular interaction with other co-regulators (4). The N-terminal transactivation domain also contains three highly polymorphic direct repeats of amino acid residues: each containing glutamine, proline, and glycine residues, respectively. The expansion of the size of the glutamine homo polymeric segment is related to the spinal and bulbar muscular atrophy (Kennedy's disease) (5), whilst the shortening of the lengths of glutamine and/or glycine repeats may be related to prostate cancer incidence (6-8) although it is not conclusive. Several studies have suggested that the change in the size of glutamine/glycine repeats alters the function of AR due to alterations in its binding affinity to its ligand, interaction with co-regulators, responsivity to phosphorylation, or changes in its AR interdomain interactions (8-11). The mechanisms as to how these changes in the homopolymeric segments result in the pathological outcome are unclear, although it has been shown that the expression of long-tract polyglutamine AR in neuronal cells results in aggregate formation (12) and progressive toxicity (13) in these cells.
The DNA binding domain encoded by exon 2 and 3 contains two “zinc finger” motifs that are hallmarks of all nuclear steroid receptor, being the most highly conserved region within this family (14). The formation of the two zinc fingers involves 8 cysteine residues. These 2 zinc-coordinated stem-loop structures are responsible for the specific interaction with the cognate DNA of target genes, and interact with the major groove of the DNA duplex. For the interaction of the AR and the androgen response element (ARE), the first zinc finger (proximal to the N-terminal domain) is associated with the determination of the sequence specificity of DNA binding, whilst the second finger helps to stabilize the DNA-receptor complex when the DNA binding site is an inverted ARE (14). When the ARE is a direct repeat, the second zinc finger and a part of the hinge region directly interact with the target DNA (15). It is worthy to note that despite their exquisite functional specificity in the physiological context, receptors for androgens, glucocorticoids, progesterone, and mineralocorticoids can recognize the same DNA response element both within in vitro binding assays and also within functional analyses using transient cell transfection (14,16). This paradox may be related to the differential recruitment of various co-regulators upon the ligand-receptor interaction, although it remains to be further investigated.
The C-terminus of the AR is the ligand-binding domain, encoded by the 3′-portion of exon 4 and exons 5-8, and is responsible for mediating the specific high-affinity ligand binding. Studies indicate that androgens interact with the ligand-binding domain mostly through hydrophobic interaction, as well as some hydrogen bonding (17,18). Although the structure of ligand and AR LBD interaction is similar to that of ER and the progesterone receptor, the functional consequences are differentiated. These differential functions may be due at least in part to the differential recruitment of co-regulators (4). The carboxyl-terminus also contains subdomains involved in dimerization and transcriptional activation. The second transactivation function (AF-2) domain of AR resides within the ligand-binding domain (19). Upon ligand binding, this AF-2 domain can interact with co-regulators such as coactivators to affect AR function (4,20). A recent study also indicates that the AR-AF2 domain may also function to stabilize the overall structure of the receptor, allowing the amino terminus to interact with appropriate coactivators (21).
Located between the DNA-binding domain and the steroid-binding domain is the hinge region which contains the nuclear translocation signal. This hinge region of all known mammalian AR also contains a PEST (praline, glutamate, serine, and threonine-rich) sequence, which may function in proteasome-mediated androgen receptor turnover (22).
The AR mediates androgen actions via directed changes in target gene expression (Figure 2). The binding of androgen on the AR results in an AR conformational change that promotes the dissociation of chaperone proteins and facilitates receptor dimerization, nuclear transportation, phosphorylation, and DNA binding (23). Upon the recruitment of co-regulators and general transcription factors, the transcription of a target-gene is either induced or inhibited, and leads ultimately to a change in androgen-target gene expression, and altered cellular or biological structures and functions (see Figure 2). Recently, a number of co-regulators have been identified which can interact with AR at various domains to either enhance or reduce androgen-AR action on target gene transcription (4). Furthermore, AR can be activated in the absence of steroid ligand by other hormones such as IGF-1 (24). Although significant progress has been made in the last decade in understanding the nature of androgen-AR action, the detailed mechanisms of the process from androgen binding on AR, to the alteration of target gene transcription is not fully understood.
Figure 2.
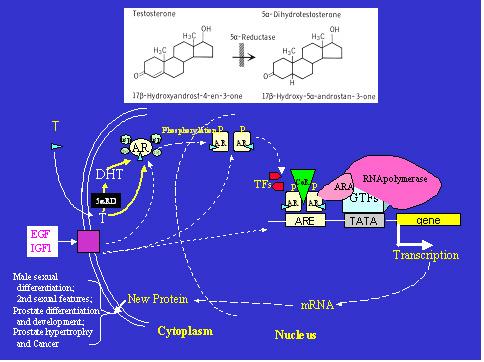
Illustrations of the molecular events of androgen-AR action in a target cell, and the 5α-reductase action. When testosterone (T) enters the cell, it can be converted to dihydrotestosterone (DHT) in the cell by 5α-reductases (5αRD) (top panel). Both T and DHT bind to androgen receptor (AR), resulting in a conformational change in AR and translocation of the receptor complex to nucleus. This complex interacts with androgen response element (ARE) on the target gene, and regulates gene expression in concert with co-regulators (CoR), transcription factors (TF) and the general transcription complex. The changes in androgen-target proteins in the cell eventually affect cellular structure and function related to male sexual differentiation, physiology, and pathophysiology. The function of AR can be activated or modified by non-ligand factors such as growth factors, EGF and IGF-1. GTFs – general transcription factors; ARA – androgen receptor associated proteins; P- phosphorylation; TFs – transcription factors; hsp – heat shock protein.
Androgen Insensitivity Syndrome
The importance of androgen-AR actions in human physiology is best illustrated by the study of androgen insensitivity syndrome, an X-linked recessive condition resulting from AR mutations (3,25). Androgen insensitivity syndrome is classified within a clinical spectrum from complete androgen insensitivity syndrome (CAIS) to partial androgen insensitivity syndrome (PAIS). 46,XY subjects with CAIS are born with female genitalia, and an absent or severely hypoplastic Wolffian ductal system in the presence of testes and normal high or increased androgen secretion. The labia are underdeveloped, the clitoris is normal or small, and the vagina ends blindly. Müllerian derived structures such as uterus and cervix are absent or rudimentary due to normal secretion of anti-Müllerian hormone by the testes (3,26). During puberty, there is a normal or augmented breast development due to an elevated estrogen level originated from the testes, the peripheral aromatization of androstenedione and testosterone by aromatase, and the unopposed estrogenic action by androgens. The pubic and axillary hair is scant or absent, and the prostate is absent in adulthood (3).
The testes of patients with CAIS are located usually within the abdomen or inguinal canal. Post pubertal histological study reveals immature tubular development with Sertoli cells, spermatogonia, and no spermatogenesis (27). Leydig cells are hyperplastic due to the elevated levels of plasma luteinizing hormone which results from unresponsiveness to androgens in the areas of the hypothalamus and/or pituitary (28). Plasma levels of testosterone are normal, high or elevated, whilst DHT levels may be decreased due to a secondary 5α-reductase deficiency (29,30). 46 XY subjects with PAIS have a wide-spectrum of clinical phenotypes ranging from near female phenotype, to severely ambiguous genitalia with gynecomastia, to mild hypospadias, gynecomastia alone, or normal male genitalia with infertility to decreased body hair in adulthood (3,25,26).
Genetic analysis has revealed that a variety of AR mutations are responsible for androgen insensitivity syndrome. To date, more than 490 different mutations throughout all 8 exons in the AR gene have been reported (3,25,26) including the mutation for the largest pedigree of CAIS, (31, see also AR gene mutation database at www.mcgill.ca/androgendb). These mutations range from a single point mutation to the deletion of an entire gene, and can result in various AR dysfunction including impaired androgen binding, impaired DNA binding; impaired cofactor interaction, blocked formation of a functional receptor (deletions, nonsense mutations, splice-junction alterations), decreased AR expression, and an unstable androgen-receptor complex. Depending upon the type of dysfunction, various degrees of functional impairment of the AR occur (3,32). A complete loss of AR function due to a mutation in AR gene results in the complete androgen insensitivity syndrome (CAIS). However, a partial loss of AR function causes the partial androgen insensitivity syndrome (PAIS). Generally mutations that delete the entire AR gene or interrupt the AR open-reading frame, thereby blocking the formation of a functional receptor due to premature termination, aberrant splicing, or even the deletion of partial or entire exon segments, are associated with CAIS. This is due to the fact that the AR DNA- and hormone-binding domain, two critical domains for AR function, are located at the carboxyl terminus of the AR protein. As a consequence, defects that truncate the receptor protein at any point during its synthesis will result in the loss of a portion of one or both of these important functional domains, resulting in a complete loss of AR function.
The majority of AR mutations related to androgen insensitivity, some 67%, are located in AR-LBD. Most of these mutations are single nucleotide substitutions resulting in defects in androgen binding. Approximately 15% of AR mutations are located within the DNA-binding domain, affecting the interaction of androgen-AR complex with the ARE of the androgen-target genes. Some AR mutations affect AR expression, resulting in decreased AR protein levels and decreased androgen binding capacity in the genital skin fibroblasts of affected individuals. These mutant ARs may have subtle differences in their function upon selected target genes when analyzed by in vitro transfection. However, the levels of AR in these individuals are significantly decreased due to altered expression levels of the AR gene arising from AR gene mutation, due to altered translation, post-translational processes or stability of the gene product (33).
Recently, a novel defect has been reported at the post-receptor level in an individual with CAIS as diagnosed based upon clinical and biochemical features (34). The AR gene in this patient is normal. However, the transmission of the activation signal from the AF-1 domain of the AR is disrupted, probably due to a defect in an AR coregulator or coactivator in this patient, indicating that defects in AR signal transmission to target gene expression can also result in androgen insensitivity despite the absence of an AR mutation. The functional significance of androgen-AR pathway in females is poorly understood since the AR gene is located within the X chromosome (35). Recently, Chang's group (36) has generated a cre-lox conditional AR knock-out in female mice, and demonstrated that androgen-AR signaling may also play a role in follicular maturation, female fertility and /or ovulation (8).
The Importance Of DHT In The Prostate
There are two potent natural androgens, testosterone and DHT, which are present in all mammals, including humans (30). Although testosterone is the primary androgen synthesized and secreted from the testes, it functions as a prohormone in certain tissues including the prostate, where it is converted to DHT, a more potent androgen, by 5α-reductase isozymes (30). Although testosterone and DHT bind to the same intracellular androgen receptor (Figure 2), they produce some distinct biological responses. The molecular mechanism for the differential activities and actions of testosterone and DHT is unclear, although differences in receptor binding (37) and DNA interaction (38) between testosterone and DHT have been reported.
5α-reductase isozymes
Steroid 5α-reductase isozymes are microsomal NADPH-dependent proteins that reduce the double bond at the 4-5 position of a variety of C19 and C21 steroids including testosterone (Figure 2). Two 5α-reductase isozymes, type 1 and type 2, encoded by two distinct genes SRD5A1 and SRD5A2 have been identified in the mammal (39-42). The characteristics of two human 5α-reducases are listed in Table 1. Both human 5α-reductase-2 and 5α-reductase-1 genes have five exons and four introns, and encode a highly hydrophobic 254 and 259 amino acid protein with a molecular weight of approximately 28.4 and 29.5 kDa, respectively. 5α-reductase-2 is mapped to the short arm of chromosome 2p23, and 5α-reductase-1 to chromosome 5p15. There is approximately a 50% homology in amino acid compositions between human type-1 and type-2 isozymes. The type-2 isozyme has a much higher affinity than type 1 isozyme for substrates such as testosterone, and is sensitive to finasteride, a 5α-reductase-2 inhibitor, while the type 1 isoform has a low binding affinity and a high capacity for substrate, and is less sensitive to finasteride. The apparent Km (3-10 μM) for the NADPH cofactor is similar for both isozymes. The type 2 isozyme has an acidic pH optimum in the enzymatic assays, while the type 1 has a broad alkaline pH optimum (40,42). However, studies with transfected Chinese hamster ovary cells suggest that the type 2 isozyme may actually have a neutral pH optimum in its native state (43). The affinity of the type 2 isozyme for steroid substrates is higher at a neutral pH than an acidic pH (pH 5.0), suggesting that this isozyme acts primary at neutral pH in the cell (43,44).
Table 1.
Comparison of human 5α-reductase isozymes
| Type 1 | Type 2 | |
|---|---|---|
| Gene structure | 5 exons, 4 introns | 5 exons, 4 introns |
| Gene, chromosome location | SRD5A1, 5p15 | SRD5A2, 2p23 |
| Size | 259 amino acids, Mr=29,462 | 254 amino acids, Mr=28,398 |
| Tissue distribution | Liver, nongenital skin, prostate, brain | Prostate, epididymis, seminal vesicle, genital skin, uterus, liver, breast, hair follicle, placenta, brain |
| pH optima | Neutral to basic | Acidic or neutral |
| Prostate level | Low | High |
| Substrate (T) affinity | Km = 1-5 μM | Km = 0.004-1 μM* |
| Activity in 5α-reductase deficiency | Normal | Mutated |
| Finasteride inhibition | Ki > 300 nM | Ki > 3-5 nM |
Note: - The Km of 5 α-reductase-2 for testosterone (T) is dependent on the assay condition (43).
The functional domains of 5α-reductase-2 have been deduced from in vitro mutagenesis-transfection analysis of natural mutations of 5α-reductase-2 gene in cultured mammalian cells (42,45,46), and mutagenesis analysis of 5α-reductase-1 isozyme (47). Mutations affecting NADPH binding map to the C-terminal half of type-2 isozyme, suggesting that the C-terminal of the isozyme appears to be a cofactor-binding domain, even though consensus adenine dinucleotide-binding sequences have not yet been identified. In contrast, 5α-reductase-2 mutations that affect substrate (testosterone) binding appear to be located at both ends of the protein. However, the amino acid determinants of the substrate binding domain appear to be mainly located at the amino terminal of the protein (42). 5α-reductase-2 is expressed in external genital tissues early in gestation (48). In adulthood, its expression in prostate, genital skin, epididymis, seminal vesicle and liver is relatively high, while it is quite low in other tissues. This isozyme also appears to be expressed in the ovary and hair follicles (49,50).
5α-reductase-1 is detected at birth in the liver and non-genital skin, and is present throughout life. Its expression in embryonic tissues, however, is quite low. In adulthood, it is expressed in non-genital skin, liver and certain brain regions; whereas its presence in the prostate, genital skin, epididymis, seminal vesicle, testis, adrenal and kidney is low. The function of 5α-reductase-1 in human physiology remains to be defined. In the human prostate, both 5α-reductase isozymes are present in epithelial cells and stromal cells, while 5α-reductase-2 is the predominant isozyme expressed within the stromal cells (42,48,51). Both isozymes are expressed in BPH and prostate cancer tissues, and in prostate tumor cells (52-56).
5α-Reductase-2 deficiency syndrome
5α-reductase-2 deficiency syndrome is an inherited autosomal recessive disease resulting from mutations in the 5α-reductase-2 gene (40). To date, over thirty-three mutations in the 5α-reductase-2 gene [(30,57-60) and our unpublished data] have been identified, including mutations in the 3 largest kindreds of male pseudohermaphrodites with 5α-reductase-2 deficiency in the world - the Dominican, New Guinean and Turkish kindred. Mutations have been identified in all five exons of the 5α-reductase-2 gene, and range from a single point defect to a deletion of the entire gene (30). These mutations result in various enzymatic dysfunction including a complete loss of enzymatic activity; impaired binding of substrate and cofactor to the isozyme; blocked formation of a functional isozyme (deletion, nonsense mutation, splice-junction alterations); and an unstable isozyme (42,45,46).
Male pseudohermaphrodites due to 5α-reductase-2 deficiency provide a natural human model to elucidate the importance of DHT in human physiology and pathophysiology. The clinical syndrome of 5α-reductase-2 deficiency was first described in a large Dominican kindred (61), and in two siblings from Dallas (62). Subsequently large cohorts in New Guinea (63) and Turkey have been described (46,64,65) as well as many other cases worldwide (30). Due to 5α-reductase deficiency and a decreased conversion of testosterone to DHT, the affected subjects have high normal to elevated levels of plasma testosterone, and low normal to decreased levels of plasma DHT, resulting in an increased testosterone to DHT ratio at baseline and/or following hCG stimulation (30,66). The levels of plasma and urinary 3α-androstanediol glucuronide, a major metabolite of DHT, are decreased. These subjects also have a global deficit in 5α-reduction as demonstrated by both decreased urinary 5α-reduced metabolites with increased 5β/5α urinary metabolite ratios of both C21 steroids and C19 steroids other than testosterone, i.e. cortisol, corticosterone, 11β-hydroxy-androstenedione and androstenedione (67,68).
Most affected 46,XY subjects with 5α-reductase-2 deficiency have ambiguous external genitalia with a clitoral-like phallus, severely bifid scrotum, pseudovaginal perineoscrotal hypospadias and a rudimentary prostate at birth (58,61,69). Wolffian duct differentiation in affected males is normal, and no Müllerian structures are present. Cryptorchidism is frequently described though it is not invariably present with testes usually found in the inguinal canal or scrotum and occasionally located in the abdomen. With the onset of puberty, the affected males have increased muscle mass and deepening of the voice. The genitalia enlarge with growth of the phallus as well as rugation and hyperpigmentation of the scrotum. The affected adult males have intact libido and are capable of erections (66). Although most subjects studied are generally oligo- or azoospermic due to undescended testes, normal sperm concentrations have been reported in subjects with descended testes (70-72), and the affected men can father children (72,73). These findings indicate that sufficient DHT activity is required for the development of male external genitalia and the prostate, and the descending of testes.
DHT action on prostate development and growth
The prostate is a ductal-acinar gland whose growth and development initiates in fetal life and completes at sexual maturity (74). Development of prostate begins when prostatic buds emerge from the urogenital sinus that derived from endoderm at tenth week in human fetuses, on day 17 in embryonic mice and on day 19 in embryonic rats. Normal prostate development requires many coordinated cellular processes and involves multiple genes and hormonal actions (75). Androgens, mainly DHT, play an essential role in the prostate development and growth as 46,XY subjects with CAIS do not have a prostate, and DHT deficient subjects with 5α-reductase deficiency have a rudimentary prostate. Studies in rabbit, rat (76), and human (77) fetuses have shown that 5α-reductase activity is present in the urogenital sinus and external genital anlage prior to prostate and external genital differentiation. However, 5α-reductase activity is not present in the Wolffian duct, at the time of epididymal, vas deferens, and seminal vesicle differentiation. Thus testosterone, and its 5α-reduced metabolite DHT have selective roles in male sexual differentiation during embryogenesis. Testosterone mediates Wolffian ductal differentiation, while DHT mediates male external genital and prostate differentiation. This hypothesis is confirmed by the study of 5α-reductase-2 deficiency syndrome.
Patients with 5α-reductase-2 deficiency syndrome have decreased circulating and prostatic DHT concentration due to decreased conversion of testosterone to DHT. In the affected male adults, the prostate is nonpalpable on rectal examination (61,78) and is found to be rudimentary on transrectal ultrasound and MRI visualization (69). Prostatic volumes are approximately a tenth of those of age-matched controls (see Figure 3). Histological analysis of prostate biopsy from these patients reveals fibrous connective tissue, smooth muscle, and no identifiable epithelial tissue, suggesting atrophic epithelium or lack of epithelial differentiation (69). Plasma PSA is low or undetectable in these patients. Administration of DHT results in enlargement of the prostate (Figure 3), and an elevation of plasma PSA levels (66,79,80). These findings provide clinical evidence that prostate differentiation and growth as well as circulating PSA level is mediated largely by DHT. However, the mere presence of a prostate in these individuals supports the notion that other growth factors are also involved in its organogenesis. Further supportive evidence is provided by animal studies using 5α-reductase-2 inhibitors and gene knockout. Administration of a 5α-reductase-2 inhibitor, finasteride, in rats (81,82) and monkeys (83) impairs male sexual differentiation and prostate development. The prostate in mice with genetic disruption of either 5α-reductase-2 or 5α-reductase-2 plus 5α-reductase-1 gene is small, but it is puzzling that these knockout animals have normal external genitalia in male offspring (84).
Figure 3.
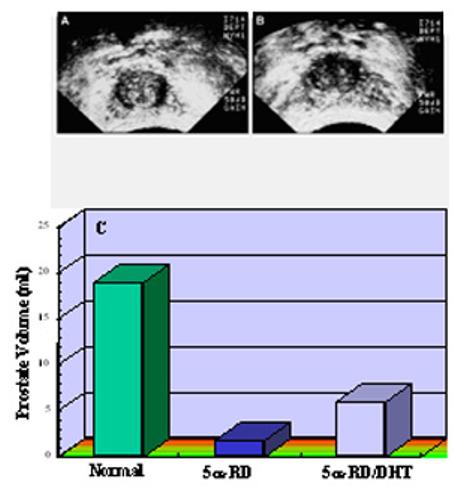
Comparison of prostate sizes between age-matched normal adult males and 5α-reductase-2 deficient patients before (5α-RD) and after (5α-RD/DHT) DHT treatment for 3 to 6 months. Panels A and B show representative sonograms of prostate in a 5α-reductase-2 deficient patient pre-DHT treatment (A) and post 2% DHT cream (B) applied to the genital area for approximately 3 months. Note the crosses at the outer edges of the prostate. Panel C shows the prostate volume as determined by sonogram in 5α-reductase-2 deficient patients and age-matched normal male controls.
DHT action on BPH
BPH is responsible for considerable morbidity due to urethral obstruction. Histological evidence of BPH is found in 50% of males by the age of 50, and 90% of males by the age of 80 (85). The development of BPH is exclusively dependent on androgens, and BPH does not occur in men castrated prior to puberty (86). Although testosterone is the major androgen from the testes, DHT is known to be the major intracellular androgen to mediate androgen actions in the prostate cells (56,87,88). Androgen withdrawal by castration leads to atrophy of prostate gland due to prostatic cell apoptosis (89). In a classic study by White in 1895, 111 men with bladder outlet obstruction were castrated (90). Nearly 87% had rapid atrophy of their enlarged prostate and almost 58% had relief of symptoms. Administration of either DHT or testosterone to castrated dogs results in an increase in intraprostatic DHT, and in BPH (91). However, the concomitant administration of testosterone with a 5α-reductase inhibitor results in a decreased DHT formation and a prevention of BPH (92,93). Administration of finasteride, a specific 5α-reductase-2 inhibitor causes a selective decrease in both circulating and intraprostatic DHT, a significant decrease in prostate size due to prostatic cell apoptosis (94,95), and an improvement of clinical symptoms in patients with BPH (96). In addition, finasteride has been shown to reduce blood flow in both the ventral and dorsal lobes of the rat prostate, which may be mediated by decreasing vascular-derived endothelial growth factor gene expression (97).
The specific 5α-reductase-2 inhibitor, finasteride has now been used for the treatment of BPH. This novel therapeutic strategy evolved from the clinical observation that adult male pseudohermaphrotides with 5α-reductase-2 deficiency and a deficiency in DHT production have rudimentary prostate (61,66,69). Recently, a specific 5α-reductase-1 inhibitor and a dual 5α-reductase-1 and 2 inhibitor have been developed. Pre-clinical and preliminary clinical studies have shown that inhibition of both 5α-reductase isozymes resulted in greater and more consistent suppression of circulating DHT than that observed with the selective inhibitor of 5α-reductase-2, finasteride (98). However, whether inhibition of both 5α-reductase isozymes is more efficacious in the treatment of BPH than 5α-reductase-2 inhibition alone remains to be defined.
DHT action on prostate cancer
Prostate cancer is the most commonly diagnosed and the second leading cause of cancer death in Western males (99,100). The importance of DHT in prostate development and growth, and the dependence of prostate cancer upon androgens indicate that DHT may play a role directly or indirectly in the onset, maintenance, or progression of prostate cancer. Epidemiological studies indicate that although the autopsy prevalence of latent non-infiltrating prostate cancer appears to be similar in many different racial groups, there is a 50-fold difference between Asian males and American black males in the incidence of clinically overt disease (101,102). Analyses of the 5α-reductase activity in these ethnic groups demonstrate that 5α-reductase activity in Asian males is lower than African-Americans and American Whites (103-106), and is increased in Asian males who live in North American compared to those in Asia (106). These data correlate with the progressive increase in clinical prostate cancer incidence in immigrants who have moved from Asia to United States, in the second-generation Asian-American males when compared to the first-generation immigrants (107,108). Furthermore, allelic variants in the 5α-reductase-2 gene may be associated with prostate cancer risk and prostate cancer progression: a variant with higher enzymatic activity is associated with a higher incidence of prostate cancer in population studies (109-111). These data suggest that increased 5α-reductase activity may relate to the pathogenesis and progress of prostate cancer.
Clinical studies of male pseudohermaphrodites with an inherited DHT deficiency due to 5α-reductase-2 gene mutations provide additional evidence to support the concept that DHT plays an important role in prostate cancer development. As stated above, patients with 5α-reductase-2 gene mutations have an underdeveloped prostate and undetectable plasma PSA levels. After following such cases for many years, neither BPH nor prostate cancer has been observed in these patients (66). Further evidence comes from studies with specific 5α-reductase inhibitors. Administration of a 5α-reductase inhibitor prevents the development of spontaneous prostate cancer in both in animals (112) and in humans (113). In a large randomized, placebo controlled chemoprevention trail (113), prophylactic use of finasteride in males over the age of fifty decreases the incidence of prostate cancer by 25% compared to those in the placebo group. However, the tumor malignancy is increased in the finasteride-treated group, a dilemma when using finasteride for the prevention of prostate cancer.
Although both type 1 and 2 5α-reductases are expressed in prostate tumor cells (56), and the administration of 5α-reductase inhibitor alone or in combination with androgen antagonist inhibits prostate tumor cell growth in culture (114,115), the efficacy of finasteride in the treatment of prostate cancer is disappointing (116). Since 5α-reductase-1 activity is 3 to 4 times greater in malignant than in benign prostate tissues, while the 5α-reductase-2 activity is similar in both forms (117), it follows that 5α-reductase-1 activity is implicated in malignancy. Whether inhibition of both isozymes by combinatorial treatment with both specific type 1 and type 2 inhibitors will be more effective in the prevention and therapy of prostate cancer is an interesting strategy, and remains to be evaluated.
Interaction Of Androgen And Estrogen In The Prostate
In vivo, the effect of a hormone is the outcome of the integration of multiple signaling events within its micro- and macro-environments (118). Interactions between androgens and estrogens typically have antagonistic effects. These interactions are documented (119) in various targets including breast (120), ovary (121), uterus (122), testis (123), and brain (124). However, androgen-estrogen interactions within the prostate are complex, and may result in either potentiating or attenuating actions (125-130).
Estrogens and estrogen receptors (ER) in the prostate
Like androgens, estrogens also act through their corresponding ERs. Both AR and ERs are members of the nuclear receptor superfamily and are ligand-dependent nuclear transcription factors (16). ERs mediate the genomic actions of estrogens via the interaction of the hormone-receptor complex with the cognate Estrogen DNA Response Element (ERE) of the target genes. Like the AR, ERs also feature ligand binding, DNA binding, dimerization and transactivation domains (Figure 4, 19). Both agonists and antagonists bind to the ligand-binding domain but produce differential conformational changes (131,132). Similar to the AR, ERs can also interact with co-regulators which modulate estrogen actions (20,133).
Figure 4.
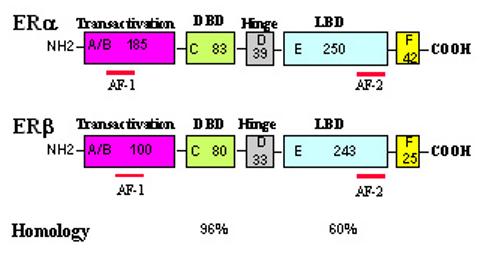
A structural comparison of human ERα and ERβ.
A schematic structural comparison of human ERα and ERβ. Receptor domains are illustrated with different colored boxes, and the approximate size of each domain is indicated.
Recently, it has been found there are at least two ER isoforms (Figure 4) encoded by two distinct genes ERα (134) and ERβ (135-137) in mammals, including humans. These two ER isoforms exhibit high amino acid homology, notably within the DNA binding domain (∼96% homology) and in the ligand binding domain (∼60% homology, 137). The N-terminal A/B domains of these two ER isoforms are more diversified with only 30% homology. ERβ, like ERα, contains a weak activation domain, AF-1, and also a repressor domain (138). In addition, both receptors contain an AF-2 element within the ligand-binding domain. Although ERβ shares many functional characteristics with ERα, it has distinct properties both in the regulation of estrogen-responsive gene expression and in its interaction with other signals (138-141). The partial agonistic activity of 4-hydroxytamoxifen upon ERα mediated changes in gene expression is not observed with ERβ (142). In co-transfection assays, SRC-1 can upregulate ERβ transactivation in the absence of estrogen, whereas SRC-1 only potentiates ERα transactivation in the presence of estrogen (142,143). Paech et al have shown that ERα and ERβ mediate opposing signals in the presence of 17β-estradiol from an AP1 site: through ERα, 17β-estradiol activates transcription, whereas via ERβ, 17β-estradiol inhibits transcription (140). Recently, we demonstrate that estrogen modulation of DHT action in prostate gene expression and prostate tumor cell growth via ER may be also ER-isoform specific (144).
ERα and ERβ can bind to ERE as homodimers which possesses transcription activity within their target genes (142,145,146). ERα and ERβ can also form a heterodimeric complex which also retains DNA-binding capacity and specificity (142,146,147). Thus the genomic actions of estrogens may be transduced through three differential pathways in vivo: as ERα homodimers, as ERβ homodimers and as ERα/ERβ heterodimers (Figure 5).
Figure 5.
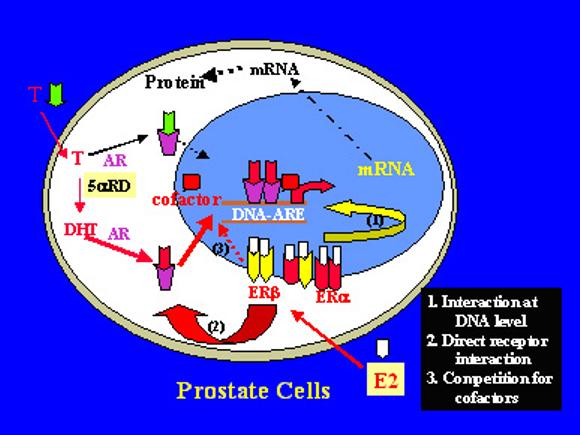
A graphic illustration of ligand-AR and ER cross-talk in the prostate cells.
A graphic illustration of ligand-AR and ER cross-talk in the prostate cells. T – testosterone, E2 – estrogens, and 5αRD - 5α-reductase.
Both ERα and ERβ are expressed in the human and animal prostate along with the AR (148-152). In prostate epithelial cells, the prominent ER-isoform expressed is ERβ, while ERα may be expressed primarily within the stromal cells. Thus, it appears that in the prostate stromal and epithelial cells, two receptors (AR and ERα, or AR and ERβ), or three receptors (AR, ERα and ERβ) may be co-expressed. In prostate cancer cells, either one or two ERs isoforms are expressed. Using RT-PCR, Lau et al have shown that among 3 prostatic cancer cell lines tested, LNCaP and DU145 cells only expressed ERβ mRNA, while PC-3 cells expressed both ERα and ERβ (149). In LAPC-4 and MDA Pca-2b prostatic tumor cells, ERβ is the major ER expressed while the level of ERα is quite low (144). ERα and ERβ are also differentially expressed in human prostate cancer tissues (153-155). These data suggest that prostate cancer cells may differentially express ER isoforms, providing a potential anatomical basis for differential direct estrogen actions in the prostate.
The physiological role of estrogens in the prostate is still obscure. Analyses by gene disruption of ERα, or ERβ, or ERα plus ERβ indicate that estrogens appear to have only minimal effect on prostate differentiation and development (156-158). However, notable changes are present in ER knockout animals (156,158,159). In ERα knockout animals the prostate is enlarged upon aging, although it remains histologically indistinguishable from that of its wild-type littermates. In ERβ knockout mice, prostate epithelial hyperplasia is observed although there appears to be some inconsistency. In ERα/ERβ double knockout mice, the prostate weight and morphology are not different from those of their wild-type littermates up to 8 months of age. These data suggest that either ERα and ERβ do not have significant effects in the prostate, and the observed prostate hyperplasia in ERβ knockout mice is due to some other unknown action; or that ERα and ERβ may possess opposing actions (140,159), resulting in a balance of ER actions when both are either present or absent.
The significance of estrogens in prostate physiology has been further analyzed in aromatase knockout (ArKO) animals. In both young and aged mice, prostate enlargement and hyperplasia, but not malignancy, are demonstrated, which may result directly from elevated androgen levels (158,160). The discrepancy of the prostate changes between ArKO and double ERα/β knockout animals suggests that other pathways in addition to ERα/β may mediate estrogen actions in the prostate. In transgenic male mice overexpressing aromatase, the prostate is rudimentary with squamous epithelial metaplasia (158,161). Taken together, these genetic analyses indicate that estrogens acting via ERs or other pathways play a role in prostate physiology and pathophysiology, which is consistent with classic studies using estrogen analogs in animal studies (128). One of the mechanisms of estrogen action in the prostate could be mediated via the modulation of androgen actions.
Direct interactions of androgens and estrogens in the prostate
In the prostate, the interactions of estrogens and androgens appear very complicated [see recent reviews (127,128)], partially due to the presence of both direct and indirect estrogen actions (Figure 6). Estrogens may either potentiate or inhibit androgen-induced biological responses and gene expression. Thus, estrogens may have either beneficial or deleterious actions with regard to BPH and prostate cancer.
Figure 6.
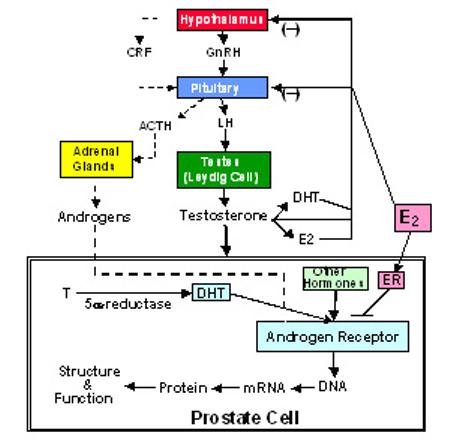
Illustration of the direct and indirect actions of estrogens in the modulation of androgen actions in the prostate cell.
Estrogens have been used as anti-androgens within androgen ablation therapy of prostate cancer (127,162), presumably acting by inhibiting testosterone biosynthesis via the negative feedback of the hypothalamus-pituitary-gonadal axis (see Figure 6). Large phase III trials in multiple clinic centers have shown a beneficial effect of the estrogen treatment of prostate cancer (127,163,164). A double-blind, randomized study of stage D2 prostate cancer patients showed that patients treated with an estrogen analog, diethylstilbestrol, had a significantly longer time to treatment failure (26.4 vs 9.7 months) and longer survival (43.2 vs 28.5 months) than flutamide (an androgen antagonist) treated patients, but with more cardiovascular complications (165). Recently, clinical studies have also shown that estrogen analogs may be effective as a second, or third-line drugs for the treatment of androgen-independent prostate cancer in the improvement of symptoms and reduction of plasma PSA levels (129,166,167). Although the mechanisms of this estrogen effect in androgen-independent prostate cancer are unknown, it is unrelated to the inhibition of androgen biosynthesis since the prostate cancer is in an androgen-independent phase. Actually, direct modulation of androgen actions in the prostate cells by estrogens has been demonstrated. Jarred et al have shown in neonatal rat prostate tissue culture that estrogen inhibits androgen-induced prostate growth and differentiation (168). Recently, we have demonstrated both within in vitro cotransfection assays and within prostate tumor cells that estrogens produce a receptor and ligand specific inhibition of DHT-induced prostate specific antigen (PSA) gene expression and tumor cell growth (144). Direct estrogen modulation of gene expression, cell growth and androgen actions in prostate tumor cells has attracted broad attention (169-171).
On the other hand, synergistic effects of estrogen and androgen interaction have been reported (126,130,151). Some epidemiological studies suggest that high plasma levels of estradiol may be a risk factor for BPH and prostate cancer (128). Animal studies have shown that estrogens can potentiate androgen-induced prostate growth and DNA biosynthesis in the dog (130) and in castrated rats (126). Administration of testosterone alone in the rat results in a low incidence of prostate cancer, but the addition of estrogens produces a significantly higher incidence of prostate cancer (128,172,173). Estrogens also potentiate the androgen-promoting effect on carcinogen-induced prostate cancer (128). Although potentiating androgen action may be a mechanism of carcinogenesis in the prostate, other estrogen effects, including DNA damage via catechol metabolites, also play a significant role (128,174).
In summary, estrogens exert complex and differential actions within the prostate, which may be dose-dependent (175), species-dependent (130), cell-specific, and receptor-isoform and ligand differentiated (144,158). Furthermore, the estrogen modulation of androgen actions in the prostate may be either indirect through the hypothalamus-pituitary gonadal axis to affect testosterone biosynthesis, or exerted directly via ERs in the prostate cells (Figure 6).
Molecular basis of direct ligand AR and ligand ER interactions in the prostate
Growing evidence supports the belief that estrogens acting via ERs directly modulate androgen actions via the AR, both in the prostate and within prostate tumor cells (144,168-170). Since both AR and ER belong to the nuclear receptor superfamily and are ligand-dependent nuclear factors, such a ligand ARER interaction could occur at multiple levels (see Figure 5) as with other nuclear receptor interactions (118,176,177). Previous studies of hormonal interactions in the nuclear receptor system indicate that ligand nuclear receptors can either interact at the DNA level via protein-DNA interaction, or at the protein level via protein-protein interaction (23,118). At the DNA level, ligand receptors may compete for the same DNA binding site, or indirectly affect protein-DNA interactions (pathway 1 of Figure 5). For example the ligand TRα1 can directly compete with ligand ERα on the DNA binding site of oxytocin gene to inhibit estrogen-induced oxytocin gene expression (178,179). At the protein level, ligand receptors may interact directly (pathway 2 of Figure 5, 170,180), or display a squelching effect to compete with common transcriptional factors or co-regulators (pathway 3 of Figure 5, 141,181), resulting in a mutual inhibition of their actions.
Using PSA and a MMTV-promoter directed reporter gene construct, the mechanisms of ligand-AR and ligand-ER interaction have been analyzed. Human PSA gene expression is induced by androgens, and multiple functional AREs have been identified in the proximal and distal promoter region of the human PSA gene (182-184). A consensus ARE is identified in the MMTV promoter (14). It is known that the consensus DNA binding elements of AR and ERs are distinct, and that ERs do not bind to a consensus ARE and vice versa (14,19). However, it is not clear whether ER can bind to the functional AREs of an androgen-target gene such as PSA. Using gel mobility shift assay, we observed that both ERα and ERβ do not directly bind to the AREs in the human PSA gene promoter (our unpublished data), suggesting that direct competition of ARE binding between ligand AR and ER in the PSA promoter is unlikely. However, this does not exclude other interactions at the DNA level. For instance, ligand ER could bind at nearby DNA sequences of a functional ARE to interfere with ligand AR binding due to spatial hindrance; or else ligand ER could affect AR binding via another intermediate factor such as AP-1, SP-1, or NFκB (23,185). Actually, it has been shown that the inhibition of DHT-induced PSA transcription by 17β-estradiol via ERα is dependent on the ERα DNA binding domain (144), indicating that the ligand ERα has a potential to interact at the PSA promoter. However, how this interaction occurs remains to be elucidated.
At the protein level, direct AR-ERα interaction has been reported using yeast and mammalian two-hybrid systems (170), resulting in a mutual inhibition of the ligand AR and ligand ERα action in regulating gene expression. However, such mutual inhibition of ligand AR and ligand ERα action has not been demonstrated in other studies although significant inhibition of androgen action by ligand ERα is present [(169,186) and our unpublished data]. The lack of mutual inhibition of ligand AR and ER actions suggests that direct receptor interaction, or squelching of common transcriptional factors or co-regulators may be not the major or the sole mechanism for estrogen modulation of androgen action in ligand AR-ER interaction. The reason for the discrepancy of ligand AR-ER interaction is unclear. One of the notable factors is the different androgen used in the experiments. Kumar et al (169) and our group induced the androgen-target gene expression by using DHT, a natural potent androgen, while Panet-Raymond et al (170) used mibolerone, a synthetic potent androgen, to stimulate target gene expression. Although both of them bind AR and stimulate target gene expression, differences are observed in AR induced conformational changes when binding to these ligands. The AR helix 12 is split into two shorter helical segments when AR binds to mibolerone (18), while the structure of AR bound to DHT shows a continuous helix 12 (17). Whether the androgenestrogen interaction via their corresponding receptors is AR-ligand specific is an interesting question in of itself.
It is interesting to note that the modulation of androgen action by estrogens is both ER-isoform and ligand specific (144). For instance, 17β-estradiol inhibits androgen induced target gene transcription via ERα, but not ERβ (144,170), while 17α-estradiol inhibits androgen action via either ERα or ERβ (144). 17α-estradiol is a stereoisomer of 17β-estradiol, which also binds to ERα and ERβ (139), and processes estrogenic activity although much weaker (187). This ER-ligand specificity may be due to the differential conformational changes in the ERs upon binding ER-ligands (185,188,189), which could result in a recruitment of various transcriptional factors or co-regulators (189,190), which would then lead to different down-stream actions as stated above. Furthermore, even though both 17β and 17α-estradiol inhibit DHT-induced PSA transcription via ERα in cotransfection assays, the mechanisms of their actions may be different (144). The inhibition of DHT-induced PSA transcription by 17β-estradiol via ERα mainly involves actions at the level of the ERα DNA binding domain, whilst 17α-estradiol action via ERα is mainly mediated through the C-terminal F domain of ERα. The F domain is highly variable within the nuclear receptor superfamily, and there is little homology between ERα and ERβ. The F domain of ERα potentially contains helix 13 and β-strand motifs based on secondary structure calculations (191,192). The F domain of ERα is required for the agonist/antagonist action of tamoxifen (193), and the estrogen activation of ERα/SP1 pathway (192), although its functional significance remains to be further elucidated.
In summary, ligand AR and ER may interact at multiple levels to regulate target gene expression and consequently cell function. Although some efforts have been made to understand the mechanisms of their interaction, further studies are necessary to elucidate in detail the nature of the ligand AR-ER interaction. It is still unclear how ligand receptors interact with their co-regulators; the functional significance of each receptor domain; and most importantly, the biological significance of this AR-ER interaction in prostate physiology and pathophysiology.
Conclusions And Perspectives
Basic and clinical studies, especially in naturally occuring human genetic models with AR and 5α-reductase-2 gene defects, have established the importance of androgen-AR signaling in prostate physiology and pathophysiology. The androgen-AR signaling pathway in the prostate cells is modulated by estrogens via estrogen receptors. It has been reported both in vivo and in vitro that estrogens can either inhibit or potentiate androgen effects within the prostate. Although the mechanisms of these apparent opposing estrogen actions are unknown, it may be related, at least in part, to distinctive receptor isoforms and the ligand specificity of their interplay (144). Since the outcome of androgen-estrogen interaction is species (130), cell-type, coregulator, ER-isoform and ligand specific, the final result can be either inhibitory or stimulatory dependent upon the ER-isoforms and co-regulators expressed in the cells, as well as the ligands used in the experiment.
Estrogens have been used for a long time in hormonal therapy of metastatic prostate cancer, presumably by inhibiting testosterone biosynthesis via the negative feedback of the hypothalamus-pituitary-gonadal axis (see Figure 6) (127,162). The identification of the ERβ isoform, which is expressed within both prostate epithelial cells and prostate tumor cells, and the demonstration of the direct action of estrogens and the direct interaction of ligand AR and ligand ER in prostate cells, provides important information for designing new strategies in the development of new estrogen analogs for prostate cancer prevention and therapy.
Treatment of estrogen analogs in prostate tumor cells has been shown to produce a dose-dependent inhibition of androgen-induced tumor cell growth (144), and to directly inhibit prostate tumor cell growth via ER (149,171). This suggests that ligand ERs within prostate cells have biological significance and clinical implications via their regulation of the androgen-AR signaling pathway and other mechanisms. As the major side-effects of estrogen therapy in prostate cancer are cardiovascular side-effects and gynecomastia, which are related to hepatic first-pass metabolites and ERα actions, the development of ERβ specific ligands with few or no side-effects may provide a new generation of effective regimens for prostate cancer prevention and therapy.
Androgen-independent prostate cancer growth and metastasis is the major cause of prostate cancer mortality. The mechanisms of androgen-independency in prostate cancer cells can be subcategorized into two subtypes: AR-dependent and AR-independent (194). Most of the androgen-independent prostate tumor cells are AR-positive, and their growth may be accounted for the aberrant AR activation by other hormones due to AR amplification, mutation, or abnormal coregulator expression, etc. The direct inhibition of ligand-AR signaling by ligand-ER in prostate cells will have the potential not only to block the effects of AR signaling by androgens originating from the testes and adrenal, but also the effects of aberrant AR activation by other hormones (see Figure 6), providing an effective therapy for patients with androgen-independent prostate cancer.
Acknowledgements
We are very grateful to Dr. Julianne Imperato-McGinley for her long-term and enthusiastic support of our study. These studies are partially supported by NIH grants (DK061004, S/C2P30 CA29502-20, and M01 RR00047), an award from USAMRAA (DAMD17-02-1-0160), and a Fellowship from the Merck Foundation.
REFERENCES
- 1.Chang C, Kokontis J, Liao S. Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science. 1988;240:324–326. doi: 10.1126/science.3353726. [DOI] [PubMed] [Google Scholar]
- 2.Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, Wilson EM. Cloning of human androgen receptor complementary DNA and localization to the X chromosome. Science. 1988;240:327–330. doi: 10.1126/science.3353727. [DOI] [PubMed] [Google Scholar]
- 3.Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS. Androgen receptor defects: historical, clinical, and molecular perspectives. Endocr Rev. 1995;16:271–321. doi: 10.1210/edrv-16-3-271. [DOI] [PubMed] [Google Scholar]
- 4.Heinlein CA, Chang C. Androgen receptor (AR) co-regulators: an overview. Endocr Rev. 2002;23:175–200. doi: 10.1210/edrv.23.2.0460. [DOI] [PubMed] [Google Scholar]
- 5.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 6.Hsing AW, Gao YT, Wu G, Wang X, Deng J, Chen YL, Sesterhenn IA, Mostofi FK, Benichou J, Chang C. Polymorphic CAG and GGN repeat lengths in the androgen receptor gene and prostate cancer risk: a population-based case-control study in China. Cancer Res. 2000;60:5111–5116. [PubMed] [Google Scholar]
- 7.Edwards SM, Badzioch MD, Minter R, Hamoudi R, Collins N, Ardern-Jones A, Dowe A, Osborne S, Kelly J, Shearer R, Easton DF, Saunders GF, Dearnaley DP, Eeles RA. Androgen receptor polymorphisms: association with prostate cancer risk, relapse and overall survival. Int J Cancer. 1999;84:458–465. doi: 10.1002/(sici)1097-0215(19991022)84:5<458::aid-ijc2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 8.Lee DK, Chang C. Endocrine mechanisms of disease: Expression and degradation of androgen receptor: mechanism and clinical implication. J Clin Endocrinol Metab. 2003;88:4043–4054. doi: 10.1210/jc.2003-030261. [DOI] [PubMed] [Google Scholar]
- 9.Chamberlain NL, Driver ED, Miesfeld RL. The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucleic Acids Res. 1994;22:3181–3186. doi: 10.1093/nar/22.15.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchanan G, Yang M, Cheong A, Harris JM, Irvine RA, Lambert PF, Moore NL, Raynor M, Neufing PJ, Coetzee GA, Tilley WD. Structural and functional consequences of glutamine tract variation in the androgen receptor. Hum Mol Genet. 2004;13:1677–1692. doi: 10.1093/hmg/ddh181. [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Udayakumar TS, Vasaitis TS, Brodie AM, Fondell JD. Mechanistic relationship between androgen receptor polyglutamine tract truncation and androgen-dependent transcriptional hyperactivity in prostate cancer cells. J Biol Chem. 2004;279:17319–17328. doi: 10.1074/jbc.M400970200. [DOI] [PubMed] [Google Scholar]
- 12.Piccioni F, Pinton P, Simeoni S, Pozzi P, Fascio U, Vismara G, Martini L, Rizzuto R, Poletti A. Androgen receptor with elongated polyglutamine tract forms aggregates that alter axonal trafficking and mitochondrial distribution in motor neuronal processes. FASEB J. 2002;16:1418–1420. doi: 10.1096/fj.01-1035fje. [DOI] [PubMed] [Google Scholar]
- 13.Avila DM, Allman DR, Gallo JM, McPhaul MJ. Androgen receptors containing expanded polyglutamine tracts exhibit progressive toxicity when stably expressed in the neuroblastoma cell line, SH-SY 5Y. Exp Biol Med (Maywood) 2003;228:982–990. doi: 10.1177/153537020322800815. [DOI] [PubMed] [Google Scholar]
- 14.Freedman LP. Anatomy of the steroid receptor zinc finger region. Endocr Rev. 1992;13:129–145. doi: 10.1210/edrv-13-2-129. [DOI] [PubMed] [Google Scholar]
- 15.Schoenmakers E, Alen P, Verrijdt G, Peeters B, Verhoeven G, Rombauts W, Claessens F. Differential DNA binding by the androgen and glucocorticoid receptors involves the second Zn-finger and a C-terminal extension of the DNA-binding domains. Biochem J. 1999;341(Pt 3):515–521. [PMC free article] [PubMed] [Google Scholar]
- 16.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sack JS, Kish KF, Wang C, Attar RM, Kiefer SE, An Y, Wu GY, Scheffler JE, Salvati ME, Krystek SR, Jr., Weinmann R, Einspahr HM. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc Natl Acad Sci U S A. 2001;98:4904–4909. doi: 10.1073/pnas.081565498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, Otto N, Joschko S, Scholz P, Wegg A, Basler S, Schafer M, Egner U, Carrondo MA. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem. 2000;275:26164–26171. doi: 10.1074/jbc.M004571200. [DOI] [PubMed] [Google Scholar]
- 19.Gronemeyer H. Transcription activation by estrogen and progesterone receptors. Ann Rev Genetics. 1991;25:89–123. doi: 10.1146/annurev.ge.25.120191.000513. review. [DOI] [PubMed] [Google Scholar]
- 20.Glass CK, Rose DW, Rosenfeld MG. Nuclear receptor coactivators. Curr Opin Cell Biol. 1997;9:222–232. doi: 10.1016/s0955-0674(97)80066-x. [DOI] [PubMed] [Google Scholar]
- 21.Chang CY, McDonnell DP. Evaluation of ligand-dependent changes in AR structure using peptide probes. Mol Endocrinol. 2002;16:647–660. doi: 10.1210/mend.16.4.0818. [DOI] [PubMed] [Google Scholar]
- 22.Sheflin L, Keegan B, Zhang W. Spaulding SW 2000 Inhibiting proteasomes in human HepG2 and LNCaP cells increases endogenous androgen receptor levels. Biochem Biophys Res Commun. 276:144–150. doi: 10.1006/bbrc.2000.3424. [DOI] [PubMed] [Google Scholar]
- 23.Aranda A, Pascual A. Nuclear hormone receptors and gene expression. Physiol Rev. 2001;81:1269–1304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- 24.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–5478. [PubMed] [Google Scholar]
- 25.Zhu YS, Imperato-McGinley J. Androgen insensitivity syndrome. In: Martini L, editor. Encyclopedia Of Endocrine Diseases. Elsevier Inc.; San Diego, CA: 2004. pp. 214–220. [Google Scholar]
- 26.Hiort O, Sinnecker GH, Holterhus PM, Nitsche EM, Kruse K. The clinical and molecular spectrum of androgen insensitivity syndromes. Am J Med Genet. 1996;63:218–222. doi: 10.1002/(SICI)1096-8628(19960503)63:1<218::AID-AJMG38>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 27.Hiort O, Holterhus PM. Androgen insensitivity and male infertility. Int J Androl. 2003;26:16–20. doi: 10.1046/j.1365-2605.2003.00369.x. [DOI] [PubMed] [Google Scholar]
- 28.Regadera J, Martinez-Garcia F, Paniagua R, Nistal M. Androgen insensitivity syndrome: an immunohistochemical, ultrastructural, and morphometric study. Arch Pathol Lab Med. 1999;123:225–234. doi: 10.5858/1999-123-0225-AIS. [DOI] [PubMed] [Google Scholar]
- 29.Imperato-McGinley J, Peterson RE, Gautier T, Cooper G, Danner R, Arthur A, Morris PL, Sweeney WJ, Shackleton CHL. Hormonal evaluation of a large kindred with complete androgen insensitivity: Evidence for secondary 5a-reductase deficiency. J Clin Endocrinol Metab. 1982;54:931–941. doi: 10.1210/jcem-54-5-931. [DOI] [PubMed] [Google Scholar]
- 30.Zhu YS, Katz MD, Imperato-McGinley J. Natural potent androgens: lessons from human genetic models. Baillieres Clin Endocrinol Metab. 1998;12:83–113. doi: 10.1016/s0950-351x(98)80478-3. [DOI] [PubMed] [Google Scholar]
- 31.Zhu YS, Cai LQ, Cordero JJ, Canovatchel WJ, Katz MD, Imperato-McGinley J. A novel mutation in the CAG triplet region of exon 1 of androgen receptor gene causes complete androgen insensitivity syndrome in a large kindred. J Clin Endocrinol Metab. 1999;84:1590–1594. doi: 10.1210/jcem.84.5.5695. [DOI] [PubMed] [Google Scholar]
- 32.Brinkmann AO. Molecular basis of androgen insensitivity. Mol Cell Endocrinol. 2001;179:105–109. doi: 10.1016/s0303-7207(01)00466-x. [DOI] [PubMed] [Google Scholar]
- 33.Avila DM, Wilson CM, Nandi N, Griffin JE, McPhaul MJ. Immunoreactive AR and genetic alterations in subjects with androgen resistance and undetectable AR levels in genital skin fibroblast ligand-binding assays. J Clin Endocrinol Metab. 2002;87:182–188. doi: 10.1210/jcem.87.1.8166. [DOI] [PubMed] [Google Scholar]
- 34.Adachi M, Takayanagi R, Tomura A, Imasaki K, Kato S, Goto K, Yanase T, Ikuyama S, Nawata H. Androgen-insensitivity syndrome as a possible coactivator disease. N Engl J Med. 2000;343:856–862. doi: 10.1056/NEJM200009213431205. [DOI] [PubMed] [Google Scholar]
- 35.Brown CJ, Goss SJ, Lubahn DB, et al. Androgen receptor locus on the human X chromosome: Regional localization to Xq11-12 and description of a DNA polymorphism. Am J Hum Genet. 1989;44:264–269. [PMC free article] [PubMed] [Google Scholar]
- 36.Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, Lin H, Yeh SD, Altuwaijri S, Zhou X, Xing L, Boyce BF, Hung MC, Zhang S, Gan L, Chang C. Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues. Proc Natl Acad Sci U S A. 2002;99:13498–13503. doi: 10.1073/pnas.212474399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilbert DM, Griffen JE, Wilson JD. Characterization of the cytosol androgen receptor of the human prostate. J Clin Endocrinol Metab. 1983;56:113–120. doi: 10.1210/jcem-56-1-113. [DOI] [PubMed] [Google Scholar]
- 38.Kovacs WJ, Griffin JE, Weaver DD, Carlson BR, Wilson JD. A mutation that causes lability of the androgen receptor under conditions that normally promote transformation to the DNA-binding state. J Clin Invest. 1984;73:1095–1104. doi: 10.1172/JCI111295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andersson S, Russell DW. Structural and biochemical properties of cloned and expressed human and rat steroid 5α-reductases. Proc Natl Acad Sci USA. 1990;87:3640–3644. doi: 10.1073/pnas.87.10.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andersson S, Berman DM, Jenkins EP, Russell DW. Deletion of steroid 5α-reductase-2 gene in male pseudohermaphroditism. Nature. 1991;354:159–161. doi: 10.1038/354159a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Labrie F, Sugimoto Y, Luu-The V, Simard J, Lachance Y, Bachvarov D, Leblanc G, Durocher F, Paquet N. Structure of human type II 5 alpha-reductase gene. Endocrinol. 1992;131:1571–1573. doi: 10.1210/endo.131.3.1505484. [DOI] [PubMed] [Google Scholar]
- 42.Russell DW, Wilson JD. Steroid 5 alpha-reductase: two genes/two enzymes. Ann Rev Biochem. 1994;63:25–61. doi: 10.1146/annurev.bi.63.070194.000325. [DOI] [PubMed] [Google Scholar]
- 43.Thigpen AE, Cala KM, Russell DW. Characterization of chinese hamster ovary cell lines expressing human steroid 5 alpha-reductase isozymes. J Biol Chem. 1993;268:17404–17412. [PubMed] [Google Scholar]
- 44.Faller B, Farley D, Nick H. Finasteride: a slow-binding 5 alpha-reductase inhibitor. Biochemistry. 1993;32:5705–5710. doi: 10.1021/bi00072a028. [DOI] [PubMed] [Google Scholar]
- 45.Wigley WC, Prihoda JS, Mowszowicz I, Mendonca BB, New MI, Wilson JD, Russell DW. Natural mutagenesis study of the human steroid 5 alpha-reductase 2 isozyme. Biochemistry. 1994;33:1265–1270. doi: 10.1021/bi00171a029. [DOI] [PubMed] [Google Scholar]
- 46.Can S, Zhu YS, Cai LQ, Ling Q, Katz MD, Akgun S, Shackleton CHL, Imperato-McGinley J. The identification of 5α-reductase-2 and 17β-hydroxysteroid dehydrogenase-3 gene defects in male pseudohermaphrodites from a Turkish kindred. J Clin Endocrinol Metab. 1998;83:560–569. doi: 10.1210/jcem.83.2.4535. [DOI] [PubMed] [Google Scholar]
- 47.Thigpen AE, Russell DW. Four amino acid segments in steroid 5α-reductase-1 confers insensitivity to finasteride, a competitive inhibitor. J Biol Chem. 1992;267:8577. [PubMed] [Google Scholar]
- 48.Thigpen AE, Silver RI, Guileyardo JM, Casey ML, McConnell JD, Russell DW. Tissue distribution and ontogeny of steroid 5α-reductase isozyme expression. J Clin Invest. 1993;92:903–910. doi: 10.1172/JCI116665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eicheler W, Tuohimaa P, Vilja P, Adermann K, Forssmann WG, Aumuller G. Immunocytochemical localization of human 5 alpha-reductase 2 with polyclonal antibodies in androgen target and non-target human tissues. J Histochem Cytochem. 1994;42:667–675. doi: 10.1177/42.5.8157936. [DOI] [PubMed] [Google Scholar]
- 50.Eicheler W, Dreher M, Hoffmann R, Happle R, Aumuller G. Immunohistochemical evidence for differential distribution of 5 alpha-reductase isoenzymes in human skin. Br J Dermatol. 1995;133:371–376. doi: 10.1111/j.1365-2133.1995.tb02663.x. [DOI] [PubMed] [Google Scholar]
- 51.Silver RI, Wiley EL, Thigpen AE, Guileyardo JM, McConnell JD, Russell DW. Cell type specific expression of steroid 5 alpha-reductase 2. J Urol. 1994;152:438–442. doi: 10.1016/s0022-5347(17)32758-1. [DOI] [PubMed] [Google Scholar]
- 52.Smith CM, Ballard SA, Wyllie MG, Masters JR. Comparison of testosterone metabolism in benign prostatic hyperplasia and human prostate cancer cell lines in vitro. J Steroid Biochem Mol Biol. 1994;50:151–159. doi: 10.1016/0960-0760(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 53.Guillemette C, Hum DW, Belanger A. Evidence for a role of glucuronosyltransferase in the regulation of androgen action in the human prostatic cancer cell line LNCaP. J Steroid Biochem Mol Biol. 1996;57:225–231. doi: 10.1016/0960-0760(95)00258-8. [DOI] [PubMed] [Google Scholar]
- 54.Delos S, Carsol JL, Fina F, Raynaud JP, Martin PM. 5alpha-reductase and 17beta-hydroxysteroid dehydrogenase expression in epithelial cells from hyperplastic and malignant human prostate. Int J Cancer. 1998;75:840–846. doi: 10.1002/(sici)1097-0215(19980316)75:6<840::aid-ijc5>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 55.Negri-Cesi P, Poletti A, Colciago A, Magni P, Martini P, Motta M. Presence of 5α-reductase isozymes and aromatase in human prostate cancer cells and in benign prostate hyperplastic tissue. Prostate. 1998;34:283–291. doi: 10.1002/(sici)1097-0045(19980301)34:4<283::aid-pros6>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 56.Zhu YS, Cai LQ, You X, Cordero JJ, Huang Y, Imperato-McGinley J. Androgen-induced PSA gene expression is mediated via DHT in LNCaP cells. J Androl. 2003;24:681–687. doi: 10.1002/j.1939-4640.2003.tb02727.x. [DOI] [PubMed] [Google Scholar]
- 57.Wilson JD, Griffin JE, Russell DW. Steroid 5α-reductase 2 deficiency. Endocr Rev. 1993;14:577–593. doi: 10.1210/edrv-14-5-577. [DOI] [PubMed] [Google Scholar]
- 58.Zhu YS, Imperato-McGinley J. Pseudohermaphroditism, male, due to 5α-reductase-2 deficiency. In: Martini L, editor. ENCYCLOPEDIA OF ENDOCRINE DISEASES. Elsevier Inc.; San Diego, CA: 2004. pp. 131–135. [Google Scholar]
- 59.Nordenskjold A, Magnus O, Aagenaes O, Knudtzon J. Homozygous mutation (A228T) in the 5alpha-reductase type 2 gene in a boy with 5alpha-reductase deficiency: genotype-phenotype correlations. Am J Med Genet. 1998;80:269–272. [PubMed] [Google Scholar]
- 60.Vilchis F, Mendez JP, Canto P, Lieberman E, Chavez B. Identification of missense mutations in the SRD5A2 gene from patients with steroid 5alpha-reductase 2 deficiency. Clin Endocrinol (Oxf) 2000;52:383–387. doi: 10.1046/j.1365-2265.2000.00941.x. [DOI] [PubMed] [Google Scholar]
- 61.Imperato-McGinley J, Guerrero L, Gautier T, Peterson RE. Steroid 5α-reductase deficiency in man: An inherited form of male pseudohermaphroditism. Science. 1974;186:1213–1216. doi: 10.1126/science.186.4170.1213. [DOI] [PubMed] [Google Scholar]
- 62.Walsh PC, Madden JD, Harrod MJ, Goldstein JL, MacDonald PC, Wilson JD. Familial incomplete male pseudohermaphroditism, type 2. Decreased dihydrotestosterone formation in pseudovaginal perineoscrotal hypospadias. N Engl J Med. 1974;291:944–949. doi: 10.1056/NEJM197410312911806. [DOI] [PubMed] [Google Scholar]
- 63.Imperato-McGinley J, Miller M, Wilson JD, Peterson RE, Shackleton CHL, Gajdusek DC. A cluster of male pseudohermaphrodites with 5 alpha-reductase deficiency in Papua New Guinea. Clin Endocrinol (Oxf) 1991;34:293–298. doi: 10.1111/j.1365-2265.1991.tb03769.x. [DOI] [PubMed] [Google Scholar]
- 64.Akgun S, Ertel N, Imperato-McGinley J, Sayli B, Shackleton CHL. Familial male pseudohermaphroditism in a Turkish village due to 5α-reductase deficiency. Am J Med. 1986;81:267–274. doi: 10.1016/0002-9343(86)90262-7. [DOI] [PubMed] [Google Scholar]
- 65.Imperato-McGinley J, Akgun S, Ertel NH, Sayli B, Shackleton CHL. The coexistence of male pseudohermaphrodites with 17-ketosteroid reductase deficiency and 5α-reductase deficiency within a Turkish kindred. Clin Endocrinol (Oxf) 1987;27:135–143. doi: 10.1111/j.1365-2265.1987.tb00849.x. [DOI] [PubMed] [Google Scholar]
- 66.Imperato-McGinley J, Zhu YS. Androgens and male physiology - The syndrome of 5α-reductase-2 deficiency. Mol Cell Endocrinol. 2002;198:51–59. doi: 10.1016/s0303-7207(02)00368-4. [DOI] [PubMed] [Google Scholar]
- 67.Imperato-McGinley J, Peterson RE, Gautier T, Shackleton CHL, Arthur A. Decreased urinary C19 and C21 steroid 5a-metabolites in parents of male pseudohermaphrodites with 5α-reductase deficiency: Detection of carriers. J Clin Endocrinol Metab. 1985;60:553–558. doi: 10.1210/jcem-60-3-553. [DOI] [PubMed] [Google Scholar]
- 68.Imperato-McGinley J, Shackleton CHL, Stoner E. C19 and C21 5β/5α-metabolite ratios in subjects treated with the 5α-reductase inhibitor finasteride: Comparison of male pseudohermaphrodites with inherited 5α-reductase deficiency. J Clin Endocrinol Metab. 1990;70(3):777–782. doi: 10.1210/jcem-70-3-777. [DOI] [PubMed] [Google Scholar]
- 69.Imperato-McGinley J, Gautier T, Zirinsky K, Hom T, Palomo O, Stein E, Vaughan ED, Markisz J, Ramirez de Arellano E, Kazam E. Prostate visualization studies in males homozygous and heterozygous for 5α-reductase deficiency. J Clin Endocrinol Metab. 1992;75:1022–1026. doi: 10.1210/jcem.75.4.1400866. [DOI] [PubMed] [Google Scholar]
- 70.Cantu JM, Hernandez-Montes H, del Castillo V, Cortés-Gallegos V, Sandoval R, Armendares S, Parra A. Potential fertility in incomplete male pseudohermaphroditism type 2. Rev Invest Clin. 1976;28:177–182. [PubMed] [Google Scholar]
- 71.Cai L-Q, Fratianni CM, Gautier T, Imperato-McGinley J. Dihydrotestosterone regulation of semen in male pseudohermaphrodites with 5 alpha-reductase-2 deficiency. J Clin Endocrinol Metab. 1994;79(2):409–414. doi: 10.1210/jcem.79.2.8045956. [DOI] [PubMed] [Google Scholar]
- 72.Katz MD, Kligman I, Cai LQ, Zhu YS, Fratianni CM, Zervoudakis I, Rosenwaks Z, Imperato-McGinley J. Paternity by intrauterine insemination with sperm from a man with 5alpha-reductase-2 deficiency. N Engl J Med. 1997;336:994–997. doi: 10.1056/NEJM199704033361404. [DOI] [PubMed] [Google Scholar]
- 73.Nordenskjold A, Ivarsson SA. Molecular characterization of 5 alpha-reductase type 2 deficiency and fertility in a Swedish family. J Clin Endocrinol Metab. 1998;83:3236–3238. doi: 10.1210/jcem.83.9.5125. [DOI] [PubMed] [Google Scholar]
- 74.Wilson JD. Sexual differentiation. Ann Rev Physiol. 1978;40:270–306. doi: 10.1146/annurev.ph.40.030178.001431. [DOI] [PubMed] [Google Scholar]
- 75.Marker PC, Donjacour AA, Dahiya R, Cunha GR. Hormonal, cellular, and molecular control of prostatic development. Dev Biol. 2003;253:165–174. doi: 10.1016/s0012-1606(02)00031-3. [DOI] [PubMed] [Google Scholar]
- 76.Frederiksen DW, Wilson JD. Partial characterization of the nuclear reduced nicotinamide adenine dinucleotide phosphate: delta 4-3-ketosteroid 5 alpha- oxidoreductase of rat prostate. J Biol Chem. 1971;246:2584–2593. [PubMed] [Google Scholar]
- 77.Siiteri P, Wilson JD. Testosterone formation and metabolism during male sexual differentiation in the human embryo. J Clin Endocrinol Metab. 1974;38:113–125. doi: 10.1210/jcem-38-1-113. [DOI] [PubMed] [Google Scholar]
- 78.Peterson RE, Imperato-McGinley J, Gautier T, Sturla E. Male pseudohermaphroditism due to steroid 5α-reductase deficiency. Am J Med. 1977;62:170–191. doi: 10.1016/0002-9343(77)90313-8. [DOI] [PubMed] [Google Scholar]
- 79.Mendonca BB, Inacio M, Costa EM, Arnhold IJ, Silva FA, Nicolau W, Bloise W, Russel DW, Wilson JD. Male pseudohermaphroditism due to steroid 5alpha-reductase 2 deficiency. diagnosis, psychological evaluation, and management. Medicine (Baltimore) 1996;75:64–76. doi: 10.1097/00005792-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 80.Zhu YS, Sung GF. 5α-reductase isozymes in the prostate. J Med Sci. 2005;25:1–11. [PMC free article] [PubMed] [Google Scholar]
- 81.Imperato-McGinley J, Sanchez RS, Spencer JR, Yee B, Vaughan ED. Comparison of the effects of the 5α-reductase inhibitor finasteride and the antiandrogen flutamide on prostate and genital differentiation: Dose-response studies. Endocrinol. 1992;131:1149–1156. doi: 10.1210/endo.131.3.1324152. [DOI] [PubMed] [Google Scholar]
- 82.Spencer JR, Torrado T, Sanchez RS, Vaughan ED, Jr., Imperato-McGinley J. Effects of flutamide and finasteride on rat testicular descent. Endocrinol. 1991;129:741–748. doi: 10.1210/endo-129-2-741. [DOI] [PubMed] [Google Scholar]
- 83.Prahalada S, Tarantal AF, Harris GS, Ellsworth KP, Clarke AP, Skiles GL, MacKenzie KI, Kruk LF, Ablin DS, Cukierski MA, Peter CP, vanZwieten MJ, Hendrickx AG. Effects of finasteride, a type 2 5-alpha reductase inhibitor, on fetal development in the rhesus monkey (Macaca mulatta) Teratology. 1997;55:119–131. doi: 10.1002/(SICI)1096-9926(199702)55:2<119::AID-TERA1>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 84.Mahendroo MS, Cala KM, Hess DL, Russell DW. Unexpected virilization in male mice lacking steroid 5 alpha-reductase enzymes. Endocrinology. 2001;142:4652–4662. doi: 10.1210/endo.142.11.8510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Berry SJ, Coffey DS, Walsh PC, Ewing LL. The development of human benign prostatic hyperplasia with age. J Urol. 1984;132:474–479. doi: 10.1016/s0022-5347(17)49698-4. [DOI] [PubMed] [Google Scholar]
- 86.Huggins C, Stevens R. The effect of castration on benign hypertrophy of the prostate in man. J Urol. 1940;43:705–714. doi: 10.1016/j.juro.2016.10.075. [DOI] [PubMed] [Google Scholar]
- 87.Bruchovsky N, Wilson JD. The conversion of testosterone to 5α-androstane-17β-ol-one by rat prostate in vivo and in vitro. J Biolumin Chemilumin. 1968;243:2012–2021. [PubMed] [Google Scholar]
- 88.Anderson KM, Liao S. Selective retention of dihydrotestosterone by prostatic nuclei. Nature. 1968;219:277–279. doi: 10.1038/219277a0. [DOI] [PubMed] [Google Scholar]
- 89.English HF, Kyprianou N, Isaacs JT. Relationship between DNA fragmentation and apoptosis in the programmed cell death in the rat prostate following castration. Prostate. 1989;15:233–251. doi: 10.1002/pros.2990150304. [DOI] [PubMed] [Google Scholar]
- 90.White JW. The results of double castration in hypertrophy of the prostate. Ann Surg. 1895;22:1–80. doi: 10.1097/00000658-189507000-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moore RJ, Gazak J, Quebbeman JF, Wilson JD. Concentration of dihydrotestosterone and 3α-androstanediol in naturally occurring and androgen-induced prostatic hyperplasia in the dog. J Clin Invest. 1979;64:1003–1010. doi: 10.1172/JCI109536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wenderoth UK, George FW, Wilson JD. The effect of a 5α-reductase inhibitor on androgen-mediated growth of the dog prostate. Endocrinol. 1983;113:569–573. doi: 10.1210/endo-113-2-569. [DOI] [PubMed] [Google Scholar]
- 93.Brooks JR, Berman C, Glitzer MS, Gordon LR, Primka RL, Reynolds GE. Effect of a new 5α-reductase inhibitor on size, histologic characteristics and androgen concentrations of the canine prostate. Prostate. 1982;3:35–44. doi: 10.1002/pros.2990030107. [DOI] [PubMed] [Google Scholar]
- 94.Rittmaster RS, Norman RW, Thomas LN, Rowden G. Evidence for atrophy and apoptosis in the prostates of men given finasteride. J Clin Endocrinol Metab. 1996;81:814–819. doi: 10.1210/jcem.81.2.8636309. [DOI] [PubMed] [Google Scholar]
- 95.Rittmaster RS, Manning AP, Wright AS, Thomas LN, Whitefield S, Norman RW, Lazier CB, Rowden G. Evidence for atrophy and apoptosis in the ventral prostate of rats given the 5 alpha-reductase inhibitor finasteride. Endocrinol. 1995;136:741–748. doi: 10.1210/endo.136.2.7835306. [DOI] [PubMed] [Google Scholar]
- 96.Gormley GJ, Stoner E, Bruskewitz RC, Imperato-McGinley J, Walsh PC, McConnell JD, Andriole GL, Geller J, Bracken BR, Tenover JS, Vaughan ED, Pappas F, Taylor A, Binkowitz B, Ng J. The effect of finasteride in men with benign prostatic hyperplasia. N Engl J Med. 1992;327:1185–1191. doi: 10.1016/s0022-5347(02)80349-4. [DOI] [PubMed] [Google Scholar]
- 97.Levine AC, Liu XH, Greenberg PD, Eliashvili M, Schiff JD, Aaronson SA, Holland JF, Kirschenbaum A. Androgens induce the expression of vascular endothelial growth factor in human fetal prostatic fibroblasts. Endocrinology. 1998;139:4672–4678. doi: 10.1210/endo.139.11.6303. [DOI] [PubMed] [Google Scholar]
- 98.Clark RV, Hermann DJ, Cunningham GR, Wilson TH, Morrill BB, Hobbs S. Marked suppression of dihydrotestosterone in men with benign prostatic hyperplasia by dutasteride, a dual 5alpha-reductase inhibitor. J Clin Endocrinol Metab. 2004;89:2179–2184. doi: 10.1210/jc.2003-030330. [DOI] [PubMed] [Google Scholar]
- 99.Weir HK, Thun MJ, Hankey BF, Ries LA, Howe HL, Wingo PA, Jemal A, Ward E, Anderson RN, Edwards BK. Annual report to the nation on the status of cancer, 1975-2000, featuring the uses of surveillance data for cancer prevention and control. J Natl Cancer Inst. 2003;95:1276–1299. doi: 10.1093/jnci/djg040. [DOI] [PubMed] [Google Scholar]
- 100.Jemal A, Tiwari RC, Murray T, Ghafoor A, Samuels A, Ward E, Feuer EJ, Thun MJ. Cancer statistics, 2004. CA Cancer J Clin. 2004;54:8–29. doi: 10.3322/canjclin.54.1.8. [DOI] [PubMed] [Google Scholar]
- 101.Boring CC, Squires TS, Tong T, Montgomery S. Cancer statistics, 1994. CA Cancer J Clin. 1994;44:7–26. doi: 10.3322/canjclin.44.1.7. [DOI] [PubMed] [Google Scholar]
- 102.Nomura AMY, Kolonel LN. Prostate cancer: a current prospective. Epidemiologic Rev. 1991;13:200–227. doi: 10.1093/oxfordjournals.epirev.a036069. [DOI] [PubMed] [Google Scholar]
- 103.Ross RK, Bernstein L, Judd H, Hanisch R, Pke M, Henderson B. Serum testosterone levels in young black and white men. J Natl Cancer Inst. 1986;76:45–48. [PubMed] [Google Scholar]
- 104.Ross RK, Bernstein L, Lobo FA, Shimizu H, Stanczyk FZ, Pike MC, Henderson BE. 5α-reductase activity and risk of prostate cancer among Japanese and U.S. white and black males. Lancet. 1992;339:887–889. doi: 10.1016/0140-6736(92)90927-u. [DOI] [PubMed] [Google Scholar]
- 105.Lookingbill DP, Demers LM, Wang C, Leung A, Rittmaster RS, Santen RJ. Clinical and biochemical parameters of androgen action in normal healthy Caucasian versus Chinese subjects. J Clin Endocrinol Metab. 1991;72:1242–1248. doi: 10.1210/jcem-72-6-1242. [DOI] [PubMed] [Google Scholar]
- 106.Wu AH, Whittemore AS, Kolonel LN, John EM, Gallagher RP, West DW, Hankin J, Teh CZ, Dreon DM, Paffenbarger RS., Jr. Serum androgens and sex hormone-binding globulins in relation to lifestyle factors in older African-American, White, and Asian men in the United States and Canada. Cancer Epidemiol Biomarkers Prev. 1995;4:735–741. [PubMed] [Google Scholar]
- 107.Haenszel W, Kurihara M. Studies of Japanese immigrants. I. Mortality from cancer and other diseases among Japanese in the United States. J Natl Cancer Inst. 1968;40:43–68. [PubMed] [Google Scholar]
- 108.Shimizu H, Ross RK, Bernstein L, Yatani R, Henderson BE, Mack TM. Cancer of the prostate and breast among Japanese and white immigrants in Los Angeles County. Br J Cancer. 1991;63:963–966. doi: 10.1038/bjc.1991.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Makridakis N, Ross RK, Pike MC, Chang L, Stanczyk FZ, Kolonel LN, Shi CY, Yu MC, Henderson BE, Reichardt JK. A prevalent missense substitution that modulates activity of prostatic steroid 5alpha-reductase. Cancer Res. 1997;57:1020–1022. [PubMed] [Google Scholar]
- 110.Makridakis NM, Ross RK, Pike MC, Crocitto LE, Kolonel LN, Pearce CL, Henderson BE, Reichardt JK. Association of mis-sense substitution in SRD5A2 gene with prostate cancer in African-American and Hispanic men in Los Angeles, USA. Lancet. 1999;354:975–978. doi: 10.1016/S0140-6736(98)11282-5. [DOI] [PubMed] [Google Scholar]
- 111.Jaffe JM, Malkowicz SB, Walker AH, MacBride S, Peschel R, Tomaszewski J, Van Arsdalen K, Wein AJ, Rebbeck TR. Association of SRD5A2 genotype and pathological characteristics of prostate tumors. Cancer Res. 2000;60:1626–1630. [PubMed] [Google Scholar]
- 112.Homma Y, Kaneko M, Kondo Y, Kawabe K, Kakizoe T. Inhibition of rat prostate carcinogenesis by a 5alpha-reductase inhibitor, FK143. J Natl Cancer Inst. 1997;89:803–807. doi: 10.1093/jnci/89.11.803. [DOI] [PubMed] [Google Scholar]
- 113.Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, Lieber MM, Cespedes RD, Atkins JN, Lippman SM, Carlin SM, Ryan A, Szczepanek CM, Crowley JJ, Coltman CA., Jr. The Influence of Finasteride on the Development of Prostate Cancer. N Engl J Med On Line. 2003 doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 114.Bologna M, Muzi P, Biordi L, Festuccia C, Vicentini C. Finasteride dose-dependently reduces the proliferation rate of the LNCaP human prostatic cancer cell line in vitro. Urology. 1995;45:282–290. doi: 10.1016/0090-4295(95)80019-0. [DOI] [PubMed] [Google Scholar]
- 115.Zaccheo T, Giudici D, Panzeri A, di Salle E. Effect of the 5 alpha-reductase inhibitor PNU 156765, alone or in combination with flutamide, in the Dunning R3327 prostatic carcinoma model in rats. Chemotherapy. 1998;44:284–292. doi: 10.1159/000007125. [DOI] [PubMed] [Google Scholar]
- 116.Gormley GJ. Role of 5 alpha-reductase inhibitors in the treatment of advanced prostatic carcinoma. Urol Clin North Am. 1991;18:93–98. [PubMed] [Google Scholar]
- 117.Iehle C, Radvanyi F, Gil Diez dM, Ouafik LH, Gerard H, Chopin D, Raynaud JP, Martin PM. Differences in steroid 5alpha-reductase isoenzymes expression between normal and pathological human prostate tissue. J Steroid Biochem Mol Biol. 1999;68:189–195. doi: 10.1016/s0960-0760(99)00030-8. [DOI] [PubMed] [Google Scholar]
- 118.Zhu YS, Dellovade TL, Pfaff DW. Interactions between hormonal and environmental signals on hypothalamic neurons: molecular mechanisms signalling environmental events. Trends Endocrinol Metab. 1997;8:111–115. doi: 10.1016/s1043-2760(97)00032-5. [DOI] [PubMed] [Google Scholar]
- 119.Kreitmann B, Bayard F. Androgen interaction with the oestrogen receptor in human tissues. J Steroid Biochem. 1979;11:1589–1595. doi: 10.1016/0022-4731(79)90354-6. [DOI] [PubMed] [Google Scholar]
- 120.Burak WE, Jr., Quinn AL, Farrar WB, Brueggemeier RW. Androgens influence estrogen-induced responses in human breast carcinoma cells through cytochrome P450 aromatase. Breast Cancer Res Treat. 1997;44:57–64. doi: 10.1023/a:1005782311558. [DOI] [PubMed] [Google Scholar]
- 121.Ghersevich S, Poutanen M, Tapanainen J, Vihko R. Hormonal regulation of rat 17 beta-hydroxysteroid dehydrogenase type 1 in cultured rat granulosa cells: effects of recombinant follicle- stimulating hormone, estrogens, androgens, and epidermal growth factor. Endocrinology. 1994;135:1963–1971. doi: 10.1210/endo.135.5.7956918. [DOI] [PubMed] [Google Scholar]
- 122.Jellinck PH, Affleck A, Newcombe AM. Estrogen-androgen interaction: selective inhibition of estrogen-induced uterine peroxidase by testosterone and 5 alpha-dihydrotestosterone. Can J Biochem Cell Biol. 1983;61:779–783. doi: 10.1139/o83-098. [DOI] [PubMed] [Google Scholar]
- 123.Moger WH. Studies on the interaction of 5 alpha-androstane-3 beta, 17 beta-diol with the testicular estrogen receptor. Can J Physiol Pharmacol. 1980;58:124–128. doi: 10.1139/y80-020. [DOI] [PubMed] [Google Scholar]
- 124.Roselli CE, Abdelgadir SE, Resko JA. Regulation of aromatase gene expression in the adult rat brain. Brain Res Bull. 1997;44:351–357. doi: 10.1016/s0361-9230(97)00214-1. [DOI] [PubMed] [Google Scholar]
- 125.Martikainen PM, Makela SI, Santti RS, Harkonen PL, Suominen JJ. Interaction of male and female sex hormones in cultured rat prostate. Prostate. 1987;11:291–303. doi: 10.1002/pros.2990110402. [DOI] [PubMed] [Google Scholar]
- 126.Suzuki K, Ito K, Suzuki T, Honma S, Yamanaka H. Synergistic effects of estrogen and androgen on the prostate: effects of estrogen on androgen- and estrogen-receptors, BrdU uptake, immunohistochemical study of AR, and responses to antiandrogens. Prostate. 1995;26:151–163. doi: 10.1002/pros.2990260307. [DOI] [PubMed] [Google Scholar]
- 127.Dowling AJ, Tannock IF. Systemic treatment for prostate cancer. Cancer Treat Rev. 1998;24:283–301. doi: 10.1016/s0305-7372(98)90062-7. [DOI] [PubMed] [Google Scholar]
- 128.Bosland MC. Chapter 2: The Role of Steroid Hormones in Prostate Carcinogenesis. J Natl Cancer Inst Monogr. 2000;2000:39–66. doi: 10.1093/oxfordjournals.jncimonographs.a024244. [DOI] [PubMed] [Google Scholar]
- 129.Farrugia D, Ansell W, Singh M, Philp T, Chinegwundoh F, Oliver RT. Stilboestrol plus adrenal suppression as salvage treatment for patients failing treatment with luteinizing hormone-releasing hormone analogues and orchidectomy. BJU Int. 2000;85:1069–1073. doi: 10.1046/j.1464-410x.2000.00673.x. [DOI] [PubMed] [Google Scholar]
- 130.Ehrlichman RJ, Isaacs JT, Coffey DS. Differences in the effects of estradiol on dihydrotestosterone induced prostatic growth of the castrate dog and rat. Invest Urol. 1981;18:466–470. [PubMed] [Google Scholar]
- 131.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 132.Katzenellenbogen JA, O'Malley BW, Katzenellenbogen BS. Tripartite steroid hormone receptor pharmacology: interaction with multiple effector sites as a basis for the cell- and promoter-specific action of these hormones. Mol Endocrinol. 1996;10:119–131. doi: 10.1210/mend.10.2.8825552. [DOI] [PubMed] [Google Scholar]
- 133.Norris JD, Fan D, Stallcup MR, McDonnell DP. Enhancement of estrogen receptor transcriptional activity by the coactivator GRIP-1 highlights the role of activation function 2 in determining estrogen receptor pharmacology. J Biol Chem. 1998;273:6679–6688. doi: 10.1074/jbc.273.12.6679. [DOI] [PubMed] [Google Scholar]
- 134.Greene GL, Gilna P, Waterfield M, Baker A, Hort Y, Shine J. Sequence and expression of human estrogen receptor complementary DNA. Science. 1986;231:1150–1154. doi: 10.1126/science.3753802. [DOI] [PubMed] [Google Scholar]
- 135.Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mosselman S, Polman J, Dijkema R. ER beta: identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996;392:49–53. doi: 10.1016/0014-5793(96)00782-x. [DOI] [PubMed] [Google Scholar]
- 137.Ogawa S, Inoue S, Watanabe T, Hiroi H, Orimo A, Hosoi T, Ouchi Y, Muramatsu M. The complete primary structure of human estrogen receptor beta (hER beta) and its heterodimerization with ER alpha in vivo and in vitro. Biochem Biophys Res Commun. 1998;243:122–126. doi: 10.1006/bbrc.1997.7893. [DOI] [PubMed] [Google Scholar]
- 138.Hall JM, McDonnell DP. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. 1999;140:5566–5578. doi: 10.1210/endo.140.12.7179. [DOI] [PubMed] [Google Scholar]
- 139.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, Gustafsson JA. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinol. 1997;138:863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 140.Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 141.Vasudevan N, Zhu YS, Daniel S, Koibuchi N, Chin WW, Pfaff D. Crosstalk between oestrogen receptors and thyroid hormone receptor isoforms results in differential regulation of the preproenkephalin gene. J Neuroendocrinol. 2001;13:779–790. doi: 10.1046/j.1365-2826.2001.00693.x. [DOI] [PubMed] [Google Scholar]
- 142.Tremblay GB, Tremblay A, Copeland NG, Gilbert DJ, Jenkins NA, Labrie F, Giguere V. Cloning,chromosomal localization, and functional analysis of the murine estrogen receptor b. Mol Endocrinol. 1997;11:353–365. doi: 10.1210/mend.11.3.9902. [DOI] [PubMed] [Google Scholar]
- 143.Onate SA, Tsai SY, Tsai M-J, O'Malley BW. Sequence and Characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354–1357. doi: 10.1126/science.270.5240.1354. [DOI] [PubMed] [Google Scholar]
- 144.Zhu YS, Cai LQ, Huang Y, Fish J, Wang L, Zhang ZK, Imperato-McGinley J. Receptor isoform and ligand specific modulation of DHT-induced PSA gene expression and prostate tumor cell growth by estrogens. J.Androl. 2005 doi: 10.2164/jandrol.05002. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schwabe JWR, Chapman L, Finch JT, Rhodes D. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell. 1993;75:567–578. doi: 10.1016/0092-8674(93)90390-c. [DOI] [PubMed] [Google Scholar]
- 146.Cowley SM, Hoare S, Mosselman S, Parker MG. Estrogen receptors alpha and beta form heterodimers on DNA. J Biol Chem. 1997;272:19858–19862. doi: 10.1074/jbc.272.32.19858. [DOI] [PubMed] [Google Scholar]
- 147.Hall JM, Chang CY, McDonnell DP. Development of peptide antagonists that target estrogen receptor beta- coactivator interactions. Mol Endocrinol. 2000;14:2010–2023. doi: 10.1210/mend.14.12.0561. [DOI] [PubMed] [Google Scholar]
- 148.Chang WY, Prins GS. Estrogen receptor-beta: implications for the prostate gland. Prostate. 1999;40:115–124. doi: 10.1002/(sici)1097-0045(19990701)40:2<115::aid-pros7>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 149.Lau KM, LaSpina M, Long J, Ho SM. Expression of estrogen receptor (ER)-alpha and ER-beta in normal and malignant prostatic epithelial cells: regulation by methylation and involvement in growth regulation. Cancer Res. 2000;60:3175–3182. [PubMed] [Google Scholar]
- 150.Pelletier G. Localization of androgen and estrogen receptors in rat and primate tissues. Histol Histopathol. 2000;15:1261–1270. doi: 10.14670/HH-15.1261. [DOI] [PubMed] [Google Scholar]
- 151.Srinivasan G, Campbell E, Bashirelahi N. Androgen, estrogen, and progesterone receptors in normal and aging prostates. Microsc Res Tech. 1995;30:293–304. doi: 10.1002/jemt.1070300405. [DOI] [PubMed] [Google Scholar]
- 152.Prins GS, Marmer M, Woodham C, Chang W, Kuiper G, Gustafsson JA, Birch L. Estrogen receptor-beta messenger ribonucleic acid ontogeny in the prostate of normal and neonatally estrogenized rats. Endocrinol. 1998;139:874–883. doi: 10.1210/endo.139.3.5827. [DOI] [PubMed] [Google Scholar]
- 153.Bonkhoff H, Fixemer T, Hunsicker I, Remberger K. Estrogen receptor gene expression and its relation to the estrogen- inducible HSP27 heat shock protein in hormone refractory prostate cancer. Prostate. 2000;45:36–41. doi: 10.1002/1097-0045(20000915)45:1<36::aid-pros4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 154.Gyftopoulos K, Sotiropoulou G, Varakis I, Barbalias GA. Cellular distribution of retinoic acid receptor-alpha in benign hyperplastic and malignant human prostates: comparison with androgen, estrogen and progesterone receptor status. Eur Urol. 2000;38:323–330. doi: 10.1159/000020301. [DOI] [PubMed] [Google Scholar]
- 155.Leav I, Lau KM, Adams JY, McNeal JE, Taplin ME, Wang J, Singh H, Ho SM. Comparative studies of the estrogen receptors beta and alpha and the androgen receptor in normal human prostate glands, dysplasia, and in primary and metastatic carcinoma. Am J Pathol. 2001;159:79–92. doi: 10.1016/s0002-9440(10)61676-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417. doi: 10.1210/edrv.20.3.0370. [DOI] [PubMed] [Google Scholar]
- 157.Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors on mouse reproductive phenotypes. Development. 2000;127:4277–4291. doi: 10.1242/dev.127.19.4277. [DOI] [PubMed] [Google Scholar]
- 158.Jarred RA, McPherson SJ, Bianco JJ, Couse JF, Korach KS, Risbridger GP. Prostate phenotypes in estrogen-modulated transgenic mice. Trends Endocrinol Metab. 2002;13:163–168. doi: 10.1016/s1043-2760(02)00575-1. [DOI] [PubMed] [Google Scholar]
- 159.Weihua Z, Warner M, Gustafsson JA. Estrogen receptor beta in the prostate. Mol Cell Endocrinol. 2002;193:1–5. doi: 10.1016/s0303-7207(02)00089-8. [DOI] [PubMed] [Google Scholar]
- 160.McPherson SJ, Wang H, Jones ME, Pedersen J, Iismaa TP, Wreford N, Simpson ER, Risbridger GP. Elevated androgens and prolactin in aromatase-deficient mice cause enlargement, but not malignancy, of the prostate gland. Endocrinology. 2001;142:2458–2467. doi: 10.1210/endo.142.6.8079. [DOI] [PubMed] [Google Scholar]
- 161.Li X, Nokkala E, Yan W, Streng T, Saarinen N, Warri A, Huhtaniemi I, Santti R, Makela S, Poutanen M. Altered structure and function of reproductive organs in transgenic male mice overexpressing human aromatase. Endocrinology. 2001;142:2435–2442. doi: 10.1210/endo.142.6.8211. [DOI] [PubMed] [Google Scholar]
- 162.Huggins C, Hodges CV. Studies on prostatic cancer I. the effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941;1:293–297. doi: 10.3322/canjclin.22.4.232. [DOI] [PubMed] [Google Scholar]
- 163.Byar DP. Proceedings: The Veterans Administration Cooperative Urological Research Group's studies of cancer of the prostate. Cancer. 1973;32:1126–1130. doi: 10.1002/1097-0142(197311)32:5<1126::aid-cncr2820320518>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 164.Leuprolide Study Group Leuprolide versus diethylstilbestrol for metastatic prostate cancer. The Leuprolide Study Group. N Engl J Med. 1984;311:1281–1286. doi: 10.1056/NEJM198411153112004. [DOI] [PubMed] [Google Scholar]
- 165.Chang A, Yeap B, Davis T, Blum R, Hahn R, Khanna O, Fisher H, Rosenthal J, Witte R, Schinella R, Trump D. Double-blind, randomized study of primary hormonal treatment of stage D2 prostate carcinoma: flutamide versus diethylstilbestrol. J Clin Oncol. 1996;14:2250–2257. doi: 10.1200/JCO.1996.14.8.2250. [DOI] [PubMed] [Google Scholar]
- 166.Ferro MA. Use of intravenous stilbestrol diphosphate in patients with prostatic carcinoma refractory to conventional hormonal manipulation. Urol Clin North Am. 1991;18:139–143. [PubMed] [Google Scholar]
- 167.Smith DC, Redman BG, Flaherty LE, Li L, Strawderman M, Pienta KJ. A phase II trial of oral diethylstilbesterol as a second-line hormonal agent in advanced prostate cancer [see comments] Urology. 1998;52:257–260. doi: 10.1016/s0090-4295(98)00173-3. [DOI] [PubMed] [Google Scholar]
- 168.Jarred RA, Cancilla B, Prins GS, Thayer KA, Cunha GR, Risbridger GP. Evidence that estrogens directly alter androgen-regulated prostate development. Endocrinology. 2000;141:3471–3477. doi: 10.1210/endo.141.9.7648. [DOI] [PubMed] [Google Scholar]
- 169.Kumar MV, Leo ME, Tindall DJ. Modulation of androgen receptor transcriptional activity by the estrogen receptor. J Androl. 1994;15:534–542. [PubMed] [Google Scholar]
- 170.Panet-Raymond V, Gottlieb B, Beitel LK, Pinsky L, Trifiro MA. Interactions between androgen and estrogen receptors and the effects on their transactivational properties. Mol Cell Endocrinol. 2000;167:139–150. doi: 10.1016/s0303-7207(00)00279-3. [DOI] [PubMed] [Google Scholar]
- 171.Ho SM. Estrogens and anti-estrogens: key mediators of prostate carcinogenesis and new therapeutic candidates. J Cell Biochem. 2004;91:491–503. doi: 10.1002/jcb.10759. [DOI] [PubMed] [Google Scholar]
- 172.Noble RL. Prostate carcinoma of the Nb rat in relation to hormones. Int Rev Exp Pathol. 1982;23:113–159. [PubMed] [Google Scholar]
- 173.Pollard M, Luckert PH, Schmidt MA. Induction of prostate adenocarcinomas in Lobund Wistar rats by testosterone. Prostate. 1982;3:563–568. doi: 10.1002/pros.2990030605. [DOI] [PubMed] [Google Scholar]
- 174.Mobley JA, Bhat AS, Brueggemeier RW. Measurement of oxidative DNA damage by catechol estrogens and analogues in vitro. Chem Res Toxicol. 1999;12:270–277. doi: 10.1021/tx980128i. [DOI] [PubMed] [Google Scholar]
- 175.vom Saal FS, Timms BG, Montano MM, Palanza P, Thayer KA, Nagel SC, Dhar MD, Ganjam VK, Parmigiani S, Welshons WV. Prostate enlargement in mice due to fetal exposure to low doses of estradiol or diethylstilbestrol and opposite effects at high doses. Proc Natl Acad Sci U S A. 1997;94:2056–2061. doi: 10.1073/pnas.94.5.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Glass CK, Holloway JM, Devary OV, Rosenfeld MG. The thyroid hormone receptor binds with opposite transcriptional effects to a common sequence motif in thyroid hormone and estrogen response elements. Cell. 1988;54:313–323. doi: 10.1016/0092-8674(88)90194-8. [DOI] [PubMed] [Google Scholar]
- 177.Zhu YS, Yen PM, Chin WW, Pfaff DW. Estrogen and thyroid hormone interaction on regulation of gene expression. Proc Natl Acad Sci USA. 1996;93:12587–12592. doi: 10.1073/pnas.93.22.12587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Adan RA, Cox JJ, Beischlag TV, Burbach JP. A composite hormone response element mediates the transactivation of the rat oxytocin gene by different classes of nuclear hormone receptors. Mol Endocrinol. 1993;7:47–57. doi: 10.1210/mend.7.1.8383287. [DOI] [PubMed] [Google Scholar]
- 179.Vasudevan N, Davidkova G, Zhu YS, Koibuchi N, Chin WW, Pfaff D. Differential interaction of estrogen receptor and thyroid hormone receptor isoforms on the rat oxytocin receptor promoter leads to differences in transcriptional regulation. Neuroendocrinology. 2001;74:309–324. doi: 10.1159/000054698. [DOI] [PubMed] [Google Scholar]
- 180.Zhang XK, Hoffmann B, Tran PB, Graupner G, Pfahl M. Retinoid X receptor is an auxiliary protein for thyroid hormone and retinoic acid receptors. Nature. 1992;355:441–446. doi: 10.1038/355441a0. [DOI] [PubMed] [Google Scholar]
- 181.Zhang X, Jeyakumar M, Bagchi MK. Ligand-dependent cross-talk between steroid and thyroid hormone receptors. Evidence for common transcriptional coactivator(s) J Biol Chem. 1996;271:14825–14833. [PubMed] [Google Scholar]
- 182.Riegman PH, Vlietstra RJ, van der Korput JA, Brinkmann AO, Trapman J. The promoter of the prostate-specific antigen gene contains a functional androgen responsive element. Mol Endocrinol. 1991;5:1921–1930. doi: 10.1210/mend-5-12-1921. [DOI] [PubMed] [Google Scholar]
- 183.Schuur ER, Henderson GA, Kmetec LA, Miller JD, Lamparski HG, Henderson DR. Prostate-specific antigen expression is regulated by an upstream enhancer. J Biol Chem. 1996;271:7043–7051. doi: 10.1074/jbc.271.12.7043. [DOI] [PubMed] [Google Scholar]
- 184.Cleutjens KB, van Eekelen CC, van der Korput HA, Brinkmann AO, Trapman J. Two androgen response regions cooperate in steroid hormone regulated activity of the prostate-specific antigen promoter. J Biol Chem. 1996;271:6379–6388. doi: 10.1074/jbc.271.11.6379. [DOI] [PubMed] [Google Scholar]
- 185.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 186.Ochiai I, Matsuda K, Nishi M, Ozawa H, Kawata M. Imaging analysis of subcellular correlation of androgen receptor and estrogen receptor alpha in single living cells using green fluorescent protein color variants. Mol Endocrinol. 2004;18:26–42. doi: 10.1210/me.2002-0262. [DOI] [PubMed] [Google Scholar]
- 187.Edwards DP, McGuire WL. 17 alpha-Estradiol is a biologically active estrogen in human breast cancer cells in tissue culture. Endocrinol. 1980;107:884–891. doi: 10.1210/endo-107-4-884. [DOI] [PubMed] [Google Scholar]
- 188.Egner U, Heinrich N, Ruff M, Gangloff M, Mueller-Fahrnow A, Wurtz JM. Different ligands-different receptor conformations: modeling of the hER alpha LBD in complex with agonists and antagonists. Med Res Rev. 2001;21:523–539. doi: 10.1002/med.1024. [DOI] [PubMed] [Google Scholar]
- 189.Margeat E, Bourdoncle A, Margueron R, Poujol N, Cavailles V, Royer C. Ligands differentially modulate the protein interactions of the human estrogen receptors alpha and beta. J Mol Biol. 2003;326:77–92. doi: 10.1016/s0022-2836(02)01355-4. [DOI] [PubMed] [Google Scholar]
- 190.Shang YF, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 191.Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. 1987;51:941–951. doi: 10.1016/0092-8674(87)90581-2. [DOI] [PubMed] [Google Scholar]
- 192.Kim K, Thu N, Saville B, Safe S. Domains of estrogen receptor alpha (ERalpha) required for ERalpha/Sp1-mediated activation of GC-rich promoters by estrogens and antiestrogens in breast cancer cells. Mol Endocrinol. 2003;17:804–817. doi: 10.1210/me.2002-0406. [DOI] [PubMed] [Google Scholar]
- 193.Schwartz JA, Zhong L, Deighton-Collins S, Zhao C, Skafar DF. Mutations targeted to a predicted helix in the extreme carboxyl-terminal region of the human estrogen receptor-alpha alter its response to estradiol and 4-hydroxytamoxifen. J Biol Chem. 2002;277:13202–13209. doi: 10.1074/jbc.M112215200. [DOI] [PubMed] [Google Scholar]
- 194.Debes JD, Tindall DJ. Mechanisms of androgen-refractory prostate cancer. N Engl J Med. 2004;351:1488–1490. doi: 10.1056/NEJMp048178. [DOI] [PubMed] [Google Scholar]