

Abstract
A central feature of heart disease is a molecular remodeling of signaling pathways in cardiac myocytes. This study focused on novel molecular elements of MAP kinase-mediated alterations in the pattern of gene expression of the protein phosphatase, PP2A. In an established model of sustained JNK activation a 70% decrease in expression of the targeting subunit of PP2A, B56α, was observed in either neonatal or adult cardiomyocytes. This loss in protein abundance was accompanied by a decrease of 69% in B56α mRNA steady-state levels. Given that the 3′-untranslated region of this transcript contains AU-rich elements known to regulate mRNA degradation, experiments explored the notion that instability of B56α mRNA accounts for the response. mRNA time course analyses with real time PCR methods showed that B56α transcript was transformed from a stable (no significant decay over 1 hr) to a labile form that rapidly degraded within minutes. These results were supported by complementary experiments that revealed that the RNA-binding protein AUF1, known to destabilize target mRNA, was increased 4-fold in JNK-activated cells. A variety of other stress-related stimuli, such as p38 MAP kinase activation and phorbol ester, upregulated AUF1 expression in cultured cardiac cells as well. In addition, gel mobility shift assays demonstrated that p37AUF1 binds with nanomolar affinity to segments of the B56α 3′-untranslated region. Thus, these studies provide new evidence that signaling-induced mRNA instability is an important mechanism that underlies the changes in the pattern of gene expression evoked by stress-activated pathways in cardiac cells.
Keywords: PP2A, MAPK, gene expression, AUF1, mRNA stability
Introduction
The tissue remodeling manifest in heart failure is accompanied by molecular remodeling, which includes changes in the balance of intracellular signaling cascades. A classic example of this is seen in the desensitization of β-adrenergic signaling which is observed in animal models and human heart failure (12). In addition, stress induced molecular remodeling involves the mitogen-activated protein kinase (MAPK) pathway which enhances hypertrophic growth (18).
It is now appreciated that there is a regulated balance between kinases and phosphatases that is crucial to normal cell function. Any disruption of this balance, including alteration in phosphatase activity, can lead to cellular dysfunction and disease. For example, overexpression of the catalytic subunit of PP2A, PP2Ac, in the cardiac myocytes causes depressed contractile function, dephosphorylation of key contractile proteins and hypertrophy (9). Targeting of this phosphatase is also important as transgenic mice expressing a dominant negative mutant of the scaffold/regulatory A subunit of PP2A, unable to bind targeting B subunits, resulted in mislocalization of PP2A and dilated cardiomyopathy (6).
In order to study molecular changes in PP2A in response to stress we exploited an established model of JNK activation (30). The main findings are that JNK activation leads to a marked loss in B56α protein expression and transcript that is associated with a striking increase in mRNA instability. Consistent with these results, JNK activation, as well as other stress-related stimuli, increased expression of AUF1, a protein known to enhance mRNA decay (2; 11). The importance of AUF1 is supported by gel mobility shift assay experiments which indicate that specific sequences within the B56α 3′-UTR bind p37AUF1. This work provides new insights into how stress-activated pathways regulate gene expression to alter signaling in cardiac cells.
Materials and Methods
Materials
The following primary antibodies were used for Western blotting: anti-HA (monoclonal, Babco), anti-PP1 (monoclonal, BD Biosciences ), anti-PP2A/A (polyclonal, Oxford Biomedical Research), anti-PP2A/C (monoclonal, BD Biosciences), anti-B56α (monoclonal, BD Biosciences), anti-AUF1 (Upstate) and the following from Cell Signaling anti-JNK, anti-phospho-JNK, anti-p38, anti-phospho-p38, anti-ERK, and anti-phospho-ERK. The following secondary antibodies were used for Western blotting: HRP conjugated goat anti-mouse and goat anti-rabbit (Jackson Immunoresearch Laboratories). The following secondary antibody was used for immunocytochemistry: Alexa Fluor-488 goat anti-mouse IgG (Molecular Probes).
The following adenoviruses were used for infection of cardiac myocytes: control, replication deficient human adenovirus type 5 mutant (Ad-dl312), constitutively active mutant of MKK7 (Ad-MKK7D) (30), constitutively active mutant of MKK6 (Ad-MKK6E) (30) or adenovirus encoding human B56α (Ad-B56α) (20). Recombinant viruses were prepared, amplified, purified, and tittered as previously described (14; 29).
Preparation of Cultured Neonatal and Adult Rat Ventricular Myocytes
Neonatal rat ventricular myocytes were routinely prepared from day-1 rats and cultured according to Gigena et al., 2005 (10). Isolation and culturing of adult rat ventricular myocytes has been previously described (10). All animal protocols were approved by the Animal Use and Care Committee of the University of Maryland School of Medicine
Adenovirus Transfection and Sample Preparation
Cultured neonatal and adult rat cardiac myocytes were transiently transfected for 1.5 hours at 37°C with one of the following adenoviruses: Ad-dl312, Ad-MKK7D, or Ad-B56α (100 particles/cell). Neonatal cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with insulin, transferrin, selenium (1%) and penicillin-streptomycin (2%) for 72 hrs. Adult cells were cultured in M199 media supplemented with insulin, transferrin, selenium (1%) and penicillin-streptomycin (2%) for 24 or 48 hours. Extracts of neonatal or adult rat myocytes were prepared by homogenizing cells in a lysis buffer (mmol/L): (20 Tris HCl pH 7.4, 1 EGTA, 1 EDTA, 150 NaCl, 0.1% β-mercaptoethanol, 1% Triton, 1:400 Protease inhibitor cocktail from Sigma) at 4°C. The cell extracts were fractionated by centrifugation at 100,000 × g for 10 min. The supernatant was removed (cytosolic fraction) and the pellet was resuspended in lysis buffer containing 1% Triton-X and recentrifuged to yield a solubilized membrane fraction. Alternatively to make whole cell extracts, lysates were homogenized in lysis buffer containing 1% Triton-X and centrifuged to obtain the supernatant.
Western blotting
Extracts were resolved on a 10% SDS polyacrylamide gel, transferred to PDVF (Immobilon-P, Millipore) or nitrocellulose (Duralose-UV, Stratagene) membranes and blocked with either 5% milk in PBS or 5% BSA in PBS with 0.1% Tween for 1 hr at room temperature. Blocked membranes were incubated overnight at 4°C with primary antibodies as indicated in Results. Following 1 hr incubations with secondary antibodies the immunoblots were developed using SuperSignal® Chemiluminescence detection method (Pierce).
Dot Blot Protocol
Neonatal rat cardiac myocytes were transiently transfected with adenovirus (Ad-MKK7D or Ad-dl312) and total RNA was isolated using Trizol reagent according to the manufacturer’s instructions (Invitrogen). RNA (2 μg) was fixed to nylon membranes (Amersham) and hybridized with 32P-labeled oligonucleotide DNA probes specific for GAPDH, atrial natiuretic factor (ANF), or α-skeletalactin (24). Results were quantified using a Storm Phosphoimager (Molecular Dynamics).
Luciferase Reporter Assay
Isolated neonatal rat cardiac myocytes (5 × 105) were seeded onto 60 mm dishes and transiently transfected with Ad-MKK7D or Ad-dl312 for 1.5 hours. Media was replaced with 5% fetal bovine serum in DMEM. Plasmids containing the human B56α gene or βGal gene (control) with either the -3500 β-myosin heavy chain-luciferase promoter (Gift from F. Haddad) or -3003 ANF-luciferase promoter (Gift from I. Farrance) were then transiently transfected overnight using FUGENE 6 (Roche). The following day the media was changed to DMEM supplemented with insulin, transferrin, selenium (1%) and penicillin-streptomycin (2%). Cells were cultured for 24 hours and then harvested in a passive lysis buffer (Promega). Promoter activity was detected with a dual luciferase kit (Promega) using thymidine kinase renilla as the normalization control according to manufacturer’s instructions.
Immunocytochemistry
Ad-d1312 or Ad-MKK7D (100 particles/cell) was transiently transfected into neonatal or adult rat cardiac myocytes on glass coverslips (1.6 cm diameter, density 4 × 105). Cells were cultured in DMEM (neonatal) or M199 medium (adult) supplemented with insulin, transferring, selenium and penicillin-streptomycin for 24 or 48 hours. Cells were fixed in 100% ethanol overnight at −20°C and then blocked for two hours at room temperature in 3% BSA and 5% normal goat serum in PBS. Cells were then incubated overnight with anti-B56α at 4°C (monoclonal, BD Biosciences). After 3 washes for 10 min each with PBS, cells were incubated for two hours with Alexa Fluor-488 goat anti-mouse IgG (Molecular Probes). Cells were examined with a laser scanning confocal microscope (63× objective; Carl Zeiss, Inc.). Control experiments were done without the primary antibody.
RT-PCR
Total RNA was isolated using Trizol reagent from neonatal or adult rat cardiac myocytes infected with Ad-dl312, Ad-MKK7D and Ad-B56α. Primers specific for B56α (forward primer: 5′-CTTGAGGCCTCTTGGCCTCACATAC-3′ and reverse primer: 5′-GCTGAGAATACTGTGCATGTTGTAAGC-3′) or B56γ1 (forward primer: 5′-AGGATGGTGGTGGATGCGG-3′ and reverse primer: 5′-CACTCCCGAGTTACTCTCTT-3′) were used to amplify a 1090 and 1032 base pair fragment respectively by reverse transcriptase-PCR (RT-PCR). A Superscript III RT-PCR with Platinum Taq (Invitrogen) kit was used for reactions at an annealing temperature of 56°C for B56α primers and 52°C for B56γ1 primers. Products were amplified up to 50 cycles.
Ribonuclease Protection Assay
RT-PCR was used to generate probes towards the C-terminal end (upstream primer: 5′-TTTCCAAAGAACACTGGAATCAGACTA-3′ and downstream primer: 5′-GCTGAGAATACTGTGCATGTTGTAAGC-3′) of the rat sequence of B56α to yield a 237 base pair fragment with the following sequence: 5′-TTTCCAAAGAACACTGGAA TCAGACTATTGTAGCTCTGGTATACAACGTGCTGAAAACCCTAATGGAGATGAACGGGAAGCTTTTTGATGACCTTACTAGTTCCTACAAGGCTGAAAGACAGAGAGAGAAGAAGAAAGAACTGGAACGCGAAGAATTATGGAAAAAGTTAGAAGAGTTGAAGCTGAAGAAGGCTCTAGAGAAACAGAACAATGCTTACAACATGCACAGTATTCTCAGC-3′. The size of the probe was verified by gel electrophoresis and the product was subcloned into the pGEM-T Easy vector (Promega). The ligated vector containing the probe was electroporated into DH5αF′ E.coli cells. These linearized probes were in vitro transcribed with SP6 RNA polymerase using MAXiscript kit (Ambion) and labeled with 32P-UTP (Perkin Elmer).
Total mRNA from cells infected with Ad-dl312 or Ad-MKK7D was isolated using Trizol reagent. The RNA pellet was resuspended in 50 μl of water and annealed to 1fmol of labeled B56α probe overnight at 42°C using RPA III kit (Ambion). The following day the unprotected RNA was digested and the protected fragment was resolved on a 5% polyacrylamide gel containing 7 mol/L urea. The gel was dried, exposed to a phosphoimager screen and analyzed. Untreated probe was run as a control. For loading control of RNA, samples were run on a formaldehyde/agarose gel and 18S was detected and quantitated by ethidium bromide staining.
Real time PCR
Total RNA was extracted using Trizol as described above. RNA was subjected to DNase treatment (DNase free kit, Ambion) and 0.5 μg of total RNA was reverse transcribed using Superscript III reverse transcriptase (Invitrogen). Primers used for the RPA probes were also used for Real time PCR. The reaction yielded a 237 bp fragment which was detected on a 1% agarose gel. 18S primers from Ambion were used for internal control for normalization. DNA was amplified using Taq Pro DNA Polymerase (Denville) and products were measured using the MJ Research real time thermocycler. SYBR Green I (Molecular Probes) was used to detect the DNA products. Both sets of primers were amplified using the same annealing temperatures using the following program: hot start to activate the polymerase at 95°C for 5 min, 95°C (30 s), 55°C (45 s), 68°C (45 s) for 50 cycles and 72°C for 2 min. A melting curve was performed to ensure that there were no primer dimers and that there was a single product. All samples were run in triplicate. The changes between the treated samples and untreated samples were based on the threshold (CT) which is set when there is a clear detectable increase in fluorescence. To measure the fold change between control and treated samples, ΔΔCT was calculated where ΔΔCT = (CTtreated-CTtreated internal) – (CTcontrol-CTcontrol internal).
Preparation of Recombinant His6-p37AUF1 and Gel Mobility Shift Assays
Recombinant p37AUF1 was synthesized in E. coli TOP10 cells as an N-terminal, His6-tagged protein using the pBAD/His system (Invitrogen) and purified by Ni2+-affinity chromatography as described previously (34). Five regions containing the 5′-end of the coding region, and four different sequences which make up the complete 3′-UTR region of the mouse B56α mRNA sequence was amplified using RT-PCR. The products were generated using the following primers: Coding region: 5′primer:GGAGAAAGTGGACGGCTTCA; 3′ primer: GCTCTTCAAGTCTGAGACAGAGTC; 3′-UTR #1: 5′ primer: TTCAGAGCAGACCTCATCAGTAT; 3′ primer: AGTGTGCAGAGGACGGCTTC; 3′-UTR #2: 5′ primer: GAAGCCGTCCTCTGCACACT; 3′ primer: TTAATGAGGCTGGGCTCTTGTCC; 3′-UTR #3: 5′ primer: GGACAAGAGCCCAGCCTCATTAA; 3′ primer: GAGGAGAGGAATGAAGGAGGTGA; 3′-UTR # 4: 5′ primer: TCACCTCCTTCATTCCTCTCCTC; 3′ primer: TCCATACAACTGCAAGGCAAGAG. Products were subcloned into the pGEM-T Easy vector (Promega) and the vector containing the probe was electroporated into DH5αF′ E.coli cells. The probes were linearized and in vitro transcribed with either the T7 or SP6 RNA polymerase using MAXiscript kit (Ambion) and labeled with 32P-UTP (Perkin Elmer) to specific activities of 8652 cpm/fmol.
Binding reactions for gel mobility shift assays were performed with 0, 2.5, 5, 10, or 25 nM (dimer concentrations) of purified recombinant His6-p37AUF1 fusion protein and 0.2 nM of the indicated 32P-labeled UTR-RNA probe in a final volume of 10 μl containing 10 mM Tris HCl (pH 8.0), 2 mM dithiothreitol, 50 mM potassium chloride, 5 mM of magnesium chloride, 1 μg/μl heparin, 0.1 μg/μl acetylated bovine serum albumin, 8 units of RNasin (Promega), and 10% glycerol. Reactions were incubated for 15 min on ice and resolved by electrophoresis through non-denaturing 4% polyacrylamide gel (60:1 acrylamide:bisacrylamide) using 0.5× TBE as described (31). Reaction products were visualized by PhosphorImager scan (Molecular Dynamics). Competition assays were performed with 25 nM (dimer concentration) His6-p37AUF1 and 2 nM of 32P-labeled #4 UTR B56α ARE probe (10-fold higher than in gel shift experiments above) with 10, 100, or 1000-fold excess of a 38-mer of ARE-containing sequence from TNF-α 3′-UTR as previously described (32). A non target RNA (31-mer) derived from the coding sequence of β-globin served as a non-target negative control (32). Competition binding reactions were performed in the same volume using the same buffer components as stated above.
Statistical Analysis
All data are reported as the means ± SE. The statistical significance of differences between the control and experimental groups were calculated by one way analysis of variance (ANOVA) followed by Newman-Keuls test or by Wilcoxon Two Group Signed Rank Test by the use of the GB-STAT statistical software program (Dynamic Microsystems, Inc.). A p-value of less than 0.05 was considered significant.
Results
Expression of MKK7D induces features of stress in cultured myocytes
In order to study the link between JNK and PP2A signaling in the heart, we used an established cellular paradigm of JNK activation, overexpression of a constitutively active upstream activator, MKK7 (30). As shown in Figures 1 and 2, overexpression of MKK7D resulted in activated JNK, seen as increases in phospho-JNK. The activation was restricted to JNK as no increases in phospho-p38 or phospho-ERK were observed (Figure 1). Expression of this recombinant mutant, MKK7D, in cultured cardiac cells is known to induce several hypertrophic features including increases in cell size, ANF expression, and sarcomere organization (30). Control studies documented that MKK7D-expressing cultured myocytes used in this study also expressed hypertrophic markers. Dot blot analyses showed that canonical marker genes, ANF and α-skeletalactin, were elevated in these cultures by 3- and 2.25-fold respectively (Figures 3A and 3B). Expression of GAPDH, a typical control gene, was also elevated in these cultures, but ANF and α-skeletalactin were increased even when normalized to this control. Other studies showed that JNK activation was accompanied by 2.5-fold and 3-fold increases in β-MHC-luciferase and ANF-luciferase activities respectively (Figure 3C). These data extend the analysis of Wang et al. (30) to show that JNK activation evokes some of the characteristic gene signals broadly associated with hypertrophic paradigms in cultured cardiac cells.
Figure 1.
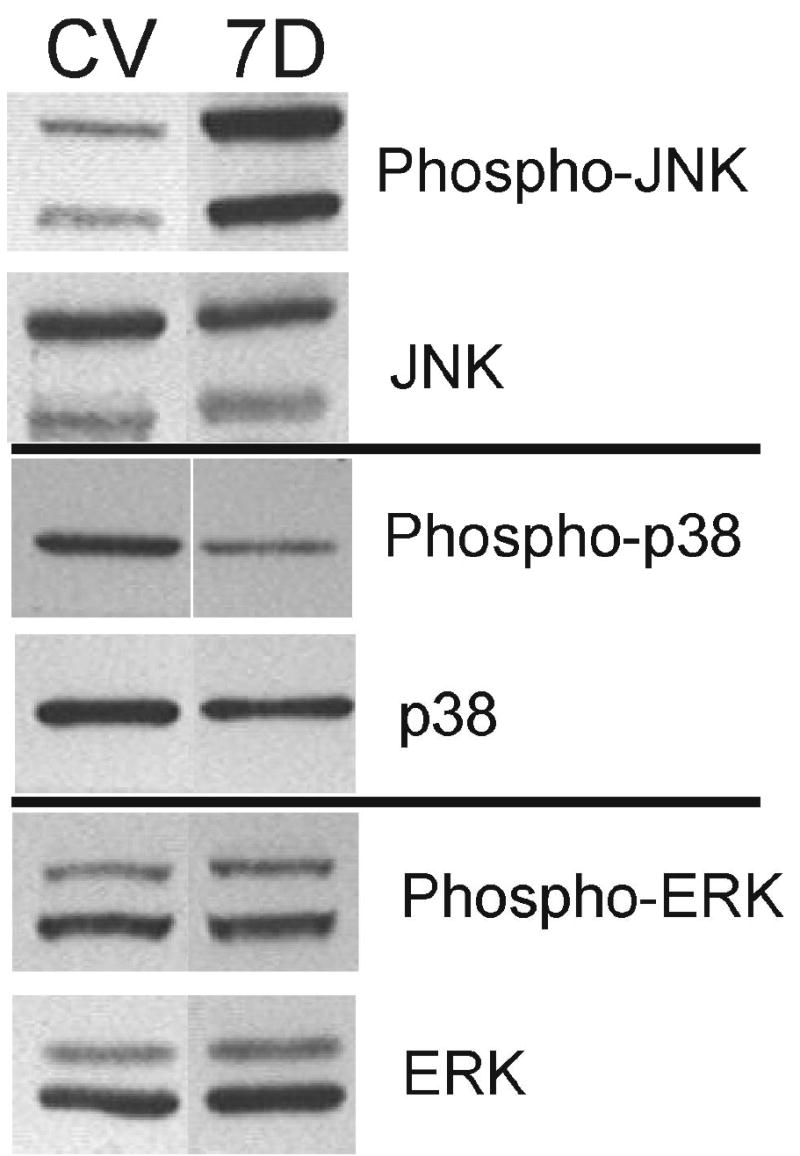
Specific activation of JNK by MKK7D in cultured neonatal cardiomyocytes. Neonatal rat cardiomyocytes were infected with Ad-MKK7D (7D) or Ad-dl312 virus (CV) for 72 hrs. Cells were homogenized in a lysis buffer containing 1% Triton-X and cell extracts were resolved by SDS-polyacrylamide gel electrophoresis and developed by Western blot approaches as described in Methods. Shown are typical Western blots probed with anti-phospho-JNK, anti-JNK, anti-phospho-p38, anti-p38, anti-phospho-ERK, and anti-ERK.
Figure 2.
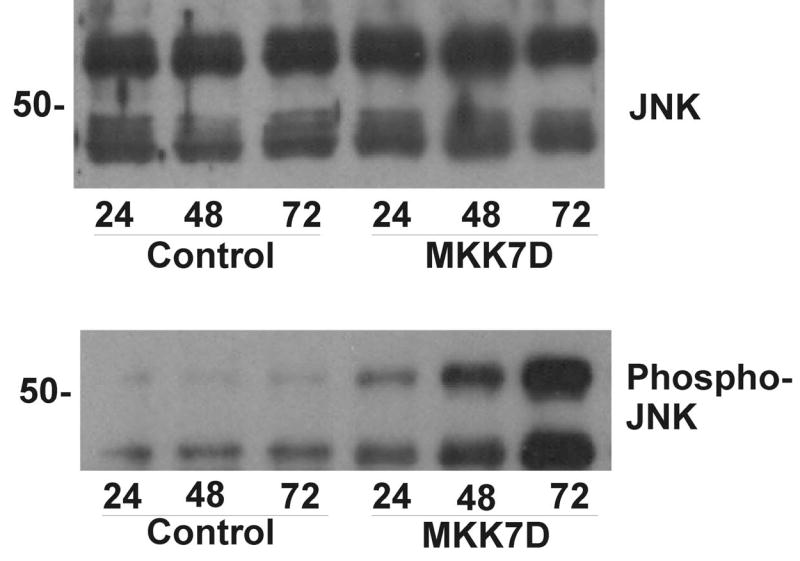
Time course of JNK activation in cultured neonatal cardiomyocytes. Cultured cardiomyocytes were infected with Ad-MKK7D or Ad-dl312 virus (Control) for 24, 48, and 72 hrs. Cell extracts were analyzed by Western blot approaches as described in Methods. Shown are typical Western blots probed with anti-phospho-JNK (bottom panel) and anti-JNK (top panel).
Figure 3.
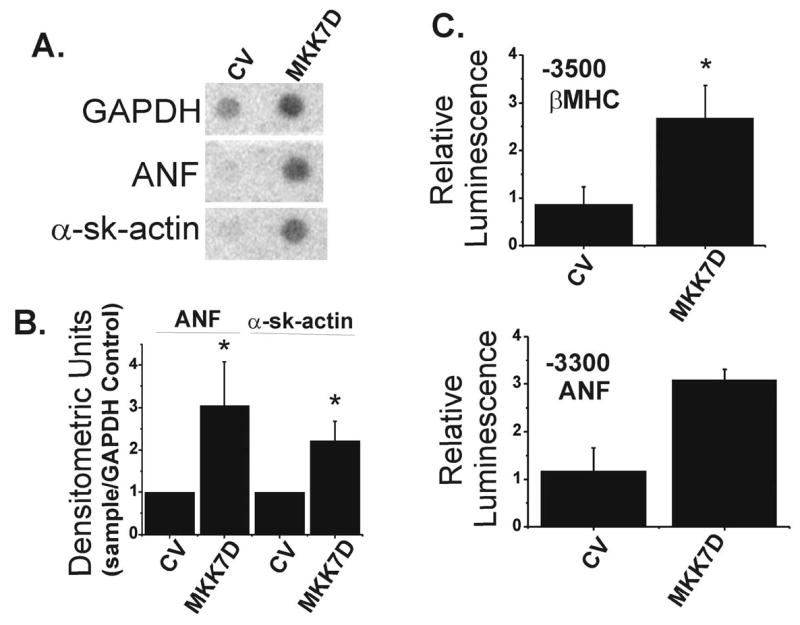
Expression of MKK7D induces hypertrophic markers in cultured myocytes. Total mRNA was isolated from neonatal rat cardiomyocytes infected for 72 hrs with Ad-dl312 (CV) or Ad-MKK7D. Levels of mRNA for ANF and α-sk-actin were quantified by dot blot analyses (see Methods). Panel A shows typical dot blots from control cultures (CV) and cultures transfected with MKK7D (MKK7D). Panel B shows summary data, normalized to control virus and corrected for GAPDH. The values are the means ± SE, n=3, performed in duplicate, * p< 0.05. In Panel C cardiomyocytes were infected with Ad-MKK7D along with expression plasmids encoding either -3500 β-MHC-luciferase or -3003 ANF-luciferase and luciferase activities were quantified as described in Methods. The values are the means ± SE for -3500 β-MHC-luciferase (n=3, * p<0.05) and -3300 ANF-luciferase (n=2, ± range/2).
Expression of MKK7D causes a decrease in B56α expression in neonatal and adult cardiac myocytes
Since MAPK signaling is known to be associated with PP2A activity, sustained JNK activation might alter PP2A at the molecular level (1; 13; 17; 28). While activation of JNK did not alter expression of PP1, or the catalytic and scaffolding subunits of PP2A, PP2Ac and PP2A/A respectively, in the cytosolic fraction, there was a marked decrease in the PP2A targeting subunit, B56α in both the cytosolic and membrane fractions (Figure 4A). As shown in summary data in Figure 4B, B56α protein decreased by 73% and 54% in the cytosolic and membrane fractions respectively. Since this B subunit has a distinct pattern of localization in cultured cardiac myocytes (10), it was possible that there was a selective loss in B56α in particular subcellular sites. However, confocal image analysis revealed that there was a global loss of B56α throughout these cells (Figure 4C). Although PP2Ac and PP2A/A increased 2-fold in the crude membranes it does not reflect a marked change in the total cell protein expression since only 5.4% of the total cellular PP2Ac and PP2A/A reside in this fraction. Thus sustained JNK activation leads to a marked decrease in B56α expression without altering levels of the catalytic or scaffolding subunits of PP2A.
Figure 4.
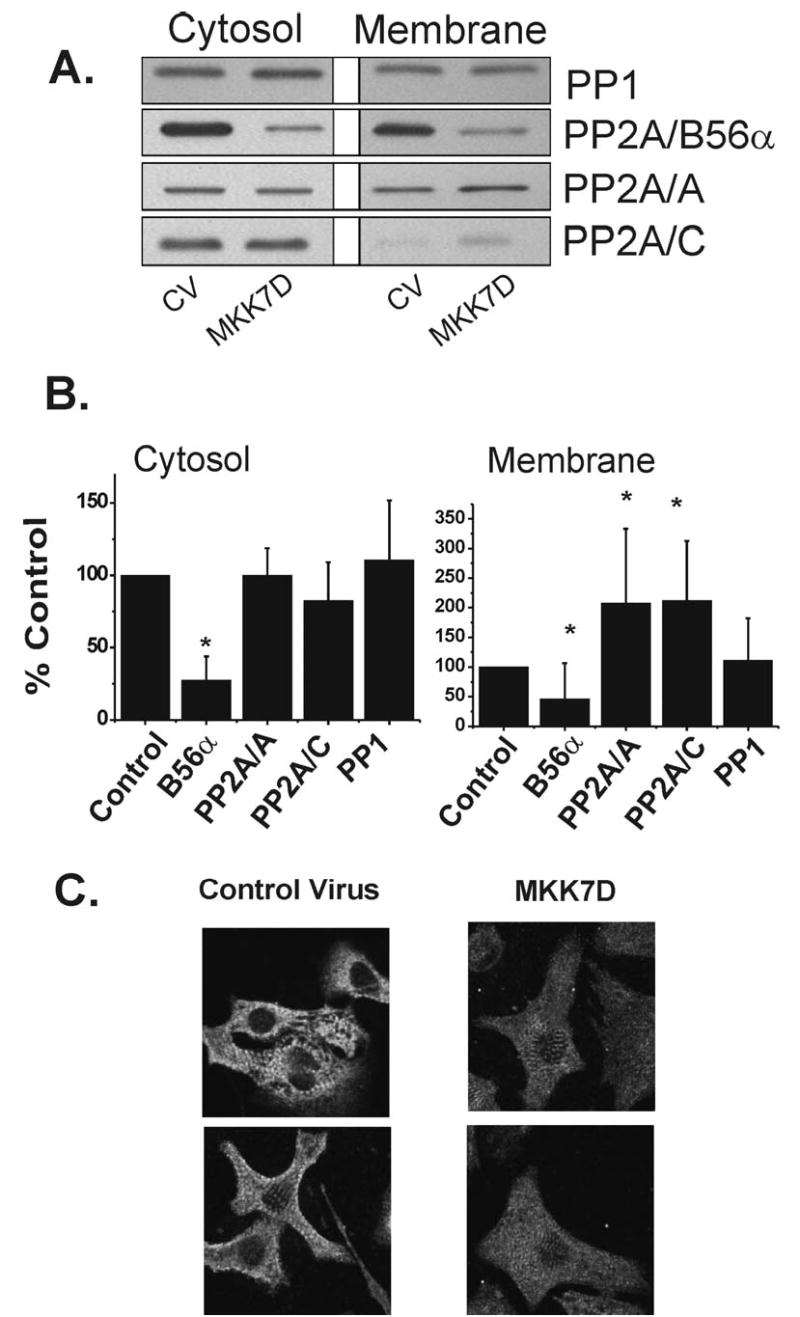
Effects of JNK activation on phosphatase subunit levels. A. Cultured cardiomyocytes were infected with Ad-MKK7D or Ad-dl312 virus (CV) for 72 hrs. Cell extracts were analyzed by Western blot approaches as described in Methods. Shown are typical Western blots probed with anti-PP1, anti-B56α, anti-PP2A/A scaffolding subunit, and anti-PP2Ac catalytic subunit. B. The histograms show the summary data in which the densities of the bands were normalized to the values in the cultures treated with control virus. The values are the means of 8–9 experiments. (mean ± SE, * p<0.05 by Wilcoxon Two Group Signed Rank Test, compared to the control) C. Neonatal cardiac myocytes were transiently transfected with Ad-dl312 (control virus) or Ad-MKK7D (MKK7D) for 48 hours. Cells were fixed in 100% ethanol and were developed for immunofluorescent analysis using anti-B56α as described in Methods. Shown are confocal images acquired in parallel with the same optical gain settings. As shown there is a global loss in B56α protein rather than decreases in particular subcellular regions.
A series of parallel experiments were performed in cultured adult ventricular myocytes. As shown in Figure 5, Western blot analyses revealed that B56α is decreased by 60% in MKK7D-transfected adult cells as well. Figure 5C shows representative transmitted light microscopic images of cells infected with virus after 24 and 48 hrs demonstrating that cells retain rod-shaped structure with no morphological indications of cell toxicity or death. Thus, the loss in B56α abundance is also seen in adult cells in response to MKK7D expression.
Figure 5.
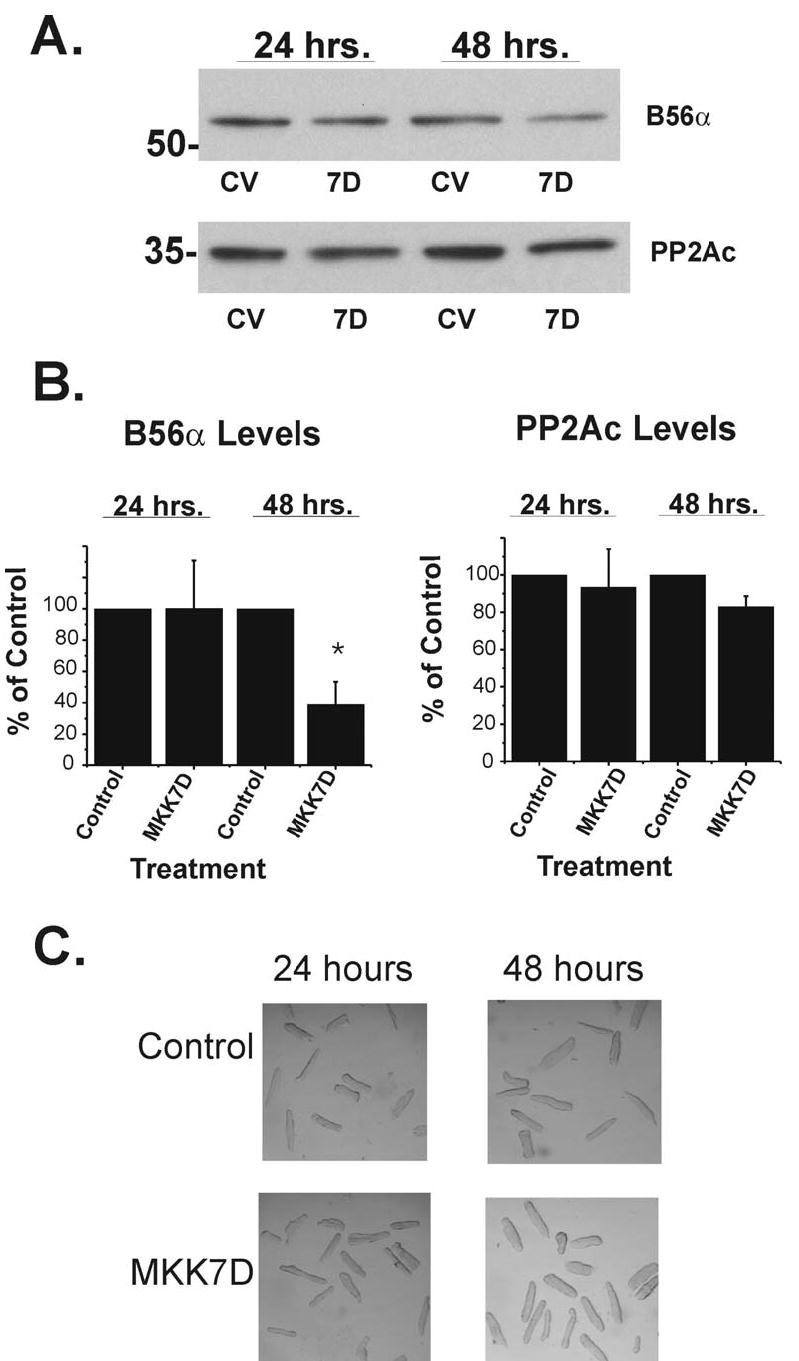
B56α expression decreases in adult cardiomyocytes. Cultured adult rat ventricular myocytes were infected with either Ad-dl312 (CV) or Ad-MKK7D (7D) for 24 and 48 hours. A. Total cell extracts were analyzed by Western blot approaches as described in Fig. 2 and Methods. Panel B shows summary data of B56α and PP2Ac Western blots (n=4 ± SE, *p< 0.05). C. Transmitted light phase contrast photomicrographs of cultures were taken at 24 and 48 hrs after infection with viruses.
Effect of JNK activation on B56α protein stability
Studies have shown that intracellular B56 proteins can be unstable unless bound to their cognate binding partners, PP2A/A and PP2Ac (27). Accordingly, studies were initiated to determine if B56α instability accounts for the decline in the protein seen in JNK-activated cells. B56α turnover in myocytes with high levels of JNK activation (Figure 2) was estimated in cycloheximide-treated cells. Important control experiments revealed that an 8 hr cycloheximide treatment alone did not stimulate JNK in control cells (Figure 6A). However, modest increases in phospho-JNK were seen in MKK7D-expressing cells at 8 hr. Importantly, Figure 6B shows that B56α is relatively stable in both control and MKK7D cells when treated with cycloheximide. The corresponding summary data in the semi-log plot in Figure 6C shows that there was no significant loss in protein over 4 hrs. However at 8 hrs B56α decreased about 40% in JNK activated-cells, a value significantly greater than that seen in control values. Longer incubations may have further resolved this increased protein instability but cycloheximide was toxic beyond 8 hrs. Overall protein instability may account for part of the decrease seen in B56α it is likely that other mechanisms are also involved.
Figure 6.
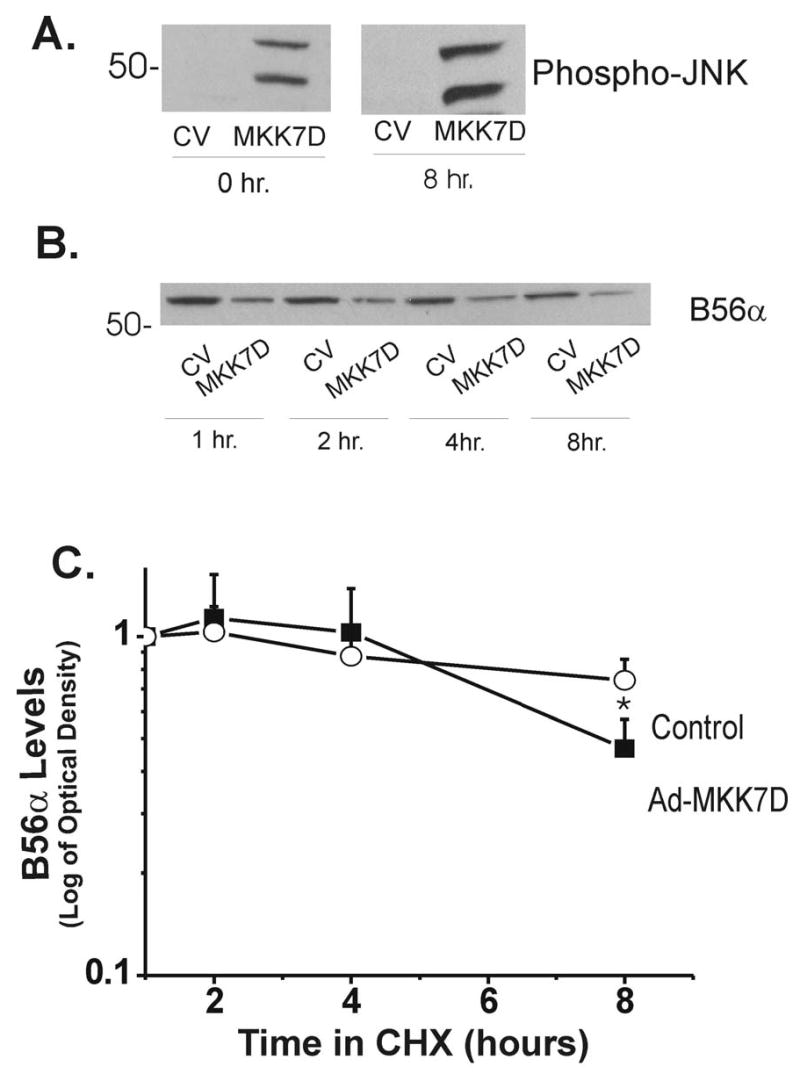
B56α protein stability in JNK-activated cardiomyocytes. Neonatal cardiomyocytes were transiently transfected with control adenovirus (Ad-dl312, CV) or Ad-MKK7D. After 72 hours in culture, cells were incubated with cycloheximide at times indicated. Panel A shows control experiments in which the effects of 8 hr incubations with cycloheximide on phospho-JNK levels were examined. Panel B shows a typical Western blot in which levels of B56α were monitored over time following addition of cycloheximide. Panel C shows a semilog plot of the time course of the decrease of B56α, where the protein levels are normalized to values at 1 hr of treatment (means ± SE, n=4, *p<0.05).
Effects of JNK activation on B56 mRNA levels
It was possible that alterations in mechanisms in protein expression prior to translation might underlie the decreases in B56α. As decreases in B56α mRNA could also contribute to loss of protein, an RT-PCR approach was developed to qualitatively examine this notion. In a positive control, mRNA amplified from cells that were infected with adenovirus that overexpresses B56α yielded an expected more abundant PCR product compared to naive cells (Figure 7A). As shown in Figure 7A, B56α mRNA appears to be markedly lower in MKK7D cells compared to B56α-transfected or even control cells. A semi-quantitative RT-PCR approach further demonstrated this decrease in B56α transcript. Aliquots were taken from the RT-PCR reaction every 5–10 cycles and as shown in Figure 7B the amount of B56α PCR product is lower in MKK7D infected cells throughout the range of cycles used in the reaction. The specificity of this effect was supported in parallel experiments in which RT-PCR amplification of mRNA with primers specific for B56γ1, a highly homologous B subunit expressed in cardiac cells (10), showed no change in response to JNK activation (Figure 7C).
Figure 7.
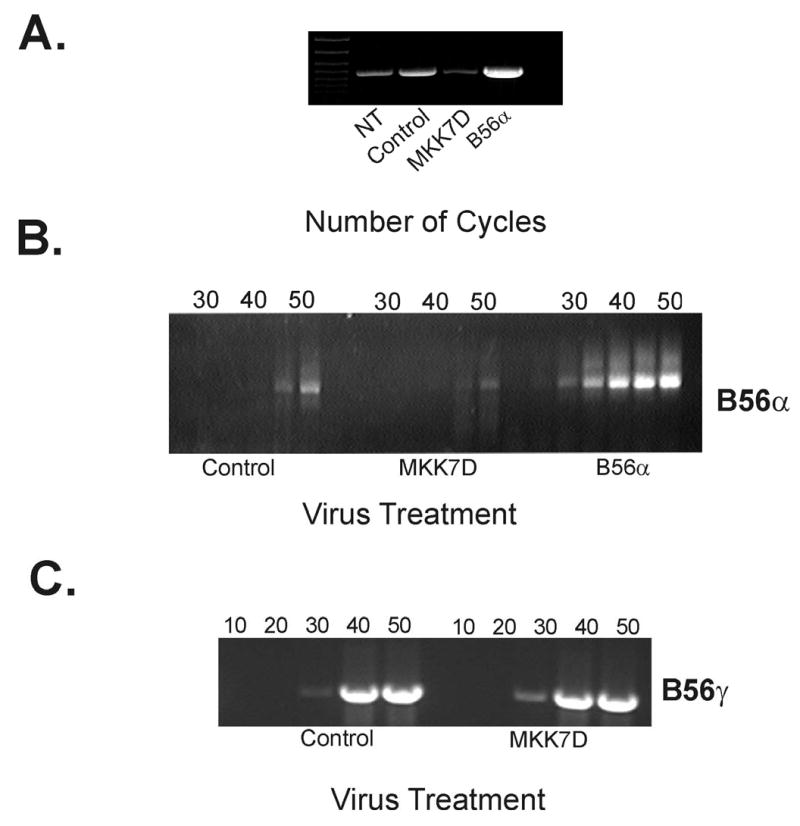
RT-PCR analysis of B56 transcript levels in JNK-activated cardiac cells. Total mRNA was isolated from neonatal rat cardiomyocytes from non-transfected cells (NT), or cells infected with Ad-dl312 (control), Ad-MKK7D, or Ad-B56α and B56α mRNA was amplified by RT-PCR as described in Methods. Shown in panel A is an ethidium bromide-stained DNA gel that resolves PCR product for each culture group. In Panel B, PCR products were analyzed every 5 cycles during the course of reaction as indicated. In panel C, primers specific for B56γ1 were used to amplify a 1000 bp fragment from total mRNA isolated from Ad-dl312 (control) and Ad-MKK7D infected rat cardiomyocytes as described in Methods. Products were collected every 10 cycles as indicated
It was important to complement the results from RT-PCR analysis with a more quantitative approach. Given the low abundance of transcript, a ribonuclease protection assay (RPA) was developed. Figure 8A is a representative RPA autoradiogram that shows the free probe (left lane) and the protected B56α fragment (right lanes). As shown in the summary data in Figure 8B, consistent with RT-PCR results, steady state B56α mRNA was decreased by 69% in JNK-activated cells. Additionally, analysis of the RPA results confirmed the low abundance of the B56α transcript, 4.9 ± 0.9 copies per cell in control cultures as determined by densitometry of the protected fragment in the RPA assay. Taken together these two complementary results demonstrate that B56α mRNA is markedly decreased in JNK-activated cells.
Figure 8.
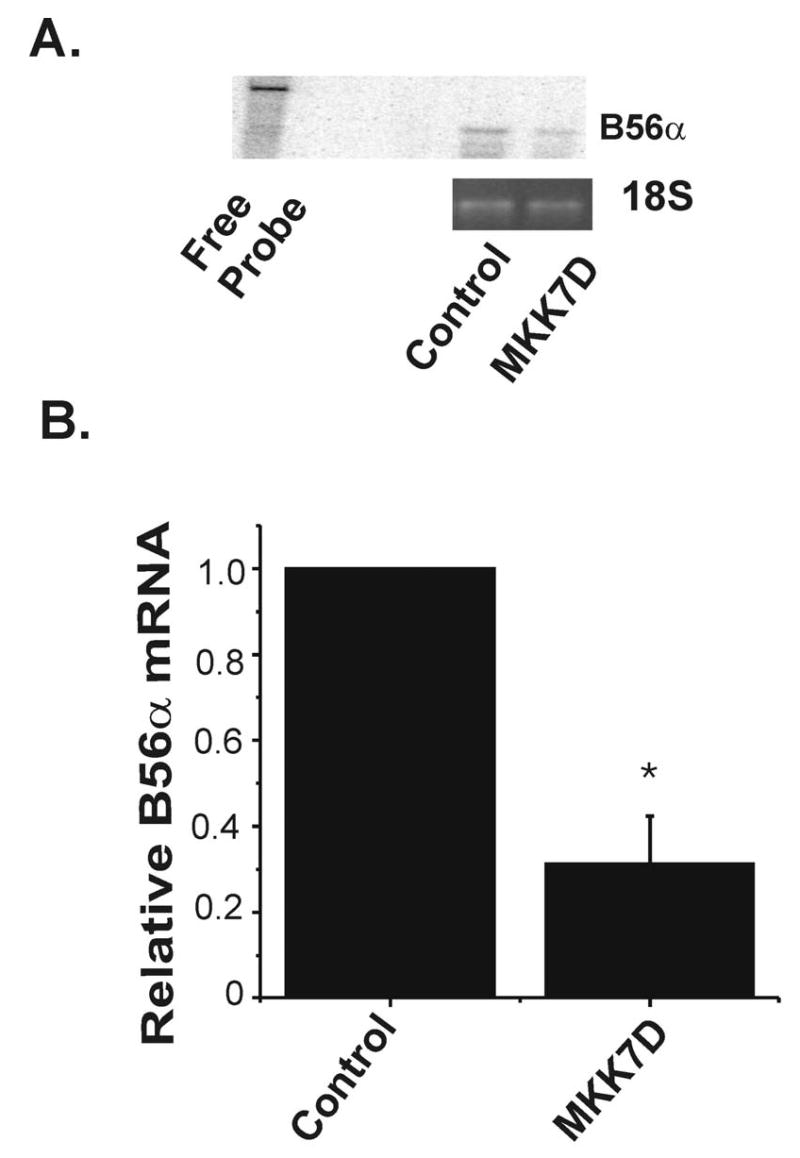
Quantification of B56α mRNA in JNK-activated cells. Panel A is a representative autoradiogram from an RPA which shows a band for the B56α probe alone (free probe, left lane) and 237 bp fragment generated in digestion reactions with mRNA isolated from cardiomyocytes infected with Ad-dl312 (control) or Ad-MKK7D. 18S ribosomal RNA, used for loading control was detected by ethidium bromide staining (lower panel). Panel B shows the summary data, normalized to control cell levels, which are the means ± SE (n=4, *p<0.01).
B56α mRNA is destabilized in JNK-activated cells
There are several possible mechanisms for the decrease in B56α transcript abundance in JNK-activated cells. An analysis of the 3′-untranslated region of B56α mRNA reveals important clues. As shown in Figure 9, such regions in mouse and human B56α contain AU-rich elements (AREs) that are known to contribute to mRNA instability through interactions with proteins such as AUF1(21; 35). The presence of these ARE elements along with the results from Figures 7 and 8 suggest that JNK activation might lead to B56α mRNA destabilization. Because of the low abundance of transcript a real time PCR method was developed to examine this notion. Initial experiments showed that B56α mRNA is decreased by 48.5% in 48 hrs in MKK7D-expressing cells so this shorter culture period was used in mRNA turnover experiments. B56α mRNA stability was estimated by quantifying the loss of mRNA. In this case cells were treated with α-amanitin under conditions that are well established to inhibit active transcription (3; 7; 10). As shown in Figure 10, whereas the transcript remained relatively stable over 1 hr in control cells, mRNA abundance was reduced to 25% of initial levels within 30 min in MKK7D-expressing cells. No further decline was seen beyond 30 min, perhaps due to the very low abundance of transcript at that point, estimated to be about 1.2 copies per cell. Thus, these results are consistent with the view that JNK-activation in cardiac cells decreases B56α protein abundance through destabilizing mRNA.
Figure 9.
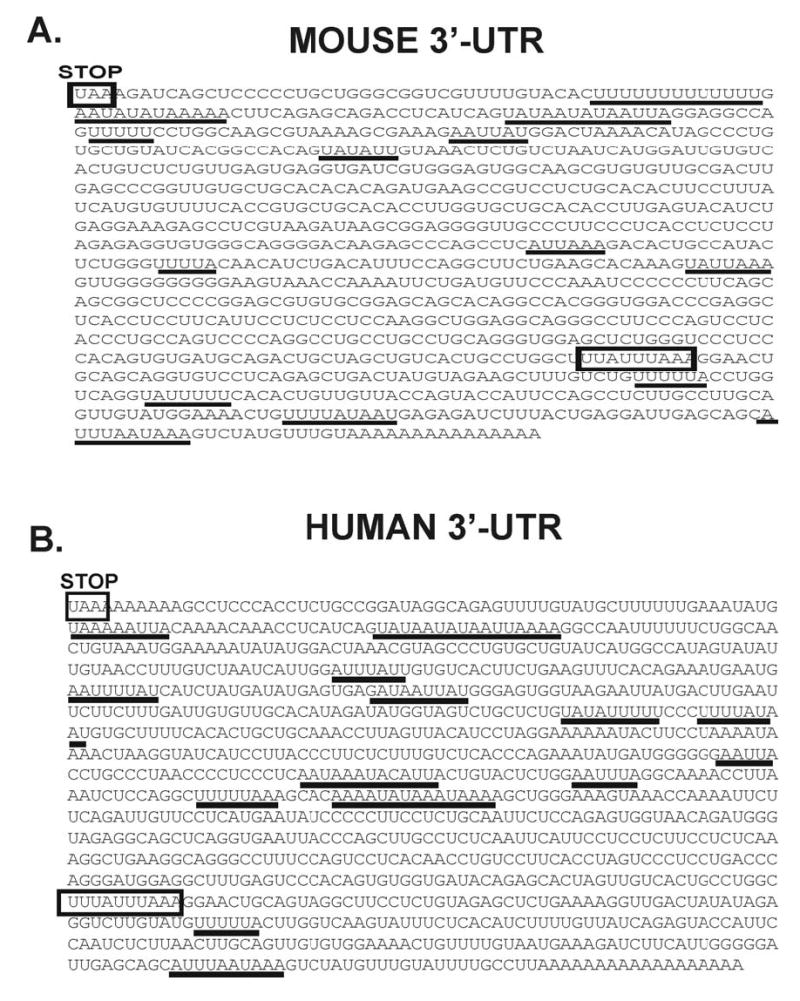
3′ UTR of B56α mRNA contains AU-rich elements. The mouse (accession number BC 023062) and the human (accession number NM 006243) nucleotide sequences for the 3′-untranslated region of B56α are displayed with regions containing canonical destabilizing motifs highlighted. The mouse nucleotide sequence begins with the stop codon (UAA) at nucleotide 656 and extends for 1005 nucleotides. The human nucleotide sequence begins with the stop codon at nucleotide 2035 and extends for 1110 nucleotides. Several AU-rich sequences that are potential AUF1 protein binding sites are underlined and a putative mRNA destabilizing sequences are highlighted in the boxed regions.
Figure 10.

Time course analysis for degradation of B56α transcript in cardiomyocytes. Cardiomyocyte groups, infected with Ad-d1312 (control, CV) or Ad-MKK7D, were treated with α-amanitin (25μg/ml) conditions that are well established to terminate transcription. At various times RNA was isolated and analyzed for B56α transcript by real time PCR as described in Methods. Shown is the summary data from time course studies of B56α transcript levels remaining after treatment with α-amanitin. The values are normalized to time 0 values for each culture group and represent means ± SE (n=4 MKK7D cells, n=3 for CV cells where each experiment was run in triplicate, * p<0.05 compared to control values).
JNK activation causes an increase in AUF1 expression
The observation that ARE segments are found in B56α mRNA combined with marked increase in B56α mRNA instability in JNK-activated cells suggested that AUF1 proteins were involved in this response. This model was explored by examining AUF1 protein expression following JNK activation. As shown in the Western blot in Figures 11A and 11B, in MKK7D-expressing cells there is a 4-fold increase of all four AUF1 protein isoforms. Interestingly, the response is also seen in another cellular stress model, p38 MAPK activation in MKK6E overexpressing cells (30) in which AUF1 increases are also accompanied by decreases in B56α protein (Figure 11B and 11C). In fact this response, where a decrease in B56α expression is accompanied by an increase in AUF1levels, is broadly seen in several stress-activated pathways including PMA, serum, and LPS treatment (Figure 11D). In other parallel experiments hypertophic-like stimuli, angiotensin II, endothelin, or phenylephrine failed to alter expression of either protein (data not shown). Thus, induction of AUF1 protein expression appears to be a central feature of some stress activation pathways and may play a role in the regulation of gene expression seen in JNK-activated cells.
Figure 11.

JNK activation causes an increase in AUF1. Neonatal rat cardiomyocytes were infected with Ad-d1312 (control, CV), Ad-MKK7D (7D), or Ad-MKK6E (6E) for 72 hours. Cell extracts were analyzed by Western blot methods using anti-AUF1 as a probe. (A) A representative Western blot is shown with the four bands corresponding to AUF1 alternatively spliced forms, p37, p40, p42, p45 (11). (B) Summary data of AUF1 protein levels in which the sum of densities of the four bands were normalized to the values in the control cultures (means ± SEM, n=4 for MKK7D cells, n=3 for MKK6E cells, * p<0.05). (C) B56α protein was quantified in parallel experiments. The summary data values are shown below as percent decrease compared to the control (means ± SE).
D. Neonatal rat cardiomyocytes were treated with 0.2 μmol/L PMA, 5% fetal bovine serum (FBS), or 1μg/ml LPS for 72 hrs. Control non-treated cells were cultured for 72 hours. Cells were homogenized in a lysis buffer containing 1% Triton-X and cell extracts were separated in a 10% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane and were probed with anti-AUF1 (top panel) or anti-B56α (bottom panel).
AUF1 Binding to 3′-UTR of B56α
The increase in AUF1 supports the idea that this protein contributes to mRNA instability in JNK-activated cells. Accordingly, a series of experiments was designed to determine if AUF1 does indeed bind to the 3′-UTR of B56α mRNA. Sequences of synthetic RNAs corresponding to the 5′-end of the coding region and four separate regions of the B56α 3′-UTR were amplified by RT-PCR (Figure 12A). The 32P-labeled probes were incubated in binding reactions with varying concentrations of purified p37AUF1 (AUF1). Distinct RNA-protein complexes were resolved from free probe by non-denaturing polyacrylamide gel electrophoresis. The results of these gel shift assays are shown in Figure 12. 3′-UTR region #4 which contains many putative AREs, including a nonamer sequence (Figures 9 and 12A), displays a significant shifted species with a concomitant loss of free probe even in the 10 nM range (Figure 12E). This binding is consistent with high affinity interactions between AUF1 and other known unstable transcripts such as c-myc (8). In addition, 3′-UTR regions #1 and 3, which contain putative AUF1 binding sequences (Figures 9 and 12A), also display significant protein binding which is most prominently seen at 25 nM (Figures 12B and 12D). There was less protein interactions evident with #2 (Figure 12C) while there was no gel shift with the negative control sequence (Figure 12F). The free probe for 3′-UTR region #2 appeared as doublet which is probably due to alternative conformations of this 175 base length probe. Note that this doublet is also evident in lane with no protein added so it is not explained as a protein-RNA complex. The diffuse banding observed for the binding complexes is likely due to the dissociation of RNA-protein complexes in the gel which unlike DNA gel shifts could occur within minutes even at low temperatures (4°C), as previously observed (33). In order to confirm the specificity of the interactions, cold competition binding assays were done with part of the TNFα 3′-UTR sequence which contains a high affinity binding site for AUF1 (Fig. 12G) (32). A segment of the coding sequence of β-globin served as a negative control (32). The results in Fig. 12G show that TNFα ARE was sufficient to significantly compete with #4 3′-UTR B56α RNA for AUF1 (Figure 12G). Further at 1000-fold excess concentration most of B56α probe exists as a free species (Figure 12G). In contrast, β-globin does not compete with B56α for AUF1 and levels of gel shifted species remained constant with increases on the non-competitor (Figure 12G). Taken together, these results indicate that p37AUF1can form high affinity complexes with specific sequences within the B56α 3′-UTR, further supporting the view that induction of AUF1 expression is a cause of B56α mRNA instability.
Figure 12.
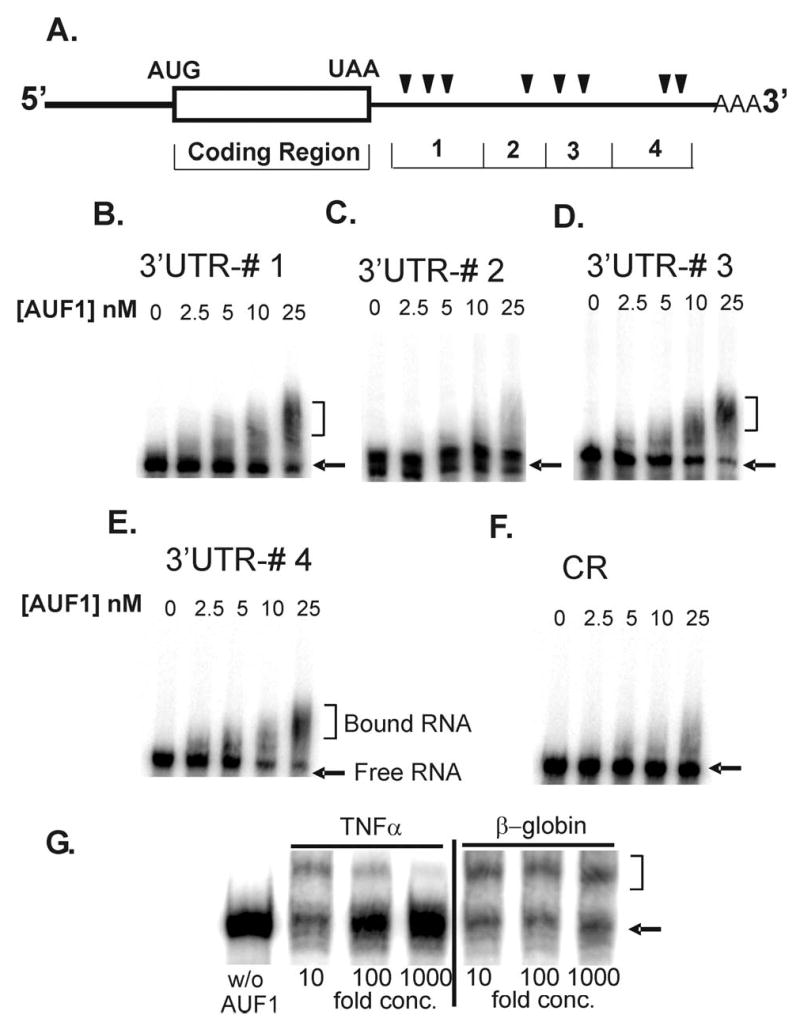
Gel mobility shift assays 3′-UTR regions of B56α mRNA and p37AUF1. A) A schematic diagram shows the regions of the B56α mRNA sequence from which locations of the synthetic RNA fragments were derived. The AU-rich motifs (AREs) are marked with triangles. Sequences of synthetic RNAs were 32P-labeled and used in binding reactions containing varying concentrations of dimeric p37AUF1 as indicated in each panel. Reaction products were then fractionated by non-denaturing polyacrylamide gel electrophoresis as described under the “Methods” section. Shown are autoradiograms of gels corresponding to B) 3′ UTR #1, C) 3′ UTR #2, D) 3′ UTR #3, E) 3′ UTR #4, and F) a 5′ end of the coding region (CR, see Methods). G) Cold competition assays were done with a sequence containing an ARE with high affinity for AUF1, a 38-mer from TNFα 3′-UTR (32), or a 31-mer from coding sequence of β-globin, which served as a negative control. Binding assays were done with 10X higher concentration of 32P-labeled RNA probe, in this case #4 B56αARE, and p37 along with increasing concentrations of either unlabeled TNFα 3′-UTR or βglobin sequence in fold-concentrations above labeled probe as indicated in lanes below gel (see Methods for details). Reaction products were fractionated by non-denaturing polyacrylamide gel electrophoresis as described above. RNA-protein complexes generated with the B56α substrates are indicated by brackets and the positions of free [32P]-RNA probe is identified by arrows.
Discussion
Heart failure is accompanied by a genetic reprogramming that underlies part of the molecular remodeling seen in cardiac myocytes. Despite the crucial importance of these processes in cardiac disease, many of the molecular elements of altered gene expression remain to be illuminated. This study focused on how stress-activated pathways bring about changes in gene expression of an important signaling phosphatase, PP2A. A new finding is that an established stress model, sustained JNK activation, causes a marked loss in a targeting/regulatory subunit of PP2A, B56α, that is accompanied by a decrease in mRNA stability and large increases in expression of the mRNA destabilizing protein, AUF1. In addition, the importance of AUF1 protein expression in cardiac cells is underscored by its upregulation in response to a range of stress-related stimuli and its high affinity binding to the 3′-UTR to B56αmRNA.
An important finding was that JNK activation led to a marked decrease in B56α protein expression in neonatal rat cardiomyocytes. This was a striking response given that the catalytic and scaffolding subunits of PP2A as well as the catalytic subunit of PP1 remained unchanged. Further, this was not an isolated response as the decrease was also seen within the broader context of adult cells as well as in other stress related stimuli, such as sustained p38 activation, phorbol ester treatment, or LPS treatment. These results raise an interesting question of whether this response is maladaptive or possibly compensatory. In this regard, B56α overexpression failed to abrogate in the stimulation of hypertrophic marker genes such as ANF, β-MHC, and α-sk-actin in JNK-activated cells (data not shown). Further studies are warranted to determine if the decrease in B56α is somehow beneficial to cardiac cells in the setting of stress.
Since little is known of the complex mechanisms that regulate cardiac gene expression following stress, an important goal here was to identify pathways that decreased B56α in JNK-activated cells. Recent studies in other cell types suggest that B subunits are quite unstable within cells unless bound to their cognate binding partners, PP2Ac and PP2A/A (27). Thus, protein turnover might be a central mechanism of B56α regulation. However, B56α was quite stable in cardiac cells with increases in decay in JNK-activated cells seen only long after translational arrest. Thus, the modest effects of JNK activation on protein turnover suggest that other mechanisms are operative.
These considerations motivated an analysis of the regulation of B56α transcript. A series of complementary studies showed that the low transcript number, 5 copies per cell, was decreased to only 2 copies in JNK-activated cells. While several mechanisms could explain this loss in steady-state mRNA, a detailed analysis of the B56α transcript revealed important clues, providing a focus for this study. In particular, mouse and human 3′UTR of B56α contain several AU-rich elements (ARE) which encode putative mRNA destabilizing sequences (35). Although these are not classical ARE motifs, studies of destabilization of the β1-adrenergic receptor mRNA in human heart failure reveal similar sequences in that transcript (23). Because of the low level of transcript, quantitative real time PCR was used to test the hypothesis that mRNA instability was important in B56α regulation. Consistent with this view, these studies revealed that JNK activation has a profound effect, transforming a transcript that is stable for at least one hour to a labile form that is substantially degraded within minutes. These results do not exclude a role of the B56α promoter, which has not been characterized at this date, and a decrease the rate of transcription. However, the presence of mRNA destabilizing sequences and the dramatic increase in mRNA turnover, provide compelling evidence that transcript destabilization is an important regulatory mechanism the decrease in B56α expression. .
If the ARE sequences were important in mediating the B56α mRNA instability then the conclusion from many studies predicts that increases in AUF1 protein expression should be seen (2; 11). The family of ARE-binding AUF1 proteins are important in regulating mRNA instability (2; 11) and are expressed as four alternatively spliced isoforms: the cytoplamsic p37AUF1 and p40AUF1 and the nuclear, and p42AUF1 and p45AUF1 (11). An important result here is that not only did JNK activation evoke a fourfold increase in AUF1 expression but the cytoplasmic isoforms were prominently increased, proteins of the AUF1 family linked to mRNA decay (2). Also, AUF1-mediated instability is consistent with the transformation of B56α mRNA to a very labile species as this protein destabilizes IL-10 mRNA to a form with a half-life of only 7 min in melanoma cells (5). Further, the significance of this result was broadened as AUF1 protein expression was markedly increased in cultured cardiac cells by other stress-linked stimuli, including LPS, p38-MAPK activation, phorbol ester, and serum. Although AUF1 is upregulated in human heart failure (23), in cultured cells agents known to stimulate hypertrophic-like responses, such as phenylephrine, cardiotrophin, angiotensin II and endothelin-1failed to decrease B56α expression or augument AUF1 (data not shown). These results underscore the importance extending this work into the context of intact heart models.
The central role of AUF1 in B56α mRNA instability was further examined in focused 3′-UTR RNA binding experiments. Not only did p37AUF1 bind to specific B56α mRNA fragments, but the interactions were of high affinity, in the 10nM range, which is consistent with the functional binding of AUF1 to other transcripts such as c-myc (8). Therefore, the binding of AUF1 to B56α combined with the observation that AUF1 protein is increased in human failing heart (23) reveal that it will be important to identify the regulatory mechanisms that stimulate AUF1 expression in cardiac cells.
The conclusion that AUF1 mediates mRNA stability seen here is strongly supported by work of many groups, where the link between AUF1 protein levels and the degradation rate of AUF1-binding mRNA substrates is observed. For example, in T lymphocytes UV-induced apoptosis is mediated by repressing synthesis of the anti-apoptotic factor Bcl-2, in part by accelerated decay of bcl-2 mRNA concomitant with increased expression of AUF1 (16). Similarly, enhanced expression of AUF1 in non-small-cell lung carcinoma cells contributes to destabilization of cyclin D1 mRNA in response to treatment with the experimental chemotherapeutic agent prostaglandin A2 (19). Conversely, loss of AUF1 expression has been linked to stabilization of several target mRNAs. Interleukin-10 mRNA is stabilized 10-fold in AUF1-deficient melanoma cells compared to normal melanocytes (5), while both RNAi-directed depletion and ectopic overexpression studies have indicated that rapid mRNA decay by the ARE from granulocyte-macrophage colony-stimulating factor mRNA is corrected with AUF1 protein levels (25; 26). The considerable evidence establishing the link between enhanced AUF1 levels and an accelerated decay of target mRNAs, strongly support the view that expression of AUF1 also contributes to the regulatory mechanism of B56α transcript decay in heart.
The balance of mRNA stability/instability is an underappreciated mechanism in the regulation of gene expression in the heart disease (21). The metabolism of several cardiac transcripts have been linked to AUF1 including the β1-adrenergic receptor, and SERCA 2A (4; 22; 23). In fact, in a recent paper the phosphorylated form of AUF1 was found to interact with the SERCA 2A 3′-UTR region (4). Yet many others such phospholamban (Accession # BC005269), the gap junction protein, connexin 43 (Accession # NM000165) and the Na-Ca exchanger (Accession # NM182933) also contain ARE motifs. It is important to note that other ARE-binding proteins such as HuR, an mRNA-stabilizing protein, have been found to compete and bind common AUF1 target transcripts (15). However, further experiments are warranted to determine if HuR has a stabilizing role in the fate of the B56α transcript here or in cardiac cells in general.
In conclusion, this study provides new insight into the mechanisms of protein expression evoked by stress-activated signaling cascades in heart. It is intriguing to speculate that signaling-induced mRNA instability, and perhaps its counterpart, mRNA stability, are associated with are range of cardiovascular pathologies including pressure overload, sepsis and ischemia.
Acknowledgments
This work was supported by grants from the National Institutes of Health (P01 HL-70709 AG-14637: TBR; R01 CA102428: GMW), and NIH Training Grants T32- GM-008181, T32-HL-072751 (YOL).
References
- 1.Avdi NJ, Malcolm KC, Nick JA, Worthen GS. A role for protein phosphatase-2A in p38 mitogen-activated protein kinase-mediated regulation of the c-Jun NH(2)-terminal kinase pathway in human neutrophils. J Biol Chem. 2002;277:40687–40696. doi: 10.1074/jbc.M204455200. [DOI] [PubMed] [Google Scholar]
- 2.Bevilacqua A, Ceriani MC, Capaccioli S, Nicolin A. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J Cell Physiol. 2003;195:356–372. doi: 10.1002/jcp.10272. [DOI] [PubMed] [Google Scholar]
- 3.Bird G, Zorio DA, Bentley DL. RNA polymerase II carboxy-terminal domain phosphorylation is required for cotransriptional pre-mRNA splicing and 3′-end formation. Mol Cell Biol. 2004;20:8963–8969. doi: 10.1128/MCB.24.20.8963-8969.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blum JL, Samarel AM, Mestril R. Phosphorylation and Binding of AUF1 to the 3′ Untranslated Region of Cardiomyocyte SERCA2a mRNA. Am J Physiol Heart Circ Physiol. 2005;289:2543–2550. doi: 10.1152/ajpheart.00545.2005. [DOI] [PubMed] [Google Scholar]
- 5.Brewer G, Saccani S, Sarkar S, Lewis A, Pestka S. Increased interleukin-10 mRNA stability in melanoma cells is associated with decreased levels of A + U-rich element binding factor AUF1. J Interferon Cytokine Res. 2003;23:553–564. doi: 10.1089/107999003322485053. [DOI] [PubMed] [Google Scholar]
- 6.Brewis N, Ohst K, Fields K, Rapacciuolo A, Chou D, Bloor C, Dillmann W, Rockman H, Walter G. Dilated cardiomyopathy in transgenic mice expressing a mutant A subunit of protein phosphatase 2A. Am J Physiol Heart Circ Physiol. 2000;279:H1307–H1318. doi: 10.1152/ajpheart.2000.279.3.H1307. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y, Weeks J, Mortin MA, Greenleaf AL. Mapping Mutations in Genes Encoding the Two Large Subunits of Drosophila RNA Polymerase II Defines Domains Essential for Basic Transcription Functions and for a Proper Expression of Developmental Genes. Mol Cell Biol. 1993;13:4214–4222. doi: 10.1128/mcb.13.7.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeMaria CT, Brewer G. AUF1 binding affinity to A + U-rich elements correlates with rapid mRNA degradation. J Biol Chem. 1996;271:12179–12184. doi: 10.1074/jbc.271.21.12179. [DOI] [PubMed] [Google Scholar]
- 9.Gergs U, Boknik P, Fabritz L, Matus M, Justus I, Hanske G, Schmitz W, Neumann J. Overexpression of the Catalytic Subunit of Protein Phosphatase 2A Impairs Cardiac Function. J Biol Chem. 2004;279:40827–40834. doi: 10.1074/jbc.M405770200. [DOI] [PubMed] [Google Scholar]
- 10.Gigena MS, Ito A, Nojima H, Rogers TB. A B56 Regulatory Subunit of Protein Phosphatase 2A Localizes to Nuclear Speckles in Cardiac Myocytes. Am J Physiol Heart Circ Physiol. 2005;289:285–294. doi: 10.1152/ajpheart.01291.2004. [DOI] [PubMed] [Google Scholar]
- 11.Guhaniyogi J, Brewer G. Regulation of mRNA stability in mammalian cells. Gene. 2001:11–23. doi: 10.1016/s0378-1119(01)00350-x. [DOI] [PubMed] [Google Scholar]
- 12.Hata JA, Williams ML, Koch WJ. Genetic manipulation of myocardial β-adrenergic receptor activation and desensitization. J Mol Cell Cardiol. 2004;37:11–21. doi: 10.1016/j.yjmcc.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 13.Ito A, Morii E, Maeyama K, Jippo T, Kim DK, Lee YM, Ogihara H, Hashimoto K, Kitamura Y, Nojima H. Systematic method to obtain novel genes that are regulated by mi transcription factor: impaired expression of granzyme B and tryptophan hydroxylase in mi/mi cultured mast cells. Blood. 1998;91:3210–3221. [PubMed] [Google Scholar]
- 14.Kohout TA, O′Brian JJ, Gaa ST, Lederer WJ, Rogers TB. Novel adenovirus component system that transfects cultured cardiac cells with high efficiency. Circ Res. 1996;78:971–977. doi: 10.1161/01.res.78.6.971. [DOI] [PubMed] [Google Scholar]
- 15.Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004;23:3092–3102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lapucci A, Donnini M, Papucci L, Witort E, Tempestini A, Bevilacqua A, Niolin A, Brewer G. AUF1 is a bcl-2 A+U-rich element-binding protein involved in bcl-2 mRNA destabilization during apoptosis. J Biol Chem. 2002;277:16139–16146. doi: 10.1074/jbc.M201377200. [DOI] [PubMed] [Google Scholar]
- 17.Lee T, Kim SJ, Sumpio BE. Role of PP2A in the regulation of p38 MAPK activation in bovine aortic endothelial cells exposed to cyclic strain. J Cell Physiol. 2003;194:349–355. doi: 10.1002/jcp.10211. [DOI] [PubMed] [Google Scholar]
- 18.Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy; dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol. 2003;35:1385–1394. doi: 10.1016/j.yjmcc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 19.Lin S, Wang W, Wilson GM, Yang X, Brewer G, Holbrook NJ, Gorospe M. Down-regulation of cyclin D1 expression by prostaglandin A2 is mediated by enhanced cyclin D1 mRNA turnover. Mol Cell Biol. 2000;20:7903–7913. doi: 10.1128/mcb.20.21.7903-7913.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCright B, Rivers AM, Audlin S, Virshup DM. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem. 1996;271:22081–22089. doi: 10.1074/jbc.271.36.22081. [DOI] [PubMed] [Google Scholar]
- 21.Misquitta CM, Iyer VR, Werstiuk ES, Grover AK. The role of 3′-untranslated region (3′-UTR) mediated mRNA stability in cardiovascular pathophysiology. Mol and Cell Biochem. 2001;224:53–67. doi: 10.1023/a:1011982932645. [DOI] [PubMed] [Google Scholar]
- 22.Misquitta CM, Mwanjewe J, Nie L, Grover AK. Sarcoplasmic reticulum Ca2+ pump mRNA stability in cardiac and smooth muscle: role of the 3′-untranslated region. Am J Physiol Cell Physiol. 2002;283:560–568. doi: 10.1152/ajpcell.00527.2001. [DOI] [PubMed] [Google Scholar]
- 23.Pende A, Tremmel KD, DeMaria CT, Blaxall BC, Minobe WA, Sherman JA, Bisognano JD, Bristow MR, Brewer G, Port J. Regulation of the mRNA-binding protein AUF1 by activation of the beta-adrenergic receptor signal transduction pathway. J Biol Chem. 1996;271:8493–8501. doi: 10.1074/jbc.271.14.8493. [DOI] [PubMed] [Google Scholar]
- 24.Petrich BG, Molkentin JD, Wang Y. Temporal activation of c-Jun N-terminal kinase in adult transgenic heart via cre-loxP-mediated DNA recombination. FASEB J. 2003;17:749–751. doi: 10.1096/fj.02-0438fje. [DOI] [PubMed] [Google Scholar]
- 25.Raineri I, Wegmueller D, Gross B, Certa U, Moroni C. Roles of AUF1 isoforms, HuR and BRF1 in ARE-dependent mRNA turnover studied by RNA interference. Nucleic Acids Res. 2004;32:1279–1288. doi: 10.1093/nar/gkh282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarkar B, Xi Q, He C, Schneider RJ. Selective degradation mRNA turnover studied by RNA interference. Mol Cell Biol. 2003;23:6685–6693. doi: 10.1128/MCB.23.18.6685-6693.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silverstein AM, Barrow CA, Davis AJ, Mumby MC. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc Natl Acad Sci U S A. 2002;99:4221–4226. doi: 10.1073/pnas.072071699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strack S. Overexpression of the protein phosphatase 2A regulatory subunit Bγ promotes neuronal differentiation by activating the MAP kinase (MAPK) cascade. J Biol Chem. 2002;277:41525–41532. doi: 10.1074/jbc.M203767200. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Krushel LA, Edelman GM. Targeted DNA recombination in vivo using an adenovirus carrying the cre recombinase gene. Proc Natl Acad Sci U S A. 1996;93:3932–3936. doi: 10.1073/pnas.93.9.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Su B, Sah VP, Brown JH, Han J, Chien KR. Cardiac hypertrophy induced by mitogen-activated protein kinase kinase 7, a specific activator for c-Jun NH2-terminal kinase in ventricular muscle cells. J Biol Chem. 1998;273:5423–5426. doi: 10.1074/jbc.273.10.5423. [DOI] [PubMed] [Google Scholar]
- 31.Wilson GM, Brewer G. Identification and Characterization of Proteins Binding A + U-Rich Elements. Methods Compan Methods Enzymol. 1999;17:74–83. doi: 10.1006/meth.1998.0709. [DOI] [PubMed] [Google Scholar]
- 32.Wilson GM, Lu J, Sutphen K, Sun Y, Huynh Y, Brewer G. Regulation of A + U-rich element-directed mRNA turnover involving reversible phosphorylation of AUF1. J Biol Chem. 2003;278:33029–33038. doi: 10.1074/jbc.M305772200. [DOI] [PubMed] [Google Scholar]
- 33.Wilson GM, Sun Y, Lu H, Brewer G. Assembly of AUF1 Oligomers on U-rich Targets by Sequential Dimer Association. J Biol Chem. 1999;274:33374–33381. doi: 10.1074/jbc.274.47.33374. [DOI] [PubMed] [Google Scholar]
- 34.Wilson GM, Sutphen K, Chuang K, Brewer G. Folding of A + U-rich RNA Elements Modulates AUF1 Binding: Potential Roles in Regulation of mRNA Turnover. J Biol Chem. 2001;276:8695–8704. doi: 10.1074/jbc.M009848200. [DOI] [PubMed] [Google Scholar]
- 35.Zubiaga A, Belasco J, Greenberg M. The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Biol Cell. 1995;15:2219–2230. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]