

Abstract
Hypoxia-inducible factors (HIFs) are heterodimeric transcription factors regulating the oxygen supply, glucose metabolism, and angiogenesis. HIF function requires the recruitment of p300/CREB-binding protein, two coactivators with histone acetyltransferase activity, by the C-terminal transactivation domain of HIF-α (HIF-αCAD). Histone deacetylase inhibitors (HDAIs) induce differentiation or apoptosis and repress tumor growth and angiogenesis, hence being explored intensively as anti-cancer agents. Using combined pharmacological, biochemical, and genetic approaches, here we show that HDAIs repress the transactivation potential of HIF-αCAD. This repression is independent of the function of tumor suppressors von Hippel-Lindau or p53 or the degradation of HIF-α. We also demonstrate the sufficiency of low concentrations of HDAIs in repression of HIF target genes in tumor cells. We further show that HDAIs induce hyperacetylation of p300 and repress the HIF-1αp300 complex in vivo. In vitro acetylation analysis reveals that the p300CH1 region, but not HIF-αCAD, is susceptible to acetylation. Taken together, our data demonstrate that a deacetylase activity is indispensable for the transactivation potential of HIF-αCAD and support a model that acetylation regulates HIF function by targeting HIF-α·p300 complex, not by direct acetylating HIF-α. The demonstration that HDAIs repress both HIF-1α and HIF-2α transactivation potential independently of von Hippel-Lindau tumor suppressor and p53 function indicates that HDAIs may have biological effects in a broad range of tissues in addition to tumors.
Hypoxia-inducible factor-1 and -2 (HIF-15 and HIF-2; collectively HIF) are transcriptional complexes that serve as the major regulators of the utilization of oxygen and nutrients and play critical roles in physiological adaptations (including angiogenesis, erythropoiesis, and adaptive energy metabolism) to hypoxia (1–3). HIF exerts its function by transcriptionally stimulating the coordinated expression of about 100 functionally related genes (2, 3). Particularly, the expression of vascular endothelial growth factor and its receptors directly contributes to angiogenesis and lymphangiogenesis (4–6). In addition, HIF-stimulated expression of glucose transporters and glycolytic enzymes ensures undisrupted energy metabolism under hypoxic or ischemic conditions. A combination of various conditions, including hypoxia, mitogenic signaling, loss of tumor suppressors, and activation of oncogenes, leads to enhanced HIF activity that promotes tumor growth. Because of the important role of HIF, blocking HIF function in tumors has become one of the major goals of cancer research (2, 7, 8), whereas enhancing HIF-1 function in ischemic lesions becomes an important topic in cardiovascular research (9, 10).
HIF-1 and HIF-2 share a common subunit HIF-β (also known as ARNT), but each has a unique α subunit. The structurally and functionally related HIF-1α and HIF-2α (collectively HIF-α) are the key determinants of the function of HIF-1 and HIF-2. HIF-α activity is controlled by two well known mechanisms (3). First, HIF-α is rapidly degraded under normoxic conditions. When oxygen is available, de novo translated HIF-α is hydroxylated at two conserved prolyl residues (Pro-402 and Pro-564 in HIF-1α) located at the oxygen-dependent degradation domain by proline hydroxylases (11–14). The hydroxylated oxygen-dependent degradation domain is recognized by the von Hippel-Lindau tumor suppressor (VHL) as a substrate for ubiquitination (13–17). The ubiquitinated HIF-α is subsequently disposed through proteasomal degradation (18, 19). Second, HIF-α activity is determined by its transactivation potential. HIF-α possesses an N-terminal and a C-terminal transactivation domain (NAD and CAD, respectively) separated by a normoxic repressive region (NRR) (20–22). HIF-1αCAD provides the major transactivation activity, which is mediated through hydrophobic and stereostatic interaction with p300/CBP (23–27). Factor inhibiting HIF-1 (FIH), an oxygen-dependent asparagine hydroxylase, hydroxylates Asn803 of HIF-1α, thus disrupting its interaction with p300/CBP (25, 28–30).
Protein acetylation has been implicated in regulation of gene expression and protein-protein interactions. Two groups of enzymes, histone acetyltransferases (HATs) and histone deacetylases (HDACs) modulate the acetylation status of histones and nonhistone proteins (31). Generally, acetylation is associated with active transcription, whereas HDACs are found in repressive complexes of transcription (31, 32). Consistently, it has been reported that HDAIs stimulate the transactivation potential of various transcription factors (TFs).
HDAIs have been shown to have anti-cancer effects by inducing growth arrest and apoptosis and repressing angiogenesis (32–37). However, the biochemical and molecular mechanisms underlying the pharmacological effects are not clear. Here we show that HDAIs at concentrations that do not affect HIF-1α levels efficiently repress the transactivation potential of HIF-αCAD and that this repression is independent of either p53 or VHL function. We further show that HDAIs repress HIF function by directly targeting the HIF-αCAD·p300 complex but do not involve a direct acetylation of HIF-1α.
MATERIALS AND METHODS
Plasmids and DNA Recombination
pFR-Luc, pRL-CMV, the pM vector, pM.H1α744–826, and pM.H1α786–826 were described previously (25). pEpo-Luc was generated by inserting a DNA fragment that carries the enhancer element and a 300-bp basic promoter derived from the erythropoietin gene in to pGL2 basic vector from Promega (19, 38). pRSV-Luc contains a Luc reporter driven by an Rous sarcoma virus promoter and enhancer elements (Promega). pcDNA3-Gal4-H1α744-826 and pcDNA3-Gal4-H1α786–826 were constructed by replacing the XhoI-XbaI fragment of pcDNA3-Gal4 (part of Gal4DBD), with XhoI-XbaI fragments from pM-H1α744–826 and pM-H1α786–826 (coding part of Gal4DBD fused with H1α) (25). pGal4-H2αCAD (aa 819–870) was provided by Dr. P. Ratcliffe (Oxford University) (21). pGal4-H1α727–826N803A was a kind gift from Drs. D. Lando and M. Whitelaw (University of Auckland, New Zealand) (28). pGEX4T-H1α530–826 was described previously (38). pGEX2T-p300aa1–596 and pGEX5X-p53 were provided by Drs. L. Puri (Burnham Institute) (39) and S. Berger (Wistar Institute) (40), respectively. pGEX4T-p300CH1 (HIF-1α binding core, aa 323–422) and p300HAT (aa 1000–1710) were constructed by inserting corresponding PCR fragments into suitable sites of pGEX4T. pCMV.MyoD and p4RTK.CAT were provided by Dr. A. Giordano (Temple University, Philadelphia, PA) (41). The pcDNA3.p53 was described previously (42). The p21waf-Luc reporter was provided by Dr. El-Deiry (University of Pennsylvania) (43). All restriction enzymes and other enzymes needed for PCR and DNA recombination were purchased from either Fisher or La Roche (Indianapolis, IN). All PCR-generated constructs were confirmed by sequencing analysis. Sequences of primers used in PCR are provided in the supplemental material.
Chemicals and Special Reagents
Common chemicals and solvents were purchased from either Fisher or Sigma. Tissue culture and transfection reagents were obtained from Invitrogen. Trichostatin A (TSA) was purchased from BioMol (Plymouth Meeting, PA). Sodium butyrate (SBT), desferrioxamine, and Me2SO were from Sigma. Suberoylanilide hydroxamic acid (SAHA) was obtained from BioVision (Mountain View, CA).
Cell Culture and Hypoxic Treatments
HeLa and Hep3B were obtained from ATCC. RCC4 (VHL−/−) and RCC4 (VHL+) were kindly provided by Dr. P. Ratcliffe (21). HCT116 (p53+/+) and HCT116 (p53−/−) were generous gifts from Dr. B. Vogelstein (John Hopkins University, Baltimore, MD) (44). The B1 cell line was described previously (19, 38). HeLa and RCC4 cells were cultured in Dulbecco’s modified Eagle’s medium. HCT116 and Hep3B were cultured in McCoy’s 5A and minimal essential medium, respectively. All culture media were supplemented with l-glutamine, sodium pyruvate, 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml). Hypoxic treatments were carried out by directly incubating cells in a Hypoxia work station equipped with a programmable controller of humidity and temperature and connected to an automatic gas mixer (InvivO2; Ruskinn Technology).
Transfection and Luciferase Assays
Transient transfection was performed with Lipofectamine Plus or Lipofectamine 2000 reagents (Invitrogen) by following the low serum protocol recommended by the manufacturer. Cells were trypsinized 24 h after transfection, pooled, divided equally, and cultured in 12-well plates for reporter assays or in 100-mm dishes for Western blot analysis. pRL-CMV (0.1 μg) was cotransfected to normalize transfection efficiency and effects of special treatments on the promoters. Luciferase and dual luciferase assays were performed in triplicate with kits from Promega. Luminescence was measured in a TD20/20 luminometer (Promega), and relative light units (RLU) were standardized to Renilla luciferase activity expressed from cotransfected pRL-CMV. Protein concentration was determined by the Bio-Rad method.
Antibodies, Cell Lysate Preparation, Immunoprecipitation, GST Pull-down, and Western Blotting
Monoclonal antibodies against Gal4DBD and p300 (NM11) were from BD Biosciences (San Diego, CA). Monoclonal antibodies against α-tubulin and GST (Sc-138) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-HIF-1α monoclonal antibodies were purchased from BD Bioscience or Novus (Littleton, CO). Anti-FIH monoclonal antibody was a kind gift from Dr. P. Ratcliffe. Horseradish peroxidase-coupled secondary antibodies were from Sigma and Invitrogen. Anti-HDAC1, anti-Hsp90, anti-acetyllysine, and anti-acetyl-H3 antibodies were from Stressgen (Victoria, Canada) and Upstate (Charlottesville, VA).
To determine protein levels, cells were lysed in urea buffer (8 m urea, 10 mm Tris, 10% glycerol, 1% SDS, 5 mm dithiothreitol, 1 mm phenyl-methylsulfonyl fluoride, 1× protease inhibitor mix, pH 6.8) on ice with an Ultra-Turrax T8 homogenizer (IKA GmbH & Co.) for 5 s followed by incubation on ice for 10 min. For GST pull-down and immunoprecipitation studies, cells were lysed on ice for 30 min in lysis buffer (50 mm Tris-HCl, 1% Triton 100, 5 mm EDTA, 50 mm NaF, 0.1 mm Na3VO4, 100 mm desferrioxamine, 1 mm phenylmethylsulfonyl fluoride, 1× protease inhibitor mix, pH 7.5) with 500 mm NaCl and special drugs as they were used in cell culture, followed by two freeze-thaw-vortex cycles. After centrifugation, supernatants were 1:1 diluted with lysis buffer without NaCl. Immunoprecipitations were performed as described previously (25). GST fusion protein expression and pull-down assays were carried out as described (45). For in vivo GST pull-down, glutathione beads were equilibrated first with lysis buffer with 250 mm NaCl and then incubated with cell lysates for 1 h on a roller at 4 °C, followed by four washes with lysis buffer. Western blot analyses were performed as previously described (25, 38). The cytoplasmic/nuclear fractionation was performed with an NE-PER nuclear and cytoplasmic extraction kit from Pierce.
Immunofluorescent Cell Staining
HeLa cells were cultured in chamber slides under normoxic or hypoxic conditions and treated with or without TSA (600 nm) for 8 h immediately before fixation in 0.3% gluteraldehyde in BRB80 buffer (80 mm PIPES, 1 mm MgCl2, 1 mM EGTA, pH 6.9). After fixation, the slides were sequentially incubated with anti-HIF-1α (1:100) and fluorescein isothiocyanate-conjugated donkey anti-mouse antibody (Jackson ImmunoResearch, 1:100) and mounted with mounting medium for fluorescein (Vector Laboratories, Burlingame, CA) with 1.5 μg/ml 4′,6-diamidino-2-phenylindole to counterstain DNA. The slides were examined under a Nikon fluorescent microscopy equipped with Zeiss lens, laser source, multiple filters, and a digital CCD camera.
On-bead Protein Acetylation Assays
Immobilized GST fusion proteins were rocked with HAT in the presence of [14C]acetyl-CoA (0.25 μCi, 4 nmol) (Amersham Biosciences), SBT (2 mm), and bovine serum albumin (2.5 mg/ml) in 300 μl of 1× HAT buffer (46) at 30 °C for 1 h. Immobilized protein substrates were collected by centrifugation and followed by four washes with 800 μl of radioimmune precipitation buffer. Finally, the samples were separated on SDS-polyacrylamide gels followed by Coomassie Blue R250 staining, destaining, and autoradiography with either PhosphorImager screen or Biomax MR film (Eastman Kodak Co.).
RNA Extraction and RT-PCR
Total RNA was extracted from cultured cells with RNeasy kits from Qiagen (Valencia, CA). Total RNA samples were digested for 2 h with RNase-free DNase (La Roche) followed by reverse transcription (RT kit from La Roche). The number of PCR cycles was optimized to exponential range for each individual mRNA target. Sequences of primers used in semiquantitative RT-PCR are described in the supplemental material.
RESULTS
A Deacetylation Event Is Required for the Transactivation Potential of HIF-1αCAD
HDACs are described as repressors of transcription. An early finding implicated HDACs in the regulation of HIF-1 transactivation (30). We therefore addressed the question if the normoxic repression of HIF-1α transactivation involved the associated HDACs. As the first step, we tested the effects of TSA, an inhibitor of class I and class II HDACs, on the transactivation potential of HIF-1αCAD, which is the major determinant of the overall transactivation activity of HIF-1α (25). To minimize the effects of TSA on expression levels of Gal4-H1α, we subcloned the Gal4-H1α fragments in a mammalian expression vector with CMV promoter/enhancer. With this expression system, we observed that TSA at concentrations as low as 75 nm efficiently repressed the transactivation potential of Gal4-H1α744–826 under both normoxic (data not shown)and hypoxic conditions(Fig.1A). Westernblot confirmed that after standardization with Renilla luciferase activity expressed from identical promoter/enhancer, the levels of Gal4-H1α744–826 in various samples were generally comparable (Fig. 1B). In initial experiments with SV40 promoter, TSA caused a dramatic increase in the levels of Gal4-H1α744–826, but to our surprise, its transactivation activity was clearly repressed (data not shown). We noticed that CMV promoter/enhancer was affected by TSA, but only 2–4 fold, contrasting with the 30–40-fold increase when SV40 promoter/enhancer was used (data not shown). TSA does not impair the DNA binding ability of Gal4, because the transactivation activity of all other Gal4 fusion constructs tested, including Gal4-VP16, was strongly enhanced by TSA under the same experimental conditions (Fig. 1C and data not shown). The decrease in transactivation activity was not caused by cytotoxicity of TSA, because the cells did not change morphologically, and RLU had been normalized to an internal standard (Renilla luciferase), and, more importantly, the same concentration range of TSA stimulated other transcription factors, including MyoD and p53, under the same experimental conditions (Fig. 1, D and E). Taken together, these data demonstrate that a deacetylase activity is indispensable for the transactivation potential of HIF-1αCAD.
FIGURE 1. TSA specifically represses HIF-1αCAD transactivation potential.
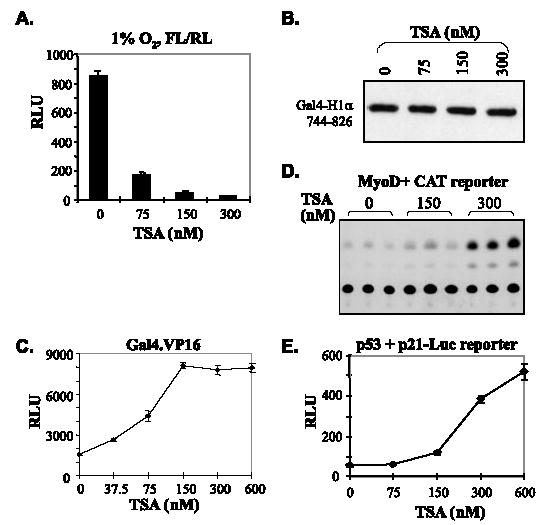
A, standardization of the TSA effects on the promoter shows more dramatic repression by TSA. pCMV-Gal4-H1α744 – 826 (3 μg), pFR-Luc (2 μg), and pRL-CMV (0.1 μg) were cotransfected into HeLa cells, which were pooled and evenly distributed into wells of 12-well culture plates. Immediately before harvesting, cells were treated with increasing concentrations of TSA and cultured with either 21 or 1% O2 for 12 h. Firefly luciferase activity (FL) was standardized to Renilla luciferase (RL) activity from cotransfected pRL-CMV. Similar repression was observed with either 21% (not shown) or 1% O2 (shown here). B, pooled HeLa cells cotransfected with pCMV-Gal4-H1α744 – 826 (5 μg) and pRL-CMV (0.1 μg) were evenly seeded in four dishes and cultured in hypoxia work station programmed with 1% O2. Immediately before harvesting, cells were treated with Me2SO or increasing doses of TSA and incubated with 1% O2 for an additional 12 h. Cells were lysed, and a small aliquot of each was used to determine Renilla luciferase activity. Total proteins (100 μg) from Me2SO-treated cells (0 nM TSA) were used as the standard. For TSA-treated samples, lysates containing RL activity equivalent to the standard were loaded. C, TSA does not repress the function of Gal4 DNA binding domain. Plasmids expressing Gal4-VP16 (3 μg) were cotransfected with pFR-Luc (2 μg) and pRL-CMV (0.1 μg) into HeLa cells. Cells were pooled, reseeded into multiwell culture plates, and treated with the indicated concentration of TSA for 12 h immediately before dual luciferase assays. RLU shown represent firefly luciferase activities corrected against RL. D, TSA stimulates MyoD transactivation activity. Plasmids expressing pcDNA3-MyoD (3 μg), pRL-CMV (0.1 μg), and plasmids of a MyoD-responsive chloramphenicol acetyltransferase reporter (2 μg) were cotransfected into HeLa cells. Transfected cells were treated with various doses of TSA for 12 h. Cell lysates were standardized with Renilla luciferase activity and assayed for chloramphenicol acetyltransferase activity as described. E, TSA stimulates p53 transactivation activity. Plasmids carrying a firefly luciferase reporter gene controlled by p21 promoter (2 μg) were cotransfected with pcDNA3-p53 (3 μg) and pRL-CMV (0.1 μg) into HeLa cells. Cotransfected cells were treated with increasing doses of TSA for 12 h before dual luciferase assays.
TSA Represses Both HIF-1αCAD and HIF-2αCAD Independently of the NRR
HIF-αNRR plays a key role in normoxic repression of the transactivation activity of HIF-αCAD (20–22). To explore whether TSA represses HIF-αCAD through HIF-αNRR, we examined effects of HDAIs on the transactivation potential of Gal4-H1α786–826, which lacks NRR, thus being constitutively active even under normoxic conditions (20, 25). We observed that TSA repressed Gal4-H1α786–826 with a similar efficiency, if not better, demonstrating that TSA-mediated repression of HIF-1αCAD does not rely on NRR (Fig. 2A). Similarly, effects of TSA on a Gal4-H2αCAD (aa 819–870) lacking NRR were tested in cotransfection assays. The repression of transactivation potential of HIF-2αCAD also was evident (Fig. 2B). These data suggest that TSA represses both HIF-1α and HIF-2α through a common mechanism that is independent of NRR.
FIGURE 2. TSA represses HIF-1αCAD and HIF-2αCAD independently of HIF-αNRR.
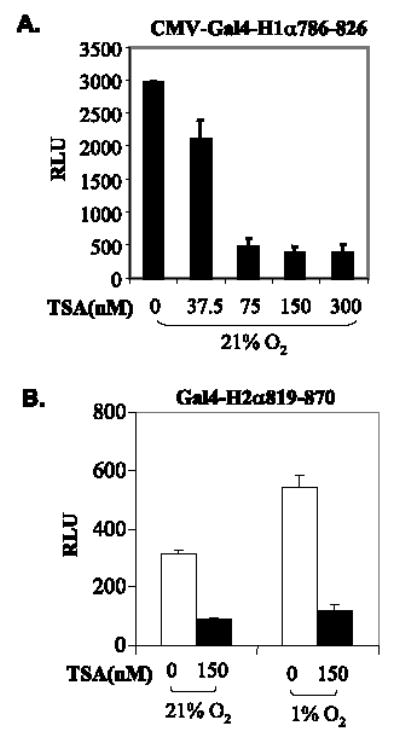
A, TSA-mediated repression of HIF-1αCAD is independent of NRR. pcDNA3-Gal4-H1α786–826 (3 μg), pFR-Luc (2 μg), and pRL-CMV (0.1 μg) were cotransfected into HeLa cells. TSA treatment and luciferase assays were performed as described above. B, TSA represses HIF-2αCAD without NRR. A plasmid expressing Gal4-H2αCAD (aa 819–870) was used in cotransfection assays. pRL-CMV (0.1 μg) was cotransfected for standardization.
Neither VHL nor p53 Function Is Indispensable for HDAIs to Repress HIF-1αCAD
VHL plays a key role in suppression of HIF activity by promoting the degradation of HIF-α. VHL also has been postulated to recruit HDACs to HIF-1α, thus repressing transactivation (30). To examine whether VHL function is required for HDAIs to repress the transactivation activity of HIF-αCAD, we cotransfected Gal4-H1α786–826 with a luciferase reporter into VHL (−/−) RCC4 cells and RCC4-derived cells where VHL function has been restored by stable transfection (43). Results shown in Fig. 3, A and B, reveal that TSA represses HIF-1αCAD in both VHL-positive and -negative cells. Since the majority of tumors carry p53 mutations, and p53 is involved in the regulation of HIF-1α function (47–49), we explored whether p53 function is required for the HDAI-mediated repression of HIF-αCAD. Using HCT116 (p53−/−) cells (double knock-out of p53) and the parental HCT116 (p53+/+) cells (44), we found that TSA repressed the transactivation potential of HIF-1α regardless of the genetic status of p53 (Fig. 3, C and D). Taken together, these data indicate that the HDAIs repress the transactivation potential of HIF-αCAD independently of either VHL or p53 function.
FIGURE 3. VHL and p53-independent repression of HIF-1 activity.
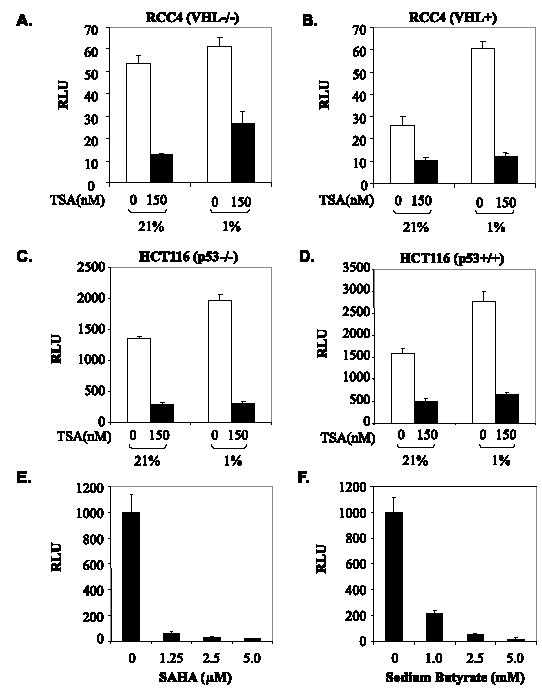
A and B, TSA-mediated repression is independent of VHL. VHL-deficient RCC4 cells or RCC4 cells with reconstituted VHL were used in cotransfection assays as in Fig. 2A. C and D, TSA-mediated repression is independent of p53. HCT116 carrying the p53 double knock-out (C) and the parental HCT116 (D) were used in cotransfection. E and F, therapeutic concentrations of HDAIs are sufficient to repress the transactivation potential of HIF-1αCAD. Cotransfection was performed as before. 24 h after transfection, cells were treated with increasing concentrations of SAHA (E) or SBT (F) for 12 h immediately before dual luciferase assays.
SAHA and SBT are among the HDAIs that are currently in clinical trials for cancers and hematological disorders. To determine whether therapeutic doses of HDAIs are sufficient to repress HIF-αCAD, we examined the concentration effects of both SAHA and SBT on Gal4-H1α786–826 and found that 1.25 μm SAHA or 1 mm SBT, a concentration achievable with normal therapeutic doses (32, 50, 51), efficiently repressed HIF-1αCAD (Fig. 3, E and F), suggesting that therapeutic doses of HDAIs may be capable of repressing HIF-1αCAD in vivo.
HDAI-mediated Repression Depends on Induction of HIF-1α and HIF Binding Site
To investigate the effects of HDAIs on endogenous HIF activity, we transfected pEPO-Luc, which carries a luciferase reporter driven by the enhancer/promoter combination of the erythropoietin (EPO) gene, into Hep3B cells. Hypoxia enhanced the activity of the EPO enhancer/promoter as shown by luciferase assays (Fig. 4, compare A and C). But under hypoxic conditions where HIF-1α was induced (Fig. 4B, last two lanes), the EPO enhancer/promoter activity was repressed by TSA (Fig. 4A). In contrast, under normoxic conditions where HIF-α was undetectable (Fig. 4B, first two lanes), TSA increased the basal level expression of the reporter (Fig. 4C). A control reporter construct, which is identical to the pEPO-Luc except for the lack of HIF binding sites, thus not responding to hypoxia (19) (data not shown), was enhanced by TSA (Fig. 4D). Since TSA did not increase endogenous HIF-1α levels as tested in time and concentration course experiments under any condition, the stimulatory effects of TSA on reporter (Fig. 4, C and D) were possibly caused by hyperacetylation of histones or other transcription regulators. Taken together, these data indicate that TSA-mediated repression of the reporter gene requires both HIF-α and HIF binding sites.
FIGURE 4. TSA represses endogenous HIF activity independently of levels of HIF-1α.
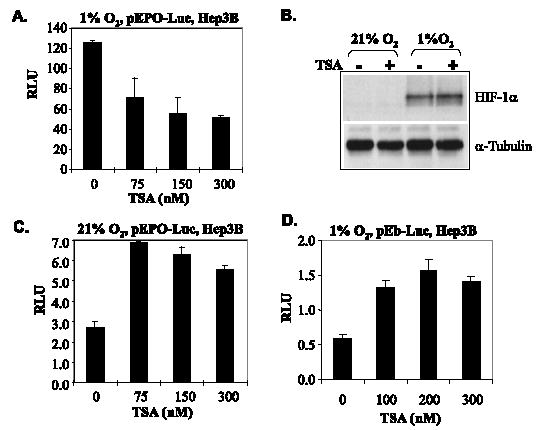
A, Hep3B cells were transfected with pEPO-Luc and treated with increasing concentrations of TSA and cultured under either normoxic or hypoxic conditions for 8 h prior to harvesting. B, Hep3B cells were treated with 150 nm TSA for 8 h, and the effects of TSA on the levels of endogenous HIF-1α were examined by Western blotting. C, TSA does not repress the expression of pEPO-Luc reporter gene in the absence of HIF-α. The pEPO-Luc plasmids were transfected into Hep3B cells. Transfected cells were treated with TSA for 12 h, and RLU was determined and normalized to total protein. D, TSA does not repress EPO promoter without hypoxia-responsive elements (HRE or HIF binding sites). The pEb-Luc (4 μg) reporter, which is identical to pEPO-Luc except for the lack of HIF binding cis-elements, was transfected into Hep3B cells, which were treated with TSA and cultured under either normoxic conditions (not shown) or hypoxic conditions for 12 h. RLU was determined and normalized to total proteins.
TSA Represses Endogenous Activity of HIF and Expression of HIF Target Genes
HDAIs promote the degradation of HIF-1α when used at higher concentrations (52–55). To study the effects of TSA on the transactivation potential of endogenous HIF, we examined concentration effects on HIF-1α stability in B1 cells that carry a stably transfected pEPO-Luc reporter (19). Western blot revealed that with concentrations up to 150 nm, TSA had no obvious effect on the levels of endogenous HIF-1α (Fig. 5A). Higher concentrations of TSA decreased the levels of HIF-1α in B1 cells (Fig. 5A), an observation similar to that made by others in HepG2 and HT1080 cells (52, 53). However, at concentrations as low as 37.5 nm, TSA repressed hypoxia-stimulated pEPO-Luc reporter expression effectively (Fig. 5B). To further distinguish the effects of HDAIs on HIF-1α levels and that on the transactivation potential, we examined various cell lines (55) and observed that up to 600 nm TSA failed to reduce HIF-1α in HeLa cells (Fig. 5C). We exposed HeLa cells to 300 nm TSA and examined the expression of vascular endothelial growth factor, CA-9, and Glut1, three endogenous HIF target genes by semiquantitative RT-PCR. We found that all of these three HIF target genes were repressed significantly by 300 nm TSA (Fig. 5D), a concentration that is not sufficient to degrade HIF-1α in HeLa cells. Taken together, these data demonstrate that TSA is able to repress endogenous HIF activity via a mechanism independent of HIF-α degradation.
FIGURE 5. Effects of TSA on endogenous HIF activity.
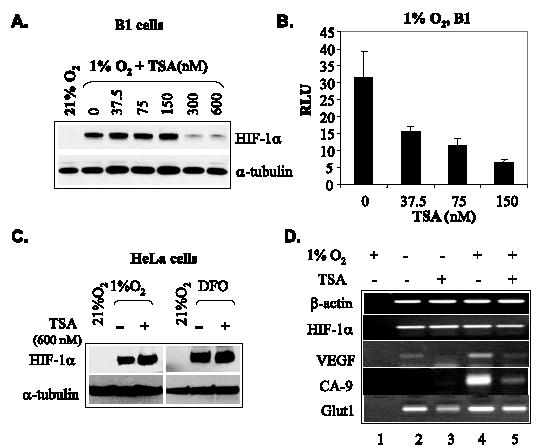
A, low concentrations of TSA show no effect on hypoxia-induced HIF-1α levels in B1 cells. B1 cells were treated with various concentrations of TSA and cultured under either normoxic or hypoxic conditions for 8 h. Endogenous HIF-1α levels were examined by Western blot. Tubulin was detected as an indicator of loading. B, degradation-independent repression of endogenous HIF-1α activity in B1 cells. B1 cells were treated with concentrations of TSA that were not sufficient to induce the degradation of HIF-1α and were cultured under normoxic or hypoxic conditions for 8 h before luciferase assays. C, TSA (600 nm) has no effect on HIF-1α levels in HeLa cells. HeLa cells were cultured with or without TSA under the indicated conditions for 8 h before harvesting. Total cell lysates were analyzed by Western blot to determine levels of HIF-1α. DFO, 130 μm desferioxamine. D, TSA represses HIF-1 target genes in HeLa cells. HeLa cells were cultured under normoxic or hypoxic conditions (1% O2) and exposed to 300 nM of TSA for 8 h before RNA extraction. The mRNA levels of vascular endothelial growth factor, CA-9, and Glut1, three HIF target genes, were examined by RT-PCR. The mRNA levels of β-actin and HIF-1α were examined as controls. In control lane 1, RNA was identical to that used in lane 4, but reverse transcriptase was omitted.
HDAIs Impair p300·HIF-α Complex
The transactivation activity of HIF-αCAD absolutely relies on its interaction with p300/CBP. To explore if HDAIs repress HIF-αCAD by altering the p300-HIF-1α interaction, HeLa cells precultured with either 21 or 1% O2 were exposed to TSA for 3 h before harvest. Western blot with whole cell lysates showed that TSA had no significant impact on the levels of HIF-1α or p300 but induced hyperacetylation of histone H3 (Fig. 6A, top). Immunoprecipitation revealed that the fraction of total HIF-1α co-precipitated with p300 was decreased (Fig. 6A, bottom), suggesting that an acetylation event affects the p300·HIF-1α complex. Whereas similar amounts of total p300 were precipitated, TSA treatment enhanced the acetylation levels of p300 (Fig. 6A, bottom). The same anti-lysine antibody and several other anti-lysine antibodies from various sources failed to recognize immunoprecipitated HIF-1α under any condition we have tested (data not shown).
FIGURE 6. TSA decreases the association of HIF-1α with p300.
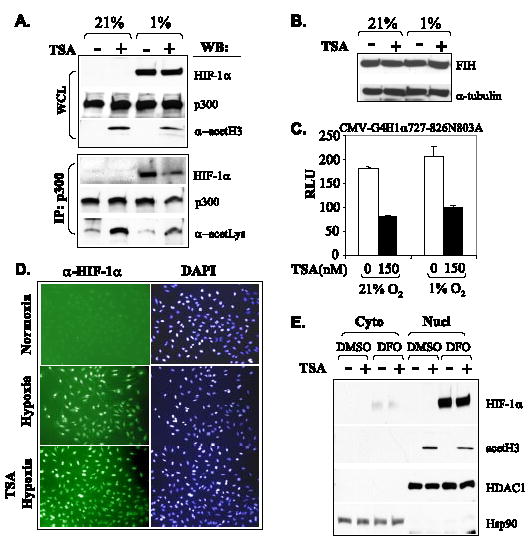
A, HeLa cells were treated with either Me2SO (−) or TSA (+) (300 nm) for 3 h and cultured under either normoxic or hypoxic conditions. Cell lysates (WCL) were prepared and used directly for Western blot (WB) (20%) to examine the levels of HIF-1α, p300, and acetylated histone H3 or used for immunoprecipitation (IP) with anti-p300 monoclonal antibody followed by Western blot to detect precipitated p300 and coimmunoprecipitated HIF-1α. The same membrane was reused to detect the levels of acetylated proteins with an anti-lysine antibody. B, TSA does not affect FIH levels. HeLa cells were cultured under the indicated conditions and treated with Me2SO or 500 nM TSA for 8 h before harvest. FIH levels were detected by Western blotting. C, TSA-mediated repression of HIF-1αCAD is independent of N803. Plasmids expressing Gal4-H1α727– 826N803A (3 μg), pFR-Luc reporter (2 μg), and pRL-CMV (0.1 μg) were cotransfected into HeLa cells. The effects of TSA were determined by luciferase assays normalized to Renilla luciferase derived from cotransfected pRL-CMV. D and E, TSA shows no effects on nuclear translocation of HIF-1α. HeLa cells were cultured in chamber slides under normoxic or hypoxic conditions and treated with or without TSA (600 nm) for 8 h. After fixation, the slides were stained with anti-HIF-1α (1:100) and fluorescein isothiocyanate-conjugated secondary antibody and examined under a fluorescent microscopy. 4′,6-Diamidino-2-phenylindole (DAPI) staining shows the location and size of nuclei (D). In addition, HeLa cells were cultured with or without desferrioxamine (130 μm) and exposed to TSA (600 nm) for 6 h immediately before harvesting. The cytoplasmic and nuclear fractionation was performed, and samples were examined by Western blot. HDAC1 and Hsp90 were detected as controls of sample loading for nuclear extracts and cytoplasmic samples, respectively (E).
Hydroxylation of the conserved asparagines (Asn803 in HIF-1α) of HIF-αCAD by FIH has been identified as a major regulation of the p300/HIF-α interaction (25, 28–30). To determine if HDAIs impair p300·HIF-1α complex via FIH-catalyzed hydroxylation of Asn803, we examined if TSA stimulated the expression levels of FIH. Western blot revealed that FIH levels were not affected by either hypoxia or exposure to TSA (Fig. 6B). In addition, we cotransfected a pFR-Luc reporter, which has four Gal4 binding sites, with Gal4-HIF-1αCAD727–826N803A mutant (Asn803 mutated to Ala), which is resistant to normoxic repression (28). Results of luciferase assays showed that the N803A mutation did not abolish TSA-mediated repression (Fig. 6C). To examine if the decreased interaction and activity of HIF-1α/p300 was resulted from reduced nuclear translocation of HIF-1α, we performed immunofluorescent staining for HIF-1α and confirmed that 600 nm TSA, a concentration sufficient to repress the transactivation potential but insufficient to destabilize HIF-1α in HeLa cells, did not change the nuclear translocation of HIF-1α (Fig. 6D). Results from cytoplasmic and nuclear fractionation of desferrioxamine-treated HeLa cells substantiated this observation (Fig. 6E).
p300HAT Acetylates p300CH1, Not HIF-1αCAD
p300 and CBP may directly acetylate transcription factors. To determine if the p300HAT activity acetylates HIF-1α and regulates its interaction with HIF-1α, we expressed p300HAT in Escherichia coli and purified the HAT activity. Similarly, we expressed and purified GST-p53, GST-p300aa1–596 and GST-p300aa323–422 fusion proteins (Fig. 7A). In vitro acetylation assays demonstrated that GST-H1α530–826 was not acetylated by p300HAT, whereas GST-p300aa1–596 and GST-p53 (a positive control) were acetylated (Fig. 7B). Additional experiments demonstrated that aa 323–422 of p300, the core region responsible for interaction with HIF-1α, contains the site(s) for p300 autoacetylation (Fig. 7B). To further illustrate that the HDAI-mediated repression of the transactivation potential does not involve a direct acetylation of HIF-α, we used GST-HIF-1α (aa 530–826) expressed in E. coli to pull down p300 from TSA-treated or -untreated cells and found that p300 in TSA-treated cells showed weaker affinity for HIF-1α (Fig. 7C). Furthermore, analysis of the primary structure of HIF-α reveals that there is no Lys residue in the highly conserved, constitutively active HIF-αCAD, which is composed of merely 41 amino acids but is sufficient to bind p300 and respond to HDAI-mediated repression (Figs. 2A and 7D). We concluded that HDAIs repress the transactivation potential of HIF-αCAD by targeting the HIF-1α·p300 complex, but not involving direct acetylation of HIF-αCAD.
FIGURE 7. HIF-1αCAD is not a target for p300 acetylation.
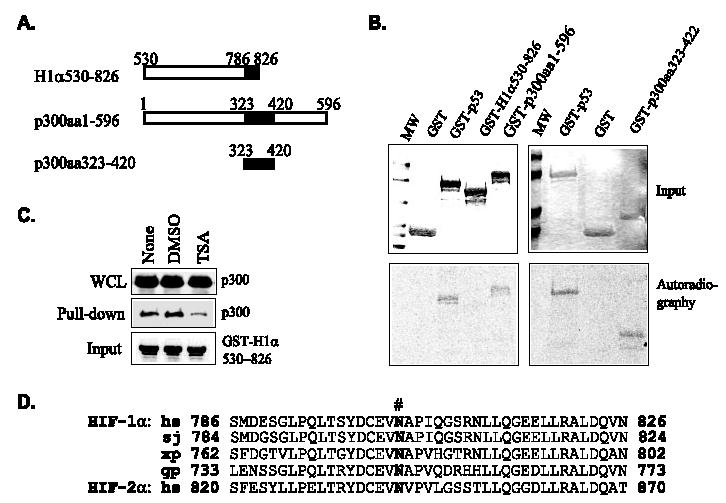
A, schematic structure of fragments of HIF-1α and p300 expressed as GST fusion proteins. B, p300CH1, but not HIF-1αCAD, is acetylated by p300HAT. Fragments covering p300CH1 and the transactivation domains of HIF-1α were used in acetylation assays with [14C]acetyl-CoA and recombinant p300HAT. GST and GST-p53 were used as negative and positive controls, respectively. Top, Coomassie Blue-stained gels showing protein input. Bottom, autoradiographic images showing the incorporation of [14C]acetyl-CoA. C, p300 in TSA-treated cell lysates (WCL) shows lower affinity for recombinant HIF-1αCAD. Aliquots of immobilized GST-H1α530–826 expressed in bacteria, thus lacking posttranslational modification, were incubated with lysates from HeLa cells treated with Me2SO or TSA. The GST-H1α530–826 pull-down complexes were separated on an SDS-polyacrylamide gel, and associated p300 was detected by Western blotting. D, alignment of amino acid sequences of HIF-αCAD shows no Lys residue. Amino acid sequences of HIF-1αCAD are identical among Homo sapiens (hs), Bos taurus, Bos grunniens, Mus musculus, Oryctolagus cuniculus, Rattus norvegicus, and Spermophilus tride-cemlineatus. Other species are as follows. sj, Spalax judaei;xl,Xenopuslaevis;gp,Gymnocyprisprzewalskii. #, Asn803.
DISCUSSION
As coactivators for a variety of TFs, p300/CBP exert their function partly by acetylating these TFs. Particularly, acetylation of p53 and MyoD has been reported to promote their ability to recruit p300/CBP and to stimulate their transactivation potential (39, 40, 56–58). In this study, we investigated the role of acetylation in the regulation of HIF-αCAD activity. Whereas it was surprising to find that HDAIs repressed the transactivation potential of HIF-αCAD, this observation is consistent with the finding that HDAIs block tumor angiogenesis (53, 59, 60). Interestingly, the HDAI-mediated repression of transactivation potential distinguishes HIF-α from other p300/CBP-dependent TFs. We notice that p53 and MyoD are involved in growth arrest and differentiation, whereas HIF activation is considered to promote cell survival and cell adaptation to adverse environments. The TF-specific response to HDAIs suggests that acetylation may differentially regulate TFs with different, sometimes opposite functions, whereas all share p300/CBP as coactivators.
The involvement of HDACs in the regulation of HIF-1 function is suggested by several studies. The demonstration that VHL interacts with both FIH and HDAC1 to -3 suggests that FIH may repress HIF-1α transactivation potential by indirectly recruiting HDAC1 to -3 through VHL (30). The findings that HDAIs repress angiogenesis and the expression of HIF target genes suggest a stimulatory, rather than repressive, role of HDACs in HIF activity (61, 62). In addition, HDAC7 was found to specifically interact with HIF-1α (63). Upon hypoxic stimulation, HDAC7 enhances HIF-1 activity by promoting its nuclear translocation.
Repression of HIF-1α activity by HDAIs has been reported by at least three groups with various explanations. The observation that TSA causes the degradation of HIF-1α (52, 53) indicates that a deacetylation event is required to stabilize HIF-1α. HDAIs also were reported to repress the DNA binding ability (59) and inhibit the nuclear translocation of HIF-1 (60). The HDAI-mediated destabilization of HIF-1α was originally explained as a result of enhanced expression of VHL and p53 (53), and direct acetylation of HIF-1α promoted its recognition and ubiquitylation by VHL (52). Recent results from our laboratory suggest that HDAIs trigger ubiquitin-independent proteasomal degradation of HIF-1α (55). In this report, we present data to show that the transactivation potential of HIF-αCAD is repressed by HDAIs, and this repression does not distinguish between cells that have normal VHL or p53 function and those that do not. Whereas at high concentrations (TSA, 300 –1000 nm; SAHA, 1–5 μm; SBT, 5–10 mm), HDAIs indeed reduced the levels of HIF-1α (52, 53, 55),6 concentrations insufficient to reduce the HIF-1α levels are sufficient to repress its transactivation potential (Fig. 3). The maximum tolerated doses of HDAIs may reach a maximum serum concentration apparently high enough to reduce HIF-α levels (32, 50, 51). However, the real concentration in solid tumors may be much lower. Furthermore, HDAIs reduce but do not completely abolish HIF-α (55) (Fig. 5A). Since repression of the transactivation potential of HIF-α needs relatively low concentrations of HDAIs that are more likely achievable in vivo (32), it should have a closer pharmacological relevance than HDAI-mediated degradation of HIF-α.
The report that mouse ARD1, the orthologue of an N-α-acetylase in yeast, serves as an N-ε-acetylase of HIF-1αK532 (52) suggested that HIF-1α might be acetylated by other acetylases in vivo. However, our group, as well as others, has failed to observe the acetylation of HIF-1α by ARD1 in vitro or any effect of ARD1 on levels of HIF-1α in vivo (64, 65). Moreover, it has been demonstrated that ARD1 has no effect on the expression of HIF target genes (66). The detection of HIF-1α in immunocomplexes precipitated with an anti-acetyllysine antibody was shown as evidence of HIF-1α acetylation in vivo (52). We believe that result could be alternatively interpreted as an acetylated protein coprecipitating with HIF-1α. At least, it has been demonstrated that p300/CBP and Hsp90 can be acetylated in vivo and interact with HIF-1α (55, 67, 68) (Fig. 6A). Whether full-length HIF-1α is acetylated in vivo remains unanswered. Nevertheless, this study demonstrates that the anti-angiogenesis effects of HDAIs are at least partly caused by repressing the transactivation potential of HIF-αCAD independently of direct acetylation of HIF-1α.
The role of acetylation in oxygen sensing and the molecular identity of the deacetylase that controls the transactivation potential of HIF-αCAD remain elusive. Based on its sensitivity to TSA, SBT, and SAHA, the deacetylase involved should be a member of the class I or class II HDACs. A previous report that hypoxia stimulated the expression of HDAC1 implicates acetylation in oxygen sensing and HDAC1 in the control of HIF transactivation activity (53). We observed that hypoxia or hypoxic mimics did not affect the HDAC1 levels in cultured cells (Fig. 6E and data not shown). However, the finding of HDAC1 in p300 complex supports a regulatory role of HDAC1 in p300-dependent transactivation (69). The report that HDAC7 interacts with HIF-1αNRR and regulates the subcellular translocation of HIF-1α upon hypoxic stimulation (63) raises the possibility that HDAC7 also regulates the transactivation potential. However, we believe that at least other HDACs are involved, because 1) HDAI-mediated repression is independent of NRR, the region required for HDAC7 binding (Fig. 2A); 2) HIF-2αCAD, which does not interact with HDAC7 (63), was repressed by HDAIs in a similar manner (Fig. 2B); and 3) TSA treatment did not change the nuclear localization of HIF-1α in HeLa cells under the experimental conditions (Fig. 6, D and E). Furthermore, since HIF-1α786–826 can interact with p300/CBP efficiently and is constitutively active under normoxic conditions, it is reasonable to believe that this deacetylase maintains a considerable level of activity even under normoxic conditions. We believe that the deacetylase activity is a prerequisite for HIF-αCAD trans-activation but is not likely to be an oxygen sensor. The availability of this deacetylase activity makes the cells ready to respond rapidly to hypoxia, oncogenic signals, and other signals directed to HIF-α.
HDAIs possess pleiotropic effects on gene expression. Whereas it is clear that HDAIs can enhance the acetylation status of p300 in vivo and p300CH1 can be acetylated by p300HAT in vitro, a role of direct CH1 acetylation in HIF-1α/p300 interaction remains to be studied further. Several possible but neither exclusive nor exhaustive models were postulated in Fig. 8. One possibility is that such acetylation directly disrupts HIF-1α/p300 interaction. Supporting this model, three highly conserved Lys residues of CH1 have been demonstrated to be essential for the HIF-αCAD/p300 interaction (26, 27). Alternatively, acetylation of p300 (CH1 and other regions) or p300-interacting partners may enhance their interactions, thus redistributing p300 to non-HIF interacting protein factors (IPFs). It is also possible that HDAI-triggered hyperacetylation of an IPF results in its interaction with HIF-αCAD. Nevertheless, our data suggest that an acetylation event provides a mechanism to regulate the distribution of p300 to different IPFs of diverse function.
FIGURE 8. Possible models of HDAI-mediated repression of HIF-αCAD·p300 complex.
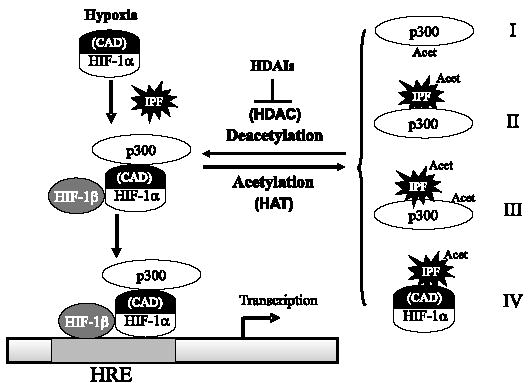
A deacetylase activity maintains p300 or a protein factor, namely IPF, at hypoacetylated status. Upon hypoxia, accumulated HIF-1α dimerizes with HIF-1β, recruits p300, and binds to HRE of HIF target genes. HDAIs repress the deacetylase(s) involved, but HIF-1αCAD (indicated) is not the direct target of acetylation. Instead, p300 or an IPF is hyperacetylated in the presence of HDAIs. In model I, it is proposed that hyperacetylation of p300 CH1 domain directly disrupts the interaction. Model II predicts that acetylation of an IPF promotes its interaction with p300. It is also possible that acetylation of both p300 and an IPF contributes to increased interaction between p300 and the IPF (model III). In the last model, an acetylated IPF may interact directly with HIF-αCAD, thus repressing its transactivation potential. HRE, hypoxia-responsive elements (i.e. HIF binding sites).
During the preparation of this manuscript, we noticed a report that a p300/CBP-independent transactivation mechanism of HIF-1 is repressed by TSA (70). Because HIF-αCAD absolutely depends on p300/CBP (23–25, 70), the deacetylase-dependent transactivation described in that report represents a CAD-independent mechanism, possibly involving the N-terminal transactivation domain (NAD) of HIF-α. Indeed, we observed that when fused with Gal4, HIF-1α NAD (aa 529–778) was repressed by TSA as well (data not shown). However, NAD overlaps the oxygen-dependent degradation domain that is sufficient for TSA-induced degradation (55), making it difficult to interpret those data. We have reexamined our data and repeated key experiments included in this paper, and concluded that HIF-αCAD·p300 complex-mediated transactivation absolutely requires a deacetylase activity. Our data described here do not conflict with that report but show a distinct mechanism by which HDAIs repress HIF function and angiogenesis.
Since HDAIs repress HIF transactivation potential independently of HIF-1α degradation and, similar to HDAI-mediated degradation of HIF-1α (55, 71), this effect is not restricted to cells with defined genetic status of VHL and p53, the HDAI-based anti-HIF therapy may be effective to a broad range of solid tumors. However, since HIF activity plays an essential role in EPO production and promotes cell survival by stimulating adaptive energy metabolism and angiogenesis in ischemic/hypoxic lesions, our findings raise the possibility that long term, systemic administration of HDAIs may adversely affect erythropoiesis and ischemic lesions, such as coronary insufficiency.
Acknowledgments
We thank Drs. S. Berger, W. el-Deiry, A. Giordano, L. Puri, P. J. Ratcliffe, and B. Vogelstein for providing essential reagents. We thank Drs. W. G. Kaelin Jr., P. J. Ratcliffe, and S. Surrey for helpful discussions and scientific advice. We thank Drs. J. San Antonio, M. Risbud, and D. Stiehl for critical review of the manuscript. We thank Dr. S. McKenzie for continuing support and encouragement.
Footnotes
This work is supported in part by NCI, National Institutes of Health (NIH), Grants K01-CA098809 (to N. S.) and RO1-CA89212 (to J. C.) and W. W. Smith Trust Cancer Research Award C#0505 (to N. S.).
The on-line version of this article (available at http://www.jbc.org) contains primer sequences.
The abbreviations used are: HIF, hypoxia-inducible factor; VHL, von Hippel-Lindau tumor suppressor; NRR, normoxic repressive region; FIH, factor inhibiting HIF-1; HAT, histone acetyltransferase; HDAC, histone deacetylase; TF, transcription factor; TSA, trichostatin A; SBT, sodium butyrate; SAHA, suberoylanilide hydroxamic acid; RLU, relative light units; PIPES, 1,4-piperazinediethanesulfonic acid; RT, reverse transcription; CMV, cytomegalovirus; aa, amino acids; EPO, erythropoietin; CBP, CREB-binding protein; CREB, cAMP-response element-binding protein; IPF, interacting protein factor; HDAI, histone deacetylase inhibitor.
D. M. Fath, X. Kong, D. Liang, Z. Lin, A. Chou, Y. Jiang, J. Fang, J. Caro, and N. Sang, unpublished data.
References
- 1.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL. Physiol. 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 4.Achen MG, McColl BK, Stacker SA. Cancer Cell. 2005;7:121–127. doi: 10.1016/j.ccr.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 5.Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Pharmacol Rev. 2004;56:549–580. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- 6.Luttun A, Carmeliet P. Curr Opin Hematol. 2004;11:262–271. doi: 10.1097/01.moh.0000126936.58889.95. [DOI] [PubMed] [Google Scholar]
- 7.Harris AL. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 8.Powis G, Kirkpatrick L. Mol Cancer Ther. 2004;3:647–654. [PubMed] [Google Scholar]
- 9.Heinl-Green A, Radke PW, Munkonge FM, Frass O, Zhu J, Vincent K, Geddes DM, Alton EW. Eur Heart J. 2005;26:1327–1332. doi: 10.1093/eurheartj/ehi223. [DOI] [PubMed] [Google Scholar]
- 10.Vincent KA, Shyu KG, Luo Y, Magner M, Tio RA, Jiang C, Goldberg MA, Akita GY, Gregory RJ, Isner JM. Circulation. 2000;102:2255–2261. doi: 10.1161/01.cir.102.18.2255. [DOI] [PubMed] [Google Scholar]
- 11.Bruick RK, McKnight SL. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 12.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 13.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 14.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim AV, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 15.Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Proc Natl Acad Sci U S A. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, Jr, Pavletich NP. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 17.Stebbins CE, Kaelin WG, Jr, Pavletich NP. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- 18.Huang LE, Gu J, Schau M, Bunn HF. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salceda S, Caro J. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- 20.Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL. J Biol Chem. 1997;272:19253–19260. doi: 10.1074/jbc.272.31.19253. [DOI] [PubMed] [Google Scholar]
- 21.O’Rourke JF, Tian YM, Ratcliffe PJ, Pugh CW. J Biol Chem. 1998;274:2060–2071. doi: 10.1074/jbc.274.4.2060. [DOI] [PubMed] [Google Scholar]
- 22.Pugh CW, O’Rourke JF, Nagao M, Gleadle JM, Ratcliffe PJ. J Biol Chem. 1997;272:11205–11214. doi: 10.1074/jbc.272.17.11205. [DOI] [PubMed] [Google Scholar]
- 23.Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingston DM. Proc Natl Acad Sci U S A. 1996;93:12969–12973. doi: 10.1073/pnas.93.23.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM. Genes Dev. 1999;13:64–75. doi: 10.1101/gad.13.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sang N, Fang J, Srinivas V, Leshchinsky I, Caro J. Mol Cell Biol. 2002;9:2984–2992. doi: 10.1128/MCB.22.9.2984-2992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dames SA, Martinez-Yamout M, DeGuzman RN, Dyson HJ, Wright PE. Proc Natl Acad Sci U S A. 2002;99:5271–5276. doi: 10.1073/pnas.082121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freedman SJ, Sun ZYJ, Poy F, Kung AL, Livingston DM, Wagner G, Eck MJ. Proc Natl Acad Sci U S A. 2002;99:5367–5372. doi: 10.1073/pnas.082117899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Science. 2002;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 29.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mahon PC, Hirota K, Semenza GL. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang XJ, Gregoire S. Mol Cell Biol. 2005;25:2873–2884. doi: 10.1128/MCB.25.8.2873-2884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Annu Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 33.Sasakawa Y, Naoe Y, Noto T, Inoue T, Sasakawa T, Matsuo M, Manda T, Mutoh S. Biochem Pharmacol. 2003;66:897–906. doi: 10.1016/s0006-2952(03)00411-8. [DOI] [PubMed] [Google Scholar]
- 34.Qian DZ, Wang X, Kachhap SK, Kato Y, Wei Y, Zhang L, Atadja P, Pili R. Cancer Res. 2004;64:6626–6634. doi: 10.1158/0008-5472.CAN-04-0540. [DOI] [PubMed] [Google Scholar]
- 35.Michaelis M, Michaelis UR, Fleming I, Suhan T, Cinatl J, Blaheta RA, Hoffmann K, Kotchetkov R, Busse R, Nau H, Cinatl J., Jr Mol Pharmacol. 2004;65:520–527. doi: 10.1124/mol.65.3.520. [DOI] [PubMed] [Google Scholar]
- 36.Minucci S, Pelicci PG. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 37.Marks PA, Richon VM, Breslow R, Rifkind RA. Curr Opin Oncol. 2001;13:477–483. doi: 10.1097/00001622-200111000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J. J Biol Chem. 2003;278:14013–14019. doi: 10.1074/jbc.M209702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puri PL, Avantaggiati ML, Balsano C, Sang N, Graessmann A, Giordano A, Levrero M. EMBO J. 1997;16:369–383. doi: 10.1093/emboj/16.2.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein R, Halazonetis TD, Berger SL. Mol Cell Biol. 1999;19:1202–1209. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yuan W, Condorelli G, Caruso M, Felsani A, Giordano A. J Biol Chem. 1996;271:9009–9013. doi: 10.1074/jbc.271.15.9009. [DOI] [PubMed] [Google Scholar]
- 42.Sang N, Avantaggiatti ML, Giordano A. J Cell Biochem. 1997;66:277–285. doi: 10.1002/(sici)1097-4644(19970901)66:3<277::aid-jcb1>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 43.el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 44.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 45.Sang N, Giordano A. J Cell Physiol. 1997;170:182–191. doi: 10.1002/(SICI)1097-4652(199702)170:2<182::AID-JCP10>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 46.Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y. Cell. 1996;87:953–959. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 47.Blagosklonny MV, An WG, Romanova LY, Trepel J, Fojo T, Neckers L. J Biol Chem. 1998;273:11995–11998. doi: 10.1074/jbc.273.20.11995. [DOI] [PubMed] [Google Scholar]
- 48.Kaluzova M, Kaluz S, Lerman MI, Stanbridge EJ. Mol Cell Biol. 2004;24:5757–5766. doi: 10.1128/MCB.24.13.5757-5766.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- 50.Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, MacGregore-Cortelli B, Tong W, Secrist JP, Schwartz L, Richardson S, Chu E, Olgac S, Marks PA, Scher H, Richon VM. J Clin Oncol. 2005;23:3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Connor OA, Heaney ML, Schwartz L, Richardson S, Willim R, MacGregor-Cortelli B, Curly T, Moskowitz C, Portlock C, Horwitz S, Zelenetz AD, Frankel S, Richon V, Marks P, Kelly WK. J Clin Oncol. 2006;24:166–173. doi: 10.1200/JCO.2005.01.9679. [DOI] [PubMed] [Google Scholar]
- 52.Jeong JW, Bae MK, Ahn MY, Kim SH, Sohn TK, Bae MH, Yoo MA, Song EJ, Lee KJ, Kim KW. Cell. 2002;111:709–720. doi: 10.1016/s0092-8674(02)01085-1. [DOI] [PubMed] [Google Scholar]
- 53.Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, Moon EJ, Kim HS, Lee SK, Chung HY, Kim CW, Kim KW. Nat Med. 2001;7:437–443. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 54.Qian DZ, Kato Y, Shabbeer S, Wei Y, Verheul HM, Salumbides B, Sanni T, Atadja P, Pili R. Clin Cancer Res. 2006;12:634–642. doi: 10.1158/1078-0432.CCR-05-1132. [DOI] [PubMed] [Google Scholar]
- 55.Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Mol Cell Biol. 2006;26:2019–2028. doi: 10.1128/MCB.26.6.2019-2028.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barlev NA, Liu L, Chehab NH, Mansfield K, Harris KG, Halazonetis TD, Berger SL. Mol Cell. 2001;8:1243–1254. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- 57.Gu W, Roeder RG. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 58.Sartorelli V, Puri PL, Hamamori Y, Ogryzko V, Chung G, Nakatani Y, Wang JYJ, Kedes L. Mol Cell. 1999;4:725–734. doi: 10.1016/s1097-2765(00)80383-4. [DOI] [PubMed] [Google Scholar]
- 59.Lee YM, Kim SH, Kim HS, Son MJ, Nakajima H, Kwon HJ, Kim KW. Biochem Biophys Res Commun. 2003;300:241–246. doi: 10.1016/s0006-291x(02)02787-0. [DOI] [PubMed] [Google Scholar]
- 60.Zgouras D, Wächtershäuser A, Frings D, Stein J. Biochem Biophys Res Commun. 2003;300:832–838. doi: 10.1016/s0006-291x(02)02916-9. [DOI] [PubMed] [Google Scholar]
- 61.Deroanne CF, Bonjean K, Servotte S, Devy L, Colige A, Clausse N, Blacher S, Verdin E, Foidart JM, Nusgens BV, Castronovo V. Oncogene. 2002;21:427–436. doi: 10.1038/sj.onc.1205108. [DOI] [PubMed] [Google Scholar]
- 62.Williams RJ. Expert Opin Investig Drugs. 2001;10:1571–1573. doi: 10.1517/13543784.10.8.1571. [DOI] [PubMed] [Google Scholar]
- 63.Kato H, Tamamizu-Kato S, Shibasaki F. J Biol Chem. 2004;279:41966–41974. doi: 10.1074/jbc.M406320200. [DOI] [PubMed] [Google Scholar]
- 64.Bilton R, Mazure N, Trottier E, Hattab M, Dery MA, Richard DE, Pouyssegur J, Brahimi-Horn MC. J Biol Chem. 2005;280:31132–31140. doi: 10.1074/jbc.M504482200. [DOI] [PubMed] [Google Scholar]
- 65.Arnesen T, Kong X, Evjenth R, Gromyko D, Varhaug JE, Lin Z, Sang N, Caro J, Lillehaug JR. FEBS Lett. 2005;579:6428–6432. doi: 10.1016/j.febslet.2005.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fisher TS, Des Etages S, Hayes L, Crimin K, Li B. J Biol Chem. 2005;280:17749–17757. doi: 10.1074/jbc.M412055200. [DOI] [PubMed] [Google Scholar]
- 67.Kovacs JJ, Murphy PJM, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 68.Yu X, Guo ZS, Marcu MG, Nechers L, Nguyen DM, Chen GA, Schrump DS. J Natl Cancer Inst. 2002;94:504–513. doi: 10.1093/jnci/94.7.504. [DOI] [PubMed] [Google Scholar]
- 69.Simone C, Stiegler P, Forcales SV, Bagella L, De Luca A, Sartorelli V, Giordano A, Puri PL. Oncogene. 2004;23:2177–2187. doi: 10.1038/sj.onc.1207327. [DOI] [PubMed] [Google Scholar]
- 70.Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, Baudino TA, Cleveland JL, Brindle PK. EMBO J. 2005;24:3846–3858. doi: 10.1038/sj.emboj.7600846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Demidenko ZN, Rapisarda A, Garayoa M, Giannakakou P, Melillo G, Blagosklonny MV. Oncogene. 2005;24:4829–4838. doi: 10.1038/sj.onc.1208636. [DOI] [PubMed] [Google Scholar]