

Abstract
It has been recently shown that recombinant adeno-associated virus serotype 8 (rAAV8) is a robust alternative serotype vector that overcomes many of the limitations of rAAV2 and transduces various tissues efficiently and globally through systemic vector administration. AAV9 is a serotype newly isolated from human tissues, but our knowledge of the biology of rAAV9 in vivo is currently limited. Here, we demonstrate by a series of comprehensive side-by-side experiments with rAAV8 and 9 vectors delivered via different routes or at various doses in mice that rAAV9 vectors share the robustness of rAAV8, i.e., (1) very high liver transduction efficiency irrespective of whether vectors are administered intravascularly or extravascularly and (2) substantial transduction in the heart, skeletal muscle, and pancreas by peripheral vein injection. Importantly, rAAV9 transduced myocardium 5- to 10-fold higher than rAAV8, resulting in over 80% cardiomyocyte transduction following tail vein injection of as low as 1.0 × 1011 particles per mouse. Thus rAAV9, as well as rAAV8, is a robust vector for gene therapy applications and rAAV9 is superior to rAAV8 specifically for cardiac gene delivery by systemic vector administration.
Keywords: adeno-associated virus, serotype, gene therapy, heart, myocardium, skeletal muscle, pancreas, liver, systemic delivery, subcutaneous injection
Introduction
Adeno-associated virus (AAV) is a small, nonpathogenic, replication-defective parvovirus with a single-stranded DNA genome. Among various serotypes, AAV serotype 2 has been most extensively investigated for the biology of AAV and explored as a gene delivery vector. Although recombinant AAV serotype 2 (rAAV2) is a promising vector, in vivo gene transfer with rAAV2 is suboptimal in many instances. To overcome such drawbacks of rAAV2 vectors, many investigators have begun to explore either naturally occurring [1,2] or artificially modified [3] novel AAV capsids, which are the major determinant of tropism and transduction efficiency. To date over a hundred naturally occurring primate AAVs have been reported [1,2,4–8]. Many in vivo studies have demonstrated that rAAV vectors packaged in alternative AAV serotype capsids exhibit serotype-specific tissue- or cell-type tropism and/ or increased transduction efficiency [1,2,9].
Recently, systemic administration of rAAV vectors derived from alternative serotypes has received much attention as a powerful strategy to deliver genes globally to a target tissue(s). Gregorevic et al. first demonstrated its proof-of-principle with rAAV6 vectors [10]. Subsequently, we demonstrated for the first time that intravenous injection of single-stranded rAAV8 vectors via the periphery can transduce not only all the hepatocytes but also all the skeletal muscles throughout the body and the entire myocardium in mice [11]. This observation was further confirmed in a study by Wang et al., in which they delivered more potent double-stranded (ds) rAAV8 vectors in adult and neonatal mice [12]. In addition to skeletal and cardiac muscles, rAAV8 can transduce pancreatic acinar cells and islet cells, smooth muscle cells in various tissues, and neurons and glial cells in the brain, with considerable efficiency, following intravenous injection of the vector [11]. Thus, a number of applications of systemic intravenous administration of rAAV8 vectors are being exploited to treat animal models of various human diseases that were previously thought to be difficult to treat by gene therapy [12,13].
We have been investigating whether any other serotype(s) achieves robust systemic transduction in various tissues in mice by intravenous vector administration via periphery and outperforms well-characterized rAAV vectors. Based on a study by Gao et al., we selected rAAV9 as the most likely candidate for a comparative study with rAAV8 [1]. AAV9 was recently isolated from human tissues and shown to have serological characteristics distinct from previously described serotypes [1]. Although it has been demonstrated that rAAV9, when administered locally, transduces murine lung and skeletal muscle better than rAAV8 [1], our knowledge about the in vivo biology of rAAV9 is currently very limited [1,14,15], and it remains to be elucidated whether rAAV9 can also systemically transduce various mouse tissues, as does rAAV8.
In the present study, we performed comprehensive side-by-side experiments with rAAV8 and 9 vectors delivered via different routes or at various doses in mice, to characterize the in vivo biology of rAAV9 vectors. The study revealed that rAAV9 vectors, as well as rAAV8, can cross vascular endothelial cell barriers very efficiently, resulting in substantial transduction in the liver irrespective of route of vector administration and efficient transduction in many nonhepatic tissues following systemic vector administration. Although rAAV9’s biological features are quite similar to those of rAAV8 vectors, detailed histological and molecular analyses uncovered differences in the vector biology between the two serotypes.
Results and Discussion
rAAV8 and 9 Vectors Efficiently Transduced Mouse Liver at Comparable Levels Irrespective of Whether Vectors Were Administered Intravascularly or Extravascularly
The route of rAAV vector administration may substantially influence liver transduction efficiency in vivo. For example, tail vein (TV) injection of rAAV1, 2, 3, 5, and 6 vectors transduces the liver 5- to 48-fold less efficiently than portal vein (PV) injection in mice [16–18]. However, interestingly, TV injection of rAAV8 vectors transduces murine liver as efficiently as PV injection [11,19], the mechanism for which has yet to be elucidated. To begin to extend our vector administration route comparison study, we first investigated how efficiently rAAV8 vectors injected into intraperitoneal (IP) space (i.e., extravascular space) can pass through various cellular barriers, entering the blood circulation to transduce the liver in adult mice. In prenatal and neonatal mice, IP injection has proven to be an efficient route for systemic gene delivery with various rAAV serotypes, presumably due to the immaturity of cellular barriers [12,20,21]. However, in adult animals, IP injection with conventional rAAV serotypes resulted in local peritoneal and diaphragm muscle transduction in most cases. One study has indicated that IP injection with rAAV8 vector in adult mice could transduce skeletal muscles systemically; however, the details of the results were not addressed in the study [12]. In the present study, to compare liver transduction efficiency with rAAV8 between TV and IP injection in adult mice, we produced AAV8-hF.IX16 vector (a liver-specific promoter-driven human factor IX (hF.IX)-expressing rAAV pseudo-serotyped with AAV8 capsid) and injected adult C57BL/6 female mice with 1.0 × 1011 vector genomes (vg) of this vector via the TV or IP route (n = 7–8 per group). As a result, plasma hF.IX levels peaked at 50.7 ± 14.8 μg/ml (TV group) and 40.0 ± 5.0 μg/ml (IP group) 1 week postinjection, followed by an ~50% decline over 16 weeks (21.2 ± 7.1 and 22.4 ± 6.7 μg/ml in TV and IP groups, respectively, 16 weeks postinjection). There was no significant difference in the plasma hF.IX levels between TV and IP groups at any time points analyzed (Student’s t test, P = 0.20–0.74). This demonstrates that the rAAV8 vector injected into IP space could be efficiently absorbed into blood circulation in adult animals, resulting in efficient liver transduction at a level comparable to that of TV injection.
Next, we compared liver transduction efficiency between rAAV8 and 9 vectors injected via various administration routes, i.e., two intravascular routes (TV and PV injections) and two extravascular routes (IP and subcutaneous (SC) injections). A recent study by Gao et al. has demonstrated that rAAV9 vector can transduce murine liver at a level comparable to that of rAAV8 when delivered via the PV route [1]. However, it is not known whether rAAV9 injection via the tail vein or other routes of administration transduces the liver as efficiently as PV or whether rAAV9 vector injected into extravascular spaces can enter the blood circulation as efficiently as rAAV8. To elucidate these points, we injected adult C57BL/6 male or female mice with 8.0 × 1010 vg of AAV8- or 9-hF.IX16 via PV, TV, IP, or SC route in eight different combinations, i.e., 9-PV-M, 9-TV-M, 9-IP-M, 9-SC-M, 9-PV-F, 8-PV-M, 8-SC-M, and 8-PV-F (note that X-Y-Z indicates X, serotype; Y, route; and Z, sex). We assessed liver transduction efficiency by monitoring plasma hF.IX levels for a period of 12 weeks (Fig. 1). The results are summarized as follows: (1) All eight different serotype–route–sex combinations resulted in hF.IX expression at high levels (>20 μg/ml in plasma). (2) rAAV9 transduced liver at levels comparable to those with rAAV8. However, rAAV9 appeared to transduce liver slightly better than rAAV8 because statistical significance (Student’s t test, P = 0.006–0.04) was observed at two to four of the eight time points in each of the comparisons between 8-PV-M and 9-PV-M, between 8-PV-F and 9-PV-F, and between 8-SC-M and 9-SC-M. (3) By PV, TV, and IP injection, hF.IX levels peaked 2–6 weeks postinjection, followed by a 25–54% decline, while this decline was not observed with SC injection. This is the case with both rAAV8 and 9 vectors. (4) PV injection of rAAV9 resulted in higher liver transduction than TV or IP injections at several earlier time points up to 4 weeks postinjection (Student’s t test, P = 0.003–0.01), although hF.IX eventually stabilized at comparable levels between the three routes by 6 weeks post-injection (one-way ANOVA, P = 0.32–0.51). (5) SC administration with rAAV8 or 9 resulted in lower liver transduction compared to PV, TV, or IP with a corresponding serotype vector at many time points (Student’s t test, P = 0.00007–0.04), but still achieved stable liver transduction at 30–50% of the levels obtained with PV, TV, or IP. (6) Liver transduction in male mice seemed to be slightly higher than in female mice. Statistical significance (Student’s t test, P = 0.0002–0.04) was observed in half of the eight time points in the comparisons between 8-PV-M and 8-PV-F and between 9-PV-M and 9-PV-F. These results demonstrate that both rAAV8 and 9 vectors efficiently transduce mouse liver at comparable levels irrespective of the route of vector administration or animal gender; however, transgene expression kinetics is influenced by how vectors are administered.
FIG. 1.
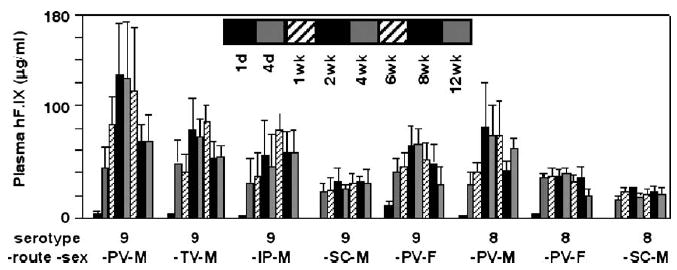
Plasma hF.IX levels in mice following injection of hF.IX-expressing rAAV8 or 9 vector via four different routes. Adult male (M) or female (F) C57BL/6 mice were injected with 8.0 × 1010 vg/ mouse of AAV8- or 9-hF.IX16 via portal vein (PV), tail vein (TV), intraperitoneal (IP), or subcutaneous (SC) injection. Combinations of serotype–vector administration route–animal sex are indicated. Plasma hF.IX levels were determined 1 day, 4 days, and 1, 2, 4, 6, 8, and 12 weeks postinjection and are shown as a bar graph with means ± standard deviations (n = 3–5 each).
It is not surprising that SC injection was less efficient than PV, TV, or IP injection in liver transduction considering the bioavailability of vectors via each administration route. Vector bioavailability via the PV and TV injections is 100%, while it is normally less than 100% when vectors are administered extravascularly. In particular, dorsal SC space has less vascularity compared to IP space, resulting in a further reduced vector bioavailability. However, interestingly, we found that vector genome-specific activity in the liver was substantially increased with SC injection compared to PV injection, compensating for the decrease in hF.IX expression due to reduced vector bioavailability via SC injection. hF.IX levels at 12 weeks postinjection in the male mice injected with AAV8-hF.IX16 at 8.0 × 1010 vg/mouse via PV and SC were 62.7 ± 8.9 and 20.7 ± 5.6 μg/ml (i.e., 3.0-fold difference), while ds vector genome copy numbers (i.e., double-stranded vector genomes per diploid genomic equivalent (ds-vg/dge)) in the livers determined by Southern blot analysis were 16.8 ± 4.1 and 1.6 ± 0.5 ds-vg/dge (i.e., 10-fold difference), respectively. In the mice injected with AAV9-hF.IX16 at the same dose, the differences in hF.IX levels and vector genome copy numbers were 2.2-fold (68.1 ± 23.3 vs. 30.6 ± 12.1 μg/ml) and 2.9-fold (29.9 ± 13.0 vs. 10.4 ± 5.8 ds-vg/dge), respectively. This observation clearly demonstrates that SC injection of AAV8-hF.IX at this dose generated vector genomes with higher specific activity than the other vector-administration route combinations. Southern blot analysis of vector genome forms in the liver samples revealed that the increased vector genome-specific activity was associated with a reduced amount of concatemeric rAAV vector genomes (Fig. 2). This observation is consistent with our proposal that concatemerization of rAAV vector genomes is in part responsible for decreasing the vector genome-specific activity [11,22]. Although we did not observe this effect with rAAV9 vector at 8.0 × 1010 vg/mouse, we might see the same effect by SC injection with rAAV9 at a lower vector dose range. Although the underlying mechanism for this effect is currently unknown, a SC injection-mediated longer hepatic exposure to a low dose rAAV vector may be responsible for the increased vector genome-specific activity. It is also intriguing that SC injection resulted in more stabilized transgene expression than the other routes.
FIG. 2.
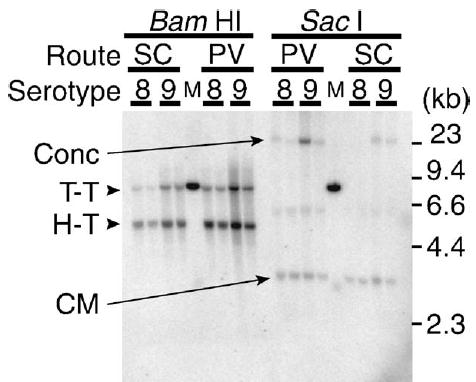
rAAV8 and 9 vector genome forms in the liver transduced via the SC or PV route. Total liver DNA extracted from AAV8- or 9-hF.IX16-transduced liver samples was subjected to Southern blot analysis with a noncutter restriction enzyme that does not cut the ds vector genomes (SacI) or a single-cutter restriction enzyme that cuts the ds vector genomes asymmetrically near the 5′ end (BamHI). Concatemers were not detected in the samples transduced with rAAV8 via the SC route. Conc, ds concatemers; CM, supercoiled ds circular monomer genomes; H-T, head-to-tail molecules; T-T, tail-to-tail molecules; M, 30 ds-vg/dge standard.
Intravascular Injection of rAAV8 and 9 Vectors Resulted in Global Transduction in Various Nonhepatic Tissues with Quite Similar Tropism
To assess transduction efficiency in various tissues following systemic administration of rAAV8 or 9 vector in mice, we injected adult C57BL/6 male mice with rAAV8 or 9 vector expressing β-galactosidase at various doses. The vectors carried either a human cytomegalovirus (CMV) immediate early gene promoter-driven cytosolic lacZ gene (CMV-lacZ) or a human elongation factor 1α (EF1α) promoter-driven nlslacZ gene (EF1α-nlslacZ). We determined transduction efficiency in various tissues 10–13 days postinjection. These time points were chosen because 10–13 days should be sufficient to reach nearly maximum transduction levels (Fig. 1) and because immune response against β-galactosidase, which may reduce transduction efficiency, should be least in the first 2 weeks following vector injection.
In the first set of side-by-side experiments, we investigated transduction efficiency in various tissues following tail vein injection of either AAV9-EF1α-nlslacZ or AAV9-CMV-lacZ at two different vector doses, i.e., 3.0 × 1011 and 1.8 × 1012 vg/mouse (n = 2 each per group). We harvested 10 major tissues (i.e., liver, brain, lung, heart, spleen, kidney, intestine, testis, pancreas, and skeletal muscle) 13 days postinjection and assessed transduction efficiency by bromo-4-chloro-3-indolyl-β-D-galactopyra-noside (X-Gal) staining. The results demonstrated a tissue distribution pattern similar to that of AAV8-EF1α-nlslacZ or AAV8-CMV-lacZ vector that we have previously published [11]. With AAV9-EF1α-nlslacZ vector at a dose of 3.0 × 1011 vg/mouse, the liver was the best transduced organ, with very few X-Gal-positive cells observed outside the liver. At 1.8 × 1012 vg/mouse, we observed a significant number of positive cells in lung, heart, brain, and smooth muscles in various tissues (data not shown). With the AAV9-CMV-lacZ vector, the heart was the best transduced organ, with nearly the entire myocardium transduced even at the lower dose. Skeletal muscle and pancreas were also very well transduced, as we have seen with the AAV8-CMV-lacZ vector [11]. Table 1 summarizes liver transduction efficiency determined by X-Gal staining in the first set of experiments.
Table 1.
Transduction efficiency in mouse livers transduced with rAAV9 via the tail vein
| Transduction efficiency (%)a | ||
|---|---|---|
| Dose (vg/mouse) | AAV9-EF1α-nlslacZ | AAV9-CMV-lacZ |
| 3.0 × 1011 | 9.4 ± 0.9b | 4.1 ± 0.8 |
| 1.8 × 1012 | 74.0 ± 11.8 | 95.9 ± 4.1 |
Transduction efficiency was determined by X-Gal staining (n = 2 each).
Mean ± |each value-mean value|.
Because the CMV-lacZ expression cassette was more sensitive than the EF1α-nlslacZ in detecting transduced cells in nonhepatic tissues, we used AAV8- and 9-CMV-lacZ vectors for the subsequent comprehensive side-by-side dose–response and tissue distribution study. In this second set of the experiments, we injected adult C57BL/6 male mice with either AAV8- or AAV9-CMV-lacZ vector at six different doses (1.0 × 1010, 3.0 × 1010, 1.0 × 1011, 3.0 × 1011, 1.0 × 1012, and 1.8 × 1012 vg/mouse) via the tail vein (n = 4 or 5 per group). We assessed transduction efficiency by X-Gal staining and determined ds vector genome copy numbers by Southern blot analysis. The results are shown in Fig. 3 and Tables 2A, 2B. We again confirmed that rAAV9 transduces the liver, skeletal muscle, heart, and pancreas with extremely high efficiency in the context of an AAV-CMV-lacZ vector. In the pancreas, ~30% of islet cells were transduced at the highest vector dose. This tissue tropism is quite similar to that of rAAV8.
FIG. 3.
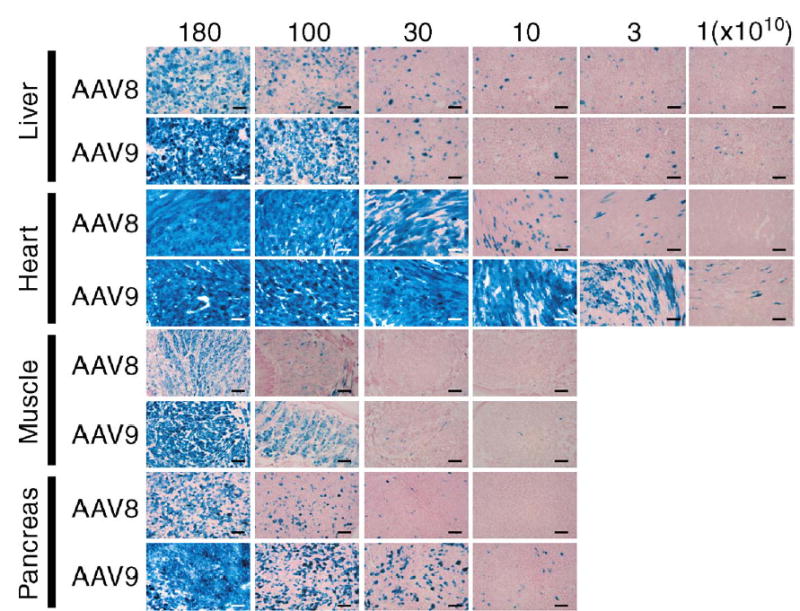
Representative photomicrographs of sections of the liver, heart, skeletal muscle, and pancreas 10 days after tail vein injection of AAV8- or 9-CMV-lacZ at various doses ranging from 1.0 × 1010 to 1.8 × 1012 vg/mouse. Each section was stained with X-Gal and nuclear fast red. Scale bars represent 100 μm.
Table 2A.
Transduction efficiency in various tissues following TV injection of various doses of AAV8- or 9-CMV-lacZ
| Transduction efficiency (%) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose (× 1010): | 180 | 100 | 30 | 10 | 3 | 1 | ||||||
| Serotype: | 8 | 9 | 8 | 9 | 8 | 9 | 8 | 9 | 8 | 9 | 8 | 9 |
| Liver | 72.3 ± 19.3 | 95.8± 3.0 | 51.8 ± 29.0 | 83.3± 8.2 | 8.3 ± 3.2 | 9.4 ± 4.8 | 7.3± 2.8 | 5.3± 2.9 | 3.5 ± 0.9 | 3.2 ± 1.3 | 2.7 ± 0.5 | 2.7 ± 0.4 |
| Muscle | 89.5 ± 6.5 | 94.0± 5.5 | 52.0 ± 29.7 | 82.3± 4.0 | 2.1 ± 3.7 | 19.3 ± 10.8 | 0.0 | 3.2± 2.7 | 0.0 | 0.0 | 0.0 | 0.0 |
| Heart | 100.0 ± 0.0 | 100.0± 0.0 | 100.0 ± 0.0 | 99.5± 3.6 | 87.2 ± 7.5 | 91.1 ± 8.2 | 14.4± 9.4 | 81.8± 15.7 | 3.3 ± 2.1 | 31.7 ± 20.2 | 0.1 ± 0.0 | 3.6 ± 2.1 |
| Pancreasa | 61.9 ± 9.8 | 91.4± 9.5 | 38.3 ± 21.8 | 60.9± 6.2 | 3.7 ± 2.3 | 18.5 ± 4.1 | 0.0 | 2.4± 0.6 | 0.0 | 0.1 ± 0.1 | 0.0 | 0.0 |
| Nb | 4 | 4 | 4 | 4 | 5 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
Only acinar cells are considered.
Number of animals analyzed.
Table 2B.
Vector genome copy numbers in various tissues following TV injection of various doses of AAV8- or 9-CMV-lacZ
| Vector copy number (ds-vg/dgea) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dose (× 1010): | 180 | 100 | 30 | 10 | 3 | 1 | ||||||
| Serotype: | 8 | 9 | 8 | 9 | 8 | 9 | 8 | 9 | 8 | 9 | 8 | 9 |
| Liver | 205.9 ± 54.4 | 296.7 ± 37.7 | 186.2 ± 30.1 | 238.2 ± 20.5 | 43.2 ± 16.3 | 76.2 ± 8.4 | 18.8 ± 3.6 | 19.5 ± 4.7 | 6.9 ± 0.9 | 6.1 ± 1.7 | 0.9 ± 0.3 | 0.3 ± 0.1 |
| Muscle | 9.9 ± 0.9 | 13.9 ± 4.3 | 5.7 ± 1.1 | 5.8 ± 0.7 | 1.7 ± 0.1 | 1.6 ± 0.5 | n.a.b | n.a. | n.a. | n.a. | n.a. | n.a. |
| Heart | 10.1 ± 5.5 | 18.4 ± 10.8 | 7.1 ± 1.6 | 9.8 ± 3.6 | 1.4 ± 0.6 | 2.7 ± 0.1 | 0.1 ± 0.1 | 0.8 ± 0.9 | 0.0 | 0.2 ± 0.1 | 0.0 | 0.0 |
| Pancreas | 0.9 ± 0.4 | 1.6 ± 0.3 | 0.4 ± 0.1 | 0.7 ± 0.3 | 0.1 ± 0.1 | 0.2 ± 0.0 | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. |
| N | 4 | 4 | 4 | 4 | 5 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
Double-stranded (ds) vector genomes (vg) per diploid genomic equivalent (dge).
Not analyzed.
In the 6 tissues brain, lung, spleen, kidney, intestine, and testis, we determined ds rAAV vector genome copy numbers by Southern blot analysis. In these tissues, the numbers of β-galactosidase-expressing cells were much less than those in the other 4 tissues (i.e., liver, skeletal muscle, heart, and pancreas) even at the highest vector dose, indicating poor transduction with AAV-CMV-lacZ vector. The ds vector copy numbers in these 6 tissues in the mice injected with AAV8-CMV-lacZ or AAV9-CMV-lacZ at a dose of 3.0 × 1011 or 1.8 × 1012 vg/mouse are summarized in Table 3. At a dose of 1.8× 1012 vg/mouse, we detected ds rAAV vector genomes in all 6 of these tissues at levels relatively high for nonhepatic tissues, i.e., ≥1.1 ds-vg/dge with rAAV8 and ≥2.2 ds-vg/dge with rAAV9. At a dose of 3.0 × 1011 vg/mouse, ds vector genomes were less than 1.0 ds-vg/dge but still detectable at levels 0.2 ds-vg/dge or greater in all 6 tissues. This demonstrates that rAAV9, as well as rAAV8, systemically infected all 10 tissues analyzed with considerable efficiency by tail vein injection. Interestingly, Southern blot analysis revealed that, despite poor transduction, a majority of the ds vector genomes in these 6 nonhepatic tissues are ds circular monomers (data not shown), which is the form primarily responsible for transgene expression among various forms of rAAV vector genomes [22–24].
Table 3.
Double-stranded vector genome copy numbers in nonhepatic tissuesa
| Dose (× 1010) | Serotype | Brain | Lung | Spleen | Kidney | Intestine | Testis |
|---|---|---|---|---|---|---|---|
| 180 | 8 | 1.1 ± 0.2b | 1.6 ± 0.2 | 1.4 ± 0.1 | 2.5 ± 0.4 | 1.5c | 1.4 ± 0.1 |
| 180 | 9 | 2.9 ± 0.3 | 4.6 ± 0.1 | 2.2 ± 0.1 | 3.1 ± 0.5 | 2.8 ± 0.2 | 3.2 ± 0.6 |
| 30 | 8 | 0.2 ± 0.0 | 0.3 ± 0.0 | 0.2 ± 0.0 | 0.4 ± 0.1 | 0.2 ± 0.0 | 0.2 ± 0.0 |
| 30 | 9 | 0.4 ± 0.1 | 0.7 ± 0.1 | 0.5 ± 0.0 | 0.6 ± 0.1 | 0.4 ± 0.0 | 0.5 ± 0.0 |
Double-stranded vector genome copy numbers were determined by Southern blot analysis (n = 2 each) and are expressed as double-stranded vector genomes per diploid genomic equivalent. Vector copy numbers in nonhepatic tissues not listed are summarized in Table 2B.
Mean ± |each value-mean value|.
Only one sample was available.
To investigate whether there is any difference in tissue tropism between rAAV8 and 9 vectors, we determined the ratios of ds vector genome copy numbers between the two vectors in each tissue. For this purpose, we employed two ratios, i.e., raw rAAV9/8 ratios and normalized rAAV9/8 ratios. The raw rAAV9/8 ratio is defined as a factor by which rAAV9 infects a tissue better than rAAV8. This ratio is calculated by an equation in each tissue; i.e., raw ratio in tissue A = (average ds rAAV9 vector copy number in tissue A)/ (average ds rAAV8 vector copy number in tissue A). To avoid bias caused by a lot-to-lot difference in vector preparations, we also used normalized rAAV9/8 ratios. The normalized ratio is defined as the ratio normalized with liver transduction efficiency and calculated by an equation in each tissue; i.e., normalized ratio in tissue B = (average ds rAAV9 vector copy number in tissue B/average ds rAAV9 vector copy number in the liver)/(average ds rAAV8 vector copy number in tissue B/average ds rAAV8 vector copy number in the liver). The rationale of the normalization by liver transduction efficiency is that both rAAV8 and rAAV9 vectors transduced liver at comparable levels.
Fig. 4 summarizes raw and normalized rAAV9/8 ratios in all 10 major tissue types analyzed. In general, rAAV8 and 9 vectors exhibited similar tropism. None of the tissues exhibited significantly decreased infection or transduction with rAAV9 compared to that with rAAV8. However, the lung and brain showed higher rAAV9/8 ratios than the other tissues. The raw and normalized rAAV9/8 ratios were 2.4–2.8 and 1.3–2.0 in the lung and 2.2–2.7 and 1.2–1.9 in the brain, respectively. These observations indicate that rAAV9 infects lung and brain better than rAAV8 by up to two- to threefold when injected via systemic circulation, although vector infection does not necessarily result in vector transduction as observed in the present study.
FIG. 4.
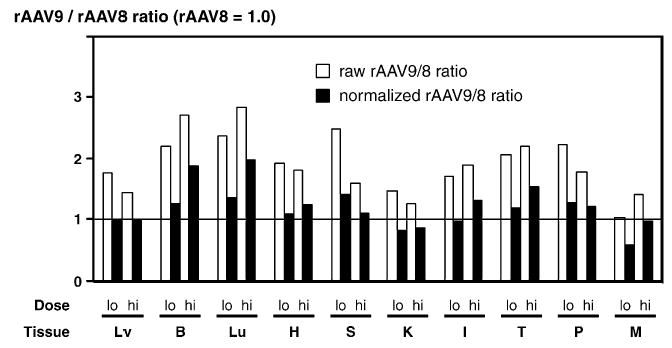
Comparison of tissue tropism between rAAV8 and rAAV9. Double-stranded vector genome copy numbers per diploid genomic equivalent (ds-vg/dge) in each tissue were used to assess the vectors’ tissue tropism. Adult male mice were injected with either AAV8-CMV-lacZ or AAV9-CMV-lacZ at a dose of 3.0 × 1011 (‘‘lo’’ or low) or 1.8 × 1012 (‘‘hi’’ or high) vg/mouse via the tail vein, and ds vector genome copy numbers in 10 major tissues (i.e., Lv, liver; B, brain; Lu, lung; H, heart; S, spleen; K, kidney; I, intestine; T, testis; P, pancreas; M, skeletal muscle) were determined 10 days postinjection by Southern blot analysis. Each bar represents a ratio of ds rAAV9 genomes to ds rAAV8 genomes (rAAV9/8 ratio) in each tissue at each vector dose (lo and hi). The ratio serves as a factor by which rAAV9 vector infects a tissue better than rAAV8. The raw ratio was determined only by rAAV8 and 9 vector copy numbers in a tissue, while the normalized ratio represents the ratio in a tissue corrected for liver transduction efficiency with each serotype vector.
All of these data have demonstrated that rAAV8 and 9 vectors share robustness and quite similar tropism in general even though they are serologically and phylogenetically distinct serotypes with 86% amino acid sequence homology in the VP1 capsid protein [1]. The mechanism(s) by which rAAV8 and 9 vectors transduce various tissues with high efficiency following intravascular injection, and transduce remote organs with high efficiency following extravascular injection, has yet to be elucidated. Wang et al. have emphasized that increased vascular permeability of rAAV8 compared to other serotypes such as rAAV1 and 2 is primarily responsible for rAAV8’s widespread skeletal muscle transduction following IP or IV injection [12]. Efficient crossing of capillary endothelial cell barriers is a prerequisite to transducing various tissues by intravascular injection and remote organs by extravascular injection; however, an alternative explanation is also possible, that not increased vascular permeability but increased stability of rAAV vectors in the bloodstream confers the ability to transduce various tissues with high efficiency. Recently, De et al. have reported that, following intrapleural administration, AAVrh.10 vector, another robust nonhuman primate rAAV serotype, efficiently transduces not only diaphragm, heart, and chest wall, but also several remote organs via the bloodstream, including the liver and skeletal muscle [15]. It is intriguing that rAAV8, 9, and rh.10, all of which exhibit robustness or very high in vivo transduction efficiency, share common properties in that they exhibit similar tropism and have the capability to transduce remote organs with high efficiency when administered extravascularly. It is presumed that they may share a common receptor(s) or coreceptor(s) and/or be more stable in the bloodstream than conventional rAAV serotypes, generating the common biological features, although the underlying mechanism(s) of these shared properties in the robust rAAV serotypes remains to be elucidated.
As we have demonstrated here, rAAV vector infection does not necessarily result in transduction even if substantial rAAV vector genomes are processed into ds circular monomers. In the present study, we found that many nonhepatic tissues other than the heart, pancreas, and skeletal muscle did not express a sufficient level of transgene products despite the presence of a substantial amount of ds circular monomer genomes. A reasonable explanation for this inconsistency is that the promoters we used were not active in these tissues. However, a rAAV2 vector carrying exactly the same AAV2-CMV-lacZ vector genome sequence as was used for producing our pseudotyped AAV8- or 9-CMV-lacZ vectors, when injected into murine kidney by intraparenchymal injection, expressed β-galactosidase in renal tubular cells around the injection site [25]. This suggests that the vector entry pathway into cells could determine vector genome activity. Although the mechanism underlying the impaired transgene expression from ds circular rAAV genomes has yet to be elucidated, a possibility exists that changing the promoter from that of CMV or EF1α to another ubiquitous or tissue-specific promoter might solve this issue.
rAAV9 Vector Transduced the Heart at a Magnitude Higher Than rAAV8 at Lower Doses
The most important finding in the present study is that rAAV9 vector was more efficient than rAAV8 in myocardial transduction. Both rAAV8 and 9 vectors transduced the heart almost entirely at doses of 3.0 × 1011 vg/mouse or more (Fig. 3 and Tables 2A, 2B). Although the difference in cardiac transduction efficiency was not clear at this dose range due to near or complete saturation of transduction, rAAV9 transduced the heart a magnitude higher than rAAV8 at nonsaturating vector doses, i.e., 1.0 × 1011 vg/mouse or lower (Fig. 3 and Tables 2A, 2B). At a dose of 1.0 × 1011 vg/mouse, rAAV9 transduced over 80% of cardiomyocytes while rAAV8 transduced only 14%. This superior cardiac transduction with rAAV9 over rAAV8 is clearly demonstrated by comparing transduction efficiency between the heart and the liver (Fig. 5). These observations indicate that we can deliver therapeutic genes to a substantial fraction of myocardium by systemic administration of a minimal dose of a rAAV9 vector. This can minimize vector genome dissemination or transduction in noncardiac tissues. A dose of 3.0 × 1010 vg of rAAV9 per mouse was enough to transduce 32% of cardiomyocytes, which should be sufficient to treat many hereditary or acquired cardiac diseases. At this dose, vector genomes were not detected in noncardiac tissues at a sensitivity of 0.03 ds-vg/dge, except for the liver, in which transduction was restricted to 3.2% hepatocytes and 6.1 ds-vg/dge. Although the mechanism underlying superior cardiac transduction with rAAV9 is currently unknown, such information will be relevant for determining an optimal vector dose for cardiac gene delivery. Very recently, Zhu et al. demonstrated proof-of-principle that systemic administration of a rAAV8 vector can successfully treat congestive heart failure, using a rodent model for muscular dystrophy [13]. With rAAV9 vector or other novel serotype vectors, it may be possible to improve further current cardiac gene transfer technologies, which are still a major challenge for cardiac gene therapy. It should be noted that superior cardiac gene transfer with rAAV9 has been recently reported by others as well (abstracts at the 8th Annual Meeting of the American Society of Gene Therapy, 2005).
FIG. 5.
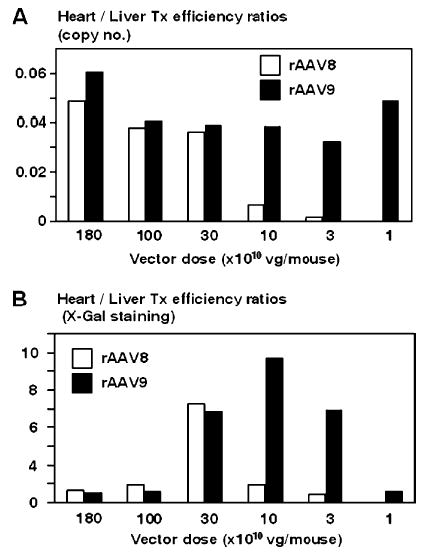
Transduction efficiency in the heart relative to that in the liver (i.e., heart/liver transduction efficiency ratios) as a function of injected vector dose. (A) Heart/liver transduction (Tx) efficiency ratios were determined based on the amount of ds vector genome copy numbers in the heart and liver. (B) Heart/liver transduction efficiency ratios were determined based on the transduction efficiencies determined by X-Gal staining. A discrepancy in the values (ratios) between white and black bars at the same vector dose represents a distinct transduction efficiency in the heart between rAAV8 and rAAV9 vectors at a given vector dose.
In summary, this study clearly outlines the biology of rAAV9 vectors as well as that of rAAV8 in mice. A number of biological features are shared between rAAV8 and rAAV9, i.e., robustness, ability to transduce tissues systemically, ability to transduce remote organs following extravascular injections, and similar tissue tropism. However, these two vectors are distinct in that rAAV9 is highly efficient in transducing the murine heart at lower doses. Further studies are warranted to elucidate the molecular basis of the shared and distinct biological features of these rAAV vectors in vivo.
Materials and Methods
Construction of rAAV vectors
The construction and production of rAAV8 vectors AAV8-hF.IX16, AAV8-EF1α-nlslacZ, and AAV8-CMV-lacZ were described elsewhere [11]. These were pseudo-serotyped vectors carrying the corresponding rAAV2 vector genomes. Briefly, AAV8-hF.IX16 carried the hF.IX minigene and the bovine growth hormone poly(A) signal, under the control of a liver-specific promoter. AAV8-EF1α-nlslacZ and AAV8-CMV-lacZ are two types of bacterial β-galactosidase-expressing rAAV8 vectors carrying either the human EF1α enhancer–promoter or the human CMV immediate early gene enhancer–promoter, either a nuclear localizing signal (nls)-harboring or a cytosolic bacterial lacZ gene, and the simian virus 40 poly(A) signal. For AAV9-hF.IX16, AAV9-EF1α-nlslacZ, and AAV9-CMV-lacZ, the corresponding rAAV2 vector genomes were cross-packaged into capsids derived from AAV9. All the vectors were produced in human embryonic kidney 293 cells by the triple-transfection method, purified by two cycles of cesium chloride gradient centrifugation, and concentrated as outlined elsewhere [16]. The final viral preparations were kept in phosphate-buffered saline containing 5% sorbitol. The physical particle titers were determined by a quantitative dot-blot assay.
Animal procedure
Six- to 8-week-old male and female C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The portal vein and tail vein injections were performed as previously described [17]. For intraperitoneal injection, mice were anesthetized by inhalation of isoflurane and the peritoneum was exposed through an abdominal skin incision. Two hundred microliters of vector preparation was then injected into the peritoneal space under direct vision to monitor the procedure. For subcutaneous injection, mice were anesthetized with isoflurane, and 200 μl of vector preparation was injected under the dorsal skin. Plasma samples were collected from the retro-orbital plexus. All the animal experiments were performed according to the guidelines for animal care at Stanford University.
Enzyme-linked immunosorbent assays
Human F.IX levels in mouse plasma were determined by an enzyme-linked immunosorbent assay specific for hF.IX as previously described [17].
Histological analysis
To determine transduction efficiency and transduced cell types, we performed X-Gal staining of various mouse tissues as previously described [11]. To quantify the transduction efficiency in the liver and pancreas, we analyzed cells corresponding to over 2000 nuclei for β-galactosidase expression in each animal. To quantify the transduction efficiency in skeletal muscle, we analyzed at least 500 skeletal myofibers in the tongue for the transgene expression in each animal. To quantify the transduction efficiency in the heart, a minimum of five 20 × fields were captured from each heart, and the percentage of the total tissue fractional area demonstrating β-galactosidase activity was determined using MetaMorph software version 6.2r3 (Molecular Devices Corp., Sunnyvale, CA, USA).
DNA analysis
We extracted total genomic DNA from each tissue by a standard phenol–chloroform extraction method. We performed Southern blot analysis to determine vector genome copy numbers and analyzed molecular forms of vector genomes using 10–30 μg of total genomic DNA as previously described [11,22]. We detected and quantified the signals with a Phosphorimager and Quantity One software (Bio-Rad, Hercules, CA, USA) or a Typhoon 9400 and ImageQuant (GE Healthcare, Giles, UK). Vector genome copy numbers in each tissue were expressed as ds vector genomes per diploid genomic equivalent with plasmids carrying corresponding ds vector genome sequences as standards.
Statistical analysis
We compared mean values from different experimental groups by a two-tailed Student’s t test or one-way ANOVA.
Acknowledgments
We thank Guang-Ping Gao and James M. Wilson (University of Pennsylvania) for providing us with AAV8 and 9 helper plasmids. This work was supported by a National Hemophilia Foundation Career Development Award to H.N. and two grants from the National Institutes of Health: DK68636 to H.N. and HL66948 to M.A.K.
References
- 1.Gao G, et al. Clades of adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao GP, et al. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muzyczka N, Warrington KH., Jr Custom adeno-associated virus capsids: the next generation of recombinant vectors with novel tropism. Hum Gene Ther. 2005;16:408–416. doi: 10.1089/hum.2005.16.408. [DOI] [PubMed] [Google Scholar]
- 4.Chiorini JA, Kim F, Yang L, Kotin RM. Cloning and characterization of adeno-associated virus type 5. J Virol. 1999;73:1309–1319. doi: 10.1128/jvi.73.2.1309-1319.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiorini JA, Yang L, Liu Y, Safer B, Kotin RM. Cloning of adeno-associated virus type 4 (AAV4) and generation of recombinant AAV4 particles. J Virol. 1997;71:6823–6833. doi: 10.1128/jvi.71.9.6823-6833.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muramatsu S, Mizukami H, Young NS, Brown KE. Nucleotide sequencing and generation of an infectious clone of adeno-associated virus 3. Virology. 1996;221:208–217. doi: 10.1006/viro.1996.0367. [DOI] [PubMed] [Google Scholar]
- 7.Rutledge EA, Halbert CL, Russell DW. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. J Virol. 1998;72:309–319. doi: 10.1128/jvi.72.1.309-319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao W, et al. Gene therapy vectors based on adeno-associated virus type 1. J Virol. 1999;73:3994–4003. doi: 10.1128/jvi.73.5.3994-4003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grimm D, Kay MA. From virus evolution to vector revolution: use of naturally occurring serotypes of adeno-associated virus (AAV) as novel vectors for human gene therapy. Curr Gene Ther. 2003;3:281–304. doi: 10.2174/1566523034578285. [DOI] [PubMed] [Google Scholar]
- 10.Gregorevic P, et al. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakai H, et al. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J Virol. 2005;79:214–224. doi: 10.1128/JVI.79.1.214-224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 13.Zhu T, et al. Sustained whole-body functional rescue in congestive heart failure and muscular dystrophy hamsters by systemic gene transfer. Circulation. 2005;112:2650–2659. doi: 10.1161/CIRCULATIONAHA.105.565598. [DOI] [PubMed] [Google Scholar]
- 14.Cearley CN, Wolfe JH. Transduction characteristics of adeno-associated virus vectors expressing cap serotypes 7, 8, 9, and rh10 in the mouse brain. Mol Ther. 2006;13:528–537. doi: 10.1016/j.ymthe.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 15.De BP, et al. High level of persistent expression of α1-antitrypsin mediated by the nonhuman primate serotype rh.10 adeno-associated virus despite preexisting immunity to common human adeno-associated viruses. Mol Ther. 2006;13:67–76. doi: 10.1016/j.ymthe.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Grimm D, et al. Preclinical in vivo evaluation of pseudotyped adeno-associated virus vectors for liver gene therapy. Blood. 2003;102:2412–2419. doi: 10.1182/blood-2003-02-0495. [DOI] [PubMed] [Google Scholar]
- 17.Nakai H, Iwaki Y, Kay MA, Couto LB. Isolation of recombinant adeno-associated virus vector–cellular DNA junctions from mouse liver. J Virol. 1999;73:5438–5447. doi: 10.1128/jvi.73.7.5438-5447.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song S, et al. Stable therapeutic serum levels of human α1-antitrypsin (AAT) after portal vein injection of recombinant adeno-associated virus (rAAV) vectors. Gene Ther. 2001;8:1299–1306. doi: 10.1038/sj.gt.3301422. [DOI] [PubMed] [Google Scholar]
- 19.Sarkar R, et al. Total correction of hemophilia A mice with canine FVIII using an AAV 8 serotype. Blood. 2004;103:1253–1260. doi: 10.1182/blood-2003-08-2954. [DOI] [PubMed] [Google Scholar]
- 20.Blankinship MJ, et al. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther. 2004;10:671–678. doi: 10.1016/j.ymthe.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 21.Lipshutz GS, et al. Comparison of gene expression after intraperitoneal delivery of AAV2 or AAV5 in utero. Mol Ther. 2003;8:90–98. doi: 10.1016/s1525-0016(03)00132-1. [DOI] [PubMed] [Google Scholar]
- 22.Nakai H, et al. A limited number of transducible hepatocytes restricts a wide-range linear vector dose response in recombinant adeno-associated virus-mediated liver transduction. J Virol. 2002;76:11343–11349. doi: 10.1128/JVI.76.22.11343-11349.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duan D, et al. Circular intermediates of recombinant adeno-associated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J Virol. 1998;72:8568–8577. doi: 10.1128/jvi.72.11.8568-8577.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakai H, et al. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol. 2001;75:6969–6976. doi: 10.1128/JVI.75.15.6969-6976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lipkowitz MS, et al. Transduction of renal cells in vitro and in vivo by adeno-associated virus gene therapy vectors. J Am Soc Nephrol. 1999;10:1908–1915. doi: 10.1681/ASN.V1091908. [DOI] [PubMed] [Google Scholar]