

Abstract
A-kinase anchoring proteins (AKAPs) function to target protein kinase A (PKA) to specific locations within the cell. AKAPs are functionally identified by their ability to bind the type II regulatory subunits (RII) of PKA in an in vitro overlay assay. We previously showed that follicle-stimulating hormone (FSH) induces the expression of an 80-kDa AKAP (AKAP 80) in ovarian granulosa cells as they mature from a preantral to a preovulatory phenotype. In this report, we identify AKAP 80 as micro-tubule-associated protein 2D (MAP2D), a low molecular weight splice variant of the neuronal MAP2 protein. MAP2D is induced in granulosa cells by dexamethasone and by FSH in a time-dependent manner that mimics that of AKAP 80, and immunoprecipitation of MAP2D depletes extracts of AKAP 80. MAP2D is the only MAP2 protein present in ovaries and is localized to granulosa cells of preovulatory follicles and to luteal cells. MAP2D is concentrated at the Golgi apparatus along with RI and RII and, based on coimmunoprecipitation results, appears to bind both RI and RII in granulosa cells. Reduced expression of MAP2D resulting from treatment of granulosa cells with antisense oligonucleotides to MAP2 inhibited the phosphorylation of cAMP-response element-binding protein. These results suggest that this classic neuronal RII AKAP is a dual RI/RII AKAP that performs unique functions in ovarian granulosa cells that contribute to the preovulatory phenotype.
Ovarian follicles house the oocyte and, upon maturation, produce steroid and protein hormones that regulate uterine receptivity and the reproductive axis. Follicles exist in a relatively dormant, preantral (PA)1 state until they are recruited to grow and differentiate to a preovulatory (PO) phenotype by the pituitary hormone follicle-stimulating hormone (FSH) (1, 2). Maturation of follicles to a PO phenotype involves not only proliferation but also differentiation of the enclosed granulosa cells. FSH triggers these events by binding to its G-protein-coupled receptor, located exclusively on granulosa cells in female mammals, and activating adenylyl cyclase, which converts ATP to cAMP. cAMP then acts as a second messenger primarily by activating protein kinase A (PKA) (3).
PKA is a tetrameric enzyme that consists of a dimeric regulatory (R) subunit and two catalytic subunits (4). Upon binding of cAMP to the R subunits, a conformational change occurs that allows for dissociation of the active catalytic subunits, which can then phosphorylate neighboring substrates. Two classes of PKA holoenzymes, PKA I and PKA II, exist based on the association of two possible RI subunits (RIα and RIβ) or two possible RII subunits (RIIα and RIIβ) with four possible catalytic subunits (Cα, Cβ1, Cβ2, and Cγ) (5). In rat granulosa cells of PA and PO follicles, PKA IIα and PKA IIβ are the predominant PKA isoforms present, whereas less than 5% of PKA holoenzyme activity is contributed by PKA Iα (6–8).
The specificity of PKA action is accomplished by the targeting of PKA to specific cellular locales by virtue of its binding to a growing family of A-kinase anchoring proteins (AKAPs). Most known AKAPs anchor RII and exhibit at least a 100-fold lower affinity for RI (9). RII subunits of PKA bind with nanomolar affinity to AKAPs (5, 10). The domain on the AKAP responsible for RII binding comprises an amphipathic helix that binds to the N termini of the RII dimer (11). A growing number of “dual” AKAPs have been identified, although they still exhibit higher affinity for RII over RI (12–15). Recent reports, however, indicate that some AKAPs can preferentially bind RI (16–19). AKAPs anchor PKA to specific cellular locations, such as the actin cytoskeleton (20, 21), plasma membrane (22), mitochondria (23, 24), Golgi apparatus (25), centrosome (26), and nuclear envelope (27). The localization of PKA to distinct regions within the cell is generally thought to allow for both specific and efficient substrate phosphorylation in response to a specific stimulus (28).
FSH receptor signaling in PA granulosa cells stimulates the PKA-dependent phosphorylation of a number of signaling intermediates including histone H3 (29), cAMP-response element-binding protein (CREB) (30, 31), and an extracellular regulated kinase (ERK)-protein-tyrosine phosphatase that leads to ERK activation (32). In addition, FSH receptor activation induces the transcription of a number of genes, including those for the luteinizing hormone (LH) receptor and inhibin-α as well as the P450 aromatase and side chain cleavage steroidogenic enzymes (33, 34). On the other hand, in granulosa cells of the PO follicle, LH receptor signaling causes an up-regulation in genes that encode for progesterone receptor and cyclooxgenase-2 while at the same time causing a down-regulation in genes that encode for the LH and FSH receptors, inhibin-α, and aromatase proteins (33, 35). Like FSH receptor signaling, LH receptor signaling also stimulates the PKA-dependent phosphorylation of key substrates such as histone H3, CREB, and an unidentified substrate upstream of ERK that leads to the activation of ERK (36). The fact that PKA plays a predominant role in the pleotrophic signaling events regulated by these hormones in PA versus PO granulosa cells led us to hypothesize that these cells may express different complements of AKAPs to localize PKA to distinct subcellular environments. In this report, we demonstrate that an 80-kDa AKAP that we previously showed is induced in ovarian granulosa cells by FSH (8, 37) is microtubule-associated protein 2D (MAP2D).
MAP2 is a microtubule- and microfilament-binding protein localized primarily to dendrites and to nonneuronal glial cells (38, 39). Most of the reports on MAP2 focus on its role in neurite outgrowth and dendrite development in the brain (40). MAP2 functions in the brain to stabilize microtubules, to stimulate microtubule assembly, and to regulate cell shape (41, 42). The MAP2 gene encodes high and low molecular weight protein isoforms that are generated by alternative splicing (43). MAP2A and MAP2B are high molecular mass isoforms (270 and 280 kDa, respectively), whereas MAP2C and MAP2D are low molecular mass isoforms (70 and 80 kDa, respectively) (44). The low molecular weight isoforms contain N- and C-terminal regions of the high molecular weight isoforms but lack the large central domain found in high molecular weight MAP2 isoforms. Each isoform contains at least three imperfect microtubule-binding domains in their C termini. MAP2D contains an additional 93-base pair insert, which comprises the fourth microtubule-binding domain that is absent from MAP2C (45, 46). MAP2 was the first protein to be recognized as an AKAP (47), and the RII-binding region was localized to amino acids 83–113 in the N-terminal region of the protein and is conserved among all MAP2 isoforms (48).
Our results show that the neuronal protein MAP2D is induced by FSH and localized to the granulosa cells of the PO follicle and that its expression is maintained in luteal cells following ovulation and corpus luteum formation. MAP2D appears to anchor both RI and RII to the Golgi apparatus in granulosa cells of PO follicles, and, based on antisense oligonucleotide experiments, MAP2D appears to participate in acute LH receptor signaling events. The function of MAP2D in ovarian granulosa cells is thus expected to be quite distinct from its established neuronal roles.
EXPERIMENTAL PROCEDURES
Materials
The following were purchased: ovine FSH (oFSH-20) from Dr. A. F. Parlow of the NIDDK, National Institutes of Health, National Hormone and Pituitary Program (Harbor-UCLA Medical Center, Torrance, CA); Profasi® hCG from Serono Laboratories Inc. (Randolph, MA); [γ-32P]ATP, ammonium salt (3000 Ci/mmol), and [32P]orthophosphate (~9000 Ci/mmol) from PerkinElmer Life Sciences; [2,8-3H]cAMP sodium salt (15–40 Ci/mmol) from ICN Chemical and Radioisotope Division (Costa Mesa, CA); DEAE-cellulose (DE-52) and P-81 cellulose phosphate paper from Whatman (Clifton, NJ); ECL reagents, rainbow molecular weight markers, and Hybond-C nitrocellulose membranes from Amersham Biosciences; SDS-PAGE reagents from Bio-Rad; X-Omat AR film from Eastman Kodak Co.; all culture media from Invitrogen; brefeldin A from LC Laboratories (San Diego, CA); MAP2D scrambled and antisense oligonucleotides from Integrated DNA Technologies (Coralville, IA); MAP2D and glyceraldehyde-3-phosphate dehydrogenase PCR primers from Northwestern University Bio-technology Laboratory (Chicago, IL); DNase and reverse transcriptase-PCR reagents and buffers from Promega (Madison, WI); actin phalloidin from Molecular Probes, Inc. (Eugene, OR); and protein A+G-agarose from Santa Cruz Biotechnologies, Inc. (Santa Cruz, CA). All other biochemical reagents were purchased from Sigma, unless otherwise indicated.
Antibodies
Inhibin-α antibody was kindly provided by Dr. Wiley Vale of the Salk Institute for Biological Studies (San Diego, CA). The following were purchased: anti-MAP2 mouse monoclonal and anti-γ-tubulin from Sigma; anti-cyclin D2 polyclonal from Santa Cruz Biotechnologies; anti-PKA RI mouse monoclonal and anti-PKA RII β mouse monoclonal from BD Biosciences; anti-PKA RII (RIIα and -β) goat polyclonal and anti-phospho-CREB (S133) from Upstate Biotechnology, Inc. (Lake Placid, NY); goat anti-mouse rhodamine, goat anti-rabbit fluorescein, goat anti-rabbit rhodamine, donkey anti-goat rhodamine, donkey anti-mouse fluorescein, and goat anti-mouse fluorescein secondary antibodies from Jackson Immunochemicals (West Grove, PA); anti-Gm 130-fluorescein conjugate from BD Biosciences (Palo Alto, CA), and anti-hemagglutinin (HA) protein of human influenza virus from Roche Applied Science.
Primary Granulosa Cell Cultures
Sprague-Dawley rats were obtained at 15–18 days of age (Sasco, Baltimore, MD) and maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Rats at 21–24 days of age were injected subcutaneously with either 1.5 mg/ml estrogen for 3 days to obtain PA ovaries or 10 IU of pregnant mares serum gonadotropin (PMSG) to obtain PO ovaries (harvested 48 –50 h postinjection). Ovaries were trimmed to remove the bursa, fat, and oviducts and incubated for 15–30 min at 37 °C in 6 mM EGTA in Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium. Ovaries were then incubated for 5–20 min in 0.5 M sucrose in Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium. Granulosa cells from PA or PO ovaries were expressed by penetration of PA or large PO follicles, respectively, with a 30-gauge needle. The cells were plated at a density of 5–7 × 106 in serum-free Dulbecco’s modified Eagle’s medium/Ham’s F-12 medium on plates coated with 0.5 μg/ml fibronectin (Invitrogen) and cultured as previously described (29, 37). Cells were treated the next day, as indicated under “Results.”
Detergent-soluble Ovarian Extracts
21–24-Day-old Sprague-Dawley rats (Sasco) were not treated or were injected subcutaneously with 25 IU PMSG 48 h prior to injection with 25 IU hCG, as specified under “Results.” Ovaries were harvested at various time points after injections; cooled to 4 °C in an iced 10 mM potassium phosphate buffer, pH 7.0; dissected free of bursa, fat, and oviducts; weighed; and homogenized (5:1 ratio of homogenization buffer/wet weight) in buffer A (10 mM potassium phosphate, pH 7.0, 1 mM EDTA, 5 mM EGTA, 10 mM MgCl2, 50 mM β-glycerol phosphate, 1 mM sodium orthovanadate, 2 mM dithiothreitol (DTT), 40 μg/ml phenylmethylsulfonyl fluoride, 10 μg/ml E64, 0.5% Nonidet P-40, 0.1% deoxycholate) using 15–20 strokes with a ground glass homogenizer. A supernatant fraction was obtained by centrifugation at 10,000 × g for 10 min at 4 °C. Protein concentrations were determined by the Lowry protein assay (49) using bovine serum albumin as the standard.
Electrophoresis and Western Blotting
Total cell extracts were made by harvesting granulosa cells into 300 μl of SDS-PAGE sample buffer (50) and boiled. Protein concentrations were controlled by plating equal numbers of cells in each dish for each experiment followed by loading equal volumes onto an SDS-PAGE gel. These cells do not divide under serum-free conditions. The proteins in the cell extracts were separated by SDS-PAGE (51), electrotransferred to Hybond C-extra nitrocellulose overnight at 4 °C, and stained for protein loading using Ponceau S. The nitrocellulose blots were incubated with primary antibody at 4 °C overnight. Antigen-antibody complexes were detected using ECL.
DEAE-cellulose Chromatography and Sucrose Density Gradient Centrifugation
DEAE-cellulose chromatography was conducted as previously described (52), collecting fractions (~0.75 ml) in tubes containing 50 μl of a concentrated mixture of protease inhibitors (buffer B) at the indicated final concentrations: 10 mM benzamidine, 2 mM DTT, 5 μg/ml pepstatin, 10 μg/ml leupeptin, 5 μg/ml aprotinin, 40 μg/ml phenylmethylsulfonyl fluoride, 5 μg/ml soybean trypsin inhibitor, 10 μg/ml E-64. Protein kinase activity in DEAE fractions was determined in the presence of 0.5 μM cAMP and 71.5 μM Leu-Arg-Arg-Ala-Ser-Leu-Gly (Kemp-tide) as substrate (8). Total [3H]cAMP binding sites on R subunits were determined by incubating sample with 0.3 μM [3H]cAMP for 30 min at 30 °C (52). When indicated, DEAE-cellulose fractions (collected in the absence of DTT) were concentrated and subjected to sucrose density gradient centrifugation, as previously described (52), in the presence of protease and phosphatase inhibitors. Following centrifugation, fractions were mixed with SDS-sample buffer, boiled, and subjected to SDS-PAGE and Western blotting.
RII Overlay Assay and cAMP-agarose Affinity Chromatography
Recombinant murine RIIα was expressed in Escherichia coli and purified by affinity chromatography on cAMP-Sepharose (53). For solid phase RII overlay assays, proteins were separated by SDS-PAGE and electrotransferred to Immobilon-P polyvinylidene difluoride (Millipore Corp.), and blots were probed with 0.5 μg of recombinant RIIα phosphorylated using [γ-32P]ATP with the catalytic subunit of PKA and then subjected to autoradiography (54). For cAMP-agarose affinity chromatography, pooled and concentrated DEAE-cellulose fractions were added to cAMP-agarose (0.2-ml packed volume; Sigma) equilibrated in buffer C (10 mM Hepes, pH 7.0, 1 mM EDTA, 5 mM EGTA, 10 mM MgCl2, 2 mM DTT, 10 μg/ml pepstatin, 10 μg/ml leupeptin, 5 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitor, 40 μg/ml phenylmethylsulfonyl fluoride, 1 mM sodium vanadate, 10 μM isobutylmethylxanthine, 20 mM benzamidine, and 10 μg/ml E-64) and incubated overnight at 4 °C on a Nutator (8). The flow-through fraction (~1.0 ml) was collected by centrifugation at 5000 × g for 5 min, an aliquot (0.1 ml) was diluted to 0.3 ml, mixed with 0.15 ml of 3-fold SDS-PAGE sample buffer, and boiled. The cAMP-agarose pellet was then washed three times by sequential centrifugations with a total of 3 ml of buffer C, three times with buffer C containing 1 M NaCl, and three times with buffer C containing 0.3 M NaCl, collecting an aliquot (0.3 ml) of the final 1.0-ml wash. cAMP-agarose was then incubated at room temperature for 30 min with 0.3 ml of 5 μM Ht31 peptide (residues 493–515) in buffer C containing 0.3 M NaCl, collecting eluate from agarose using a syringe followed by boiling after the addition of 3-fold SDS-PAGE sample buffer (0.15 ml). Finally, cAMP-agarose was incubated at room temperature for 30 min with 0.3 ml of 75 mM cAMP in buffer C containing 0.3 M NaCl, collecting eluate as described above.
RNA Isolation and Reverse Transcriptase-PCR
RNA was collected by lysing cultured granulosa cells in 1 ml of Trizol® reagent (Invitrogen) per 3 ml of medium according to the manufacturer’s instructions. RNA (5 μg) was treated with DNase I (Promega) for 15 min at room temperature and heat-inactivated at 65 °C for 10 min. The reverse transcription reaction was performed using 2.5 μg of DNase-treated RNA and avian myeloblastosis virus reverse transcriptase (Promega) as described previously (55). PCR was performed using 500 ng of DNA using primers for MAP2 (forward, 5′-CAC AAG GAT CAG CCT GCA GCT CTG-3′; reverse, 5′-CAC CTG GCC TGT GAC GGA TGT TCT-3′) that generate a 756-base pair PCR product (45) or primers for glyceraldehyde-3-phosphate dehydrogenase (forward, 5′-CCCTTCATTGACCTCAACTA-3′; reverse, 5′-CCAAAGTTGTCATGGATGAC-3′) that generate a 350-bp PCR product (56). The PCR consisted of 24 cycles of 94 °C denaturation, 55 °C annealing, and 72 °C elongation steps. The reactions were analyzed on a 1.5% agarose gel containing ethidium bromide.
Immunofluorescence and Immunohistochemistry
For immunofluorescence analysis, granulosa cells from PA follicles were cultured on fibronectin-coated glass coverslips and treated as indicated under “Results.” The cells were washed in phosphate-buffered saline (PBS), fixed for 10 min in 4% formaldehyde in PBS, and permeabilized with 0.1% Triton X-100. Coverslips were then washed three times with PBS and blocked for 1 h in 1% bovine serum albumin in PBS. Coverslips were incubated overnight at 4 °C in a humidified chamber with primary antibody at a 1:200 dilution (unless otherwise indicated). Coverslips were then washed three times with PBS, incubated for 2 h at 37 °C with secondary antibody conjugated to rhodamine or flourescein, as indicated, and washed three times with PBS. Coverslips were then allowed to dry and mounted onto glass slides using Vectashield® mounting medium (Vector Laboratories, Burlingame, CA). The slides were analyzed using a Zeiss LSM510 laser-scanning microscope.
For immunohistochemistry, whole ovaries were harvested in 4% paraformaldehyde, paraffin-embedded, sectioned at 4 μm onto glass slides, and subjected to enzyme-induced epitope retrieval immunohistochemistry according to the protocol developed by Northwestern University Robert H. Lurie Cancer Center Pathology Core (Chicago IL).
Immunoprecipitation
For immunoprecipitation of MAP2 from ovarian and brain extracts (see Fig. 3), extracts were prepared in buffer A and incubated with 10 μl of anti-MAP2 antibody and 30 μl of protein A+G-agarose for 4 h at 4 °C on a Nutator. The agarose was then washed with buffer D (20 mM Hepes, 150 mM NaCl, 10% glycerol, and 0.1% Triton-X). Proteins bound to the agarose were eluted by boiling in 50 μl of SDS-PAGE sample buffer. For PKA R subunit immunoprecipitations from ovarian extracts and DEAE fractions, samples (containing 500 –800 μg of protein in ovarian extracts or 1 ml from pooled DEAE fractions collected in the absence of DTT) were incubated with 1 μM dithiobis[succinimidylpropionate] (DSP) (Pierce) in dimethyl sulfoxide for 15 min at room temperature. The cross-linking reaction was stopped by adding 1 M Tris-HCl, pH 7.5, to a final concentration of 25 mM and incubating samples at room temperature for 15 min. Antibody (10 μl of anti-MAP2 (Sigma), 25 μl of anti-RI (BD Biosciences), 20 μl of anti-RII (Upstate Biotechnology), 10 μl of anti-HA as a nonimmune (NI) control, 10 μl of anti-RIIβ (BD Biosciences Transduction Laboratories)) and 30 μl of protein A+G-agarose was added and incubated for 4 h at 4 °C. The agarose was then washed with buffer D. When indicated, proteins were eluted using Immunopure® elution buffer, pH 2.8, that contains a primary amine (Pierce) in three elution steps at 50 μl each for a total volume of 150 μl. Remaining proteins bound to agarose were eluted by boiling in 100 μl of SDS-PAGE sample buffer.
Fig. 3. MAP2 isoform expression in rat ovary compared with brain.
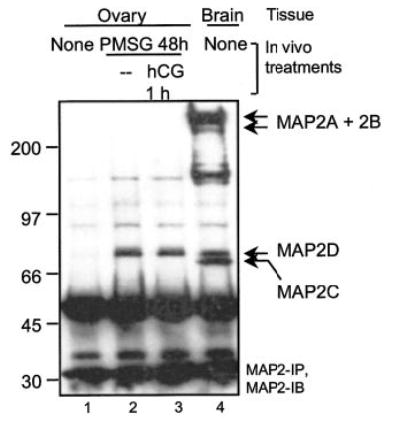
Rats were injected with vehicle (Veh) or 25 IU of PMSG, and ovaries were harvested 48 h later. For hCG treatment, rats were injected with 25 IU of hCG 48 h post-PMSG injection, and ovaries were obtained 1 h post-hCG. Brain tissue was collected 48 h post-PMSG injection. Soluble extracts were made by homogenizing ovaries or brain in buffer A and centrifugation (10 min, 10,000 × g, 4 °C). MAP2 was immunoprecipitated from soluble extracts (500 μg of protein in a total volume of 500 μl) as described under “Experimental Procedures.” Results are representative of three separate experiments.
For MAP2D phosphorylation, PO granulosa cells from PMSG-treated rats were incubated for 1 h in phosphate-free medium and then incubated for 2 h in 1.25 mCi of 32Pi/6 × 106 cells. Cells were then treated with vehicle or hCG for 15 min and lysed in buffer A, and insoluble cell debris was removed by centrifugation at 10,000 × g for 2 min. Cell extracts were then incubated overnight with 10 μl of anti-MAP2 antibody or NI antibody plus 30 μl of protein A+G-agarose on a Nutator. Immunoprecipitates were washed with buffer D, mixed with SDS-PAGE sample buffer, and boiled, and proteins were separated by SDS-PAGE and subjected to autoradiography.
Antisense Oligonucleotide Transfection
Granulosa cells from PA follicles were plated in either 6-well or 60-mm plates coated with 100 ng/ml poly-L-lysine. Prior to hormone treatment, either a MAP2 anti-sense oligonucleotide (18-mer; CTC GTC AGC CAT CCT TCA) (41, 44) or a scrambled oligonucleotide (18-mer; ATT CCA GTA CGC TCC CCT), diluted in 10 mM Tris-HCl, pH 7.5, 1 mM EDTA buffer, was added to the cells at a concentration of 10 μg/ml. Oligonucleotides were added every 12 h during the 72-h incubation period. Total cell extracts were collected in SDS-PAGE sample buffer and analyzed by gel electrophoresis and Western blotting.
Purification of AKAP 80
Ovaries from 110 PMSG-primed rats were flash frozen in liquid nitrogen; ground with a mortar and pestle; scraped into buffer E (20 mM Hepes, pH 7.4, 20 mM NaCl, 5 mM EDTA, 2 mM DTT, 1 mM EGTA, 5 μg/ml pepsatin and aprotinin, 10 μg/ml leupeptin and E64, 50 μg/ml soybean trypsin inhibitor, 10 mM benzamidine), which was maintained at 100 °C; homogenized using a Polytron® homogenizer (Brinkman Instruments, Westbury, NY); and boiled for 20 min. The homogenate was chilled to 4 °C, fresh protease inhibitors as well as 10 μM isomethylbutylxanthine and 1 mM sodium vanadate were added, and a soluble extract was obtained upon centrifugation at 5000 × g for 10 min at 4 °C. Boiling was performed based on evidence that AKAP 80 was heat-stable (data not shown). The soluble extract was applied to 15 g of DEAE-cellulose, and AKAP 80 was batch-eluted with 0.1 M potassium phosphate, pH 7.0. The DEAE eluate was then incubated with 2 ml of cAMP-agarose beads complexed with 20 μg of recombinant RIIα and 35 μg of recombinant RIIβ subunits of PKA overnight at 4 °C. The cAMP-agarose was then washed extensively, and the bound AKAPs were eluted with 50 ml of 75 mM cAMP in buffer C containing 0.3 M NaCl. The proteins in the cAMP eluate were precipitated with 15% trichloroacetic acid, and the trichloroacetic acid pellet was washed with ethanol, resuspended in 100 μl of SDS-PAGE sample buffer, and boiled. Following confirmation (by RII overlay assay) that AKAP 80 was present in 0.5 μl of the reconstituted trichloroacetic acid pellet, the remaining 99 μl was subjected to SDS-PAGE. The portion of the gel ~80 kDa was cut and sent to the Harvard Microchemistry Facility for sequence analysis by microcapillary reverse-phase high pressure liquid chromatography nanoelectrospray tandem mass spectroscopy on a Finnigan LQC DECA quadrupole ion trap mass spectrometer. Peptide sequence identification was based on the Sequest algorithm.
RESULTS
FSH and Dexamethasone Induce the Expression of MAP2D
We previously reported that FSH induces the expression of an 80-kDa heat-stable AKAP as granulosa cells mature from a PA to a PO phenotype (8, 37, 58). AKAP 80 is also induced by dexamethasone (DEX) (58). AKAP 80 was partially purified by cAMP-agarose affinity chromatography from boiled ovarian extracts of PMSG-primed rats and subjected to SDS-PAGE, and peptides from ~80-kDa proteins were separated by mass spectrometry, as detailed under “Experimental Procedures.” A BLAST search revealed sequence identity to a peptide (KIDFSK) unique to the fourth micro-tubule binding domain of the 80-kDa protein MAP2D. The high molecular weight MAP2s were the first AKAPs to be identified and contain an N-terminal 31-amino acid RII binding domain that is conserved in the MAP2D splice variant (46).
We therefore hypothesized that AKAP 80 corresponded to MAP2D. To test this hypothesis, granulosa cells from PA follicles were treated for 72 h with vehicle, FSH, or DEX, and total cell extracts were subjected to an RII overlay assay and Western blotting using an antibody to MAP2 that detects MAP2A, -2B, -2C, and -2D (45). The RII overlay detects induction of AKAP 80 by FSH and an even greater induction by DEX (Fig. 1A). The corresponding Western blot shows that FSH and DEX induce a similar pattern of expression of a MAP2 isoform at 80 kDa (Fig. 1B). Additionally, two-dimensional gel electrophoresis showed an equivalent migration pattern for AKAP 80 (8) and MAP2D as detected by Western blotting (data not shown).
Fig. 1. Effect of FSH and dexam-ethasone on the expression of AKAP 80/MAP2D.
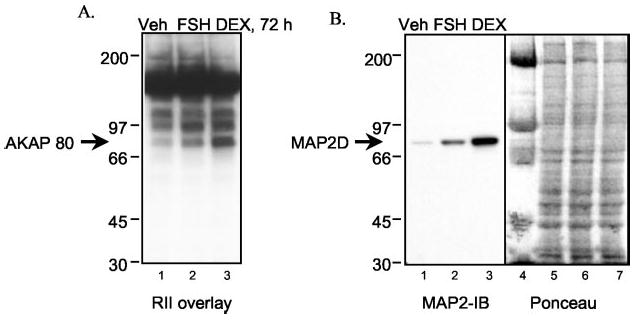
Granulosa cells from estrogen-primed rats were treated for 72 h with vehicle (Veh), 50 ng/ml FSH, or 10 nM dexamethasone (DEX). Total cell extracts were subjected to either RII overlay assay (A) or immunoblotting (IB) using a mouse monoclonal MAP2 antibody at 1:1000 dilution (B). Ponceau S staining of the protein in B (lanes 1–3) is shown in lanes 5–7, respectively. Lane 4, molecular weight markers. See “Experimental Procedures” for details. Results are representative of two separate experiments.
We compared the time course of expression of MAP2D and AKAP 80. Granulosa cells were treated with FSH for 0, 8, 24, 48, or 72 h. Total cell lysates were then subjected to both an RII overlay assay and an immunoblot with the MAP2 antibody (Fig. 2A). FSH treatment for 48 h promotes an increase in MAP2D expression that corresponds to the increase in AKAP 80. Both MAP2D and AKAP 80 expression remain elevated at 72 h post-FSH (Fig. 2A, lanes 4 and 5). Since heat stability is a defining characteristic of AKAP 80, we tested the heat stability of MAP2D. Granulosa cells treated for 72 h with FSH were collected in buffer A and boiled for 15 min. Soluble heat-stable proteins were obtained by centrifuging the boiled extract at 10,000 × g for 10 min at 4 °C. Results show that MAP2D, like AKAP 80, is heat-stable (Fig. 2A, lane 6), consistent with reports for MAP2 (59). To confirm that the 80-kDa AKAP induced by FSH as cells mature to the PO phenotype is MAP2D, ovarian extracts enriched in PO follicles were subjected to immunoprecipitation using the MAP2 antibody and a mouse monoclonal antibody as a NI control. The resulting immunoprecipitations were subjected to an RII overlay assay. As shown in Fig. 2B, the MAP2 antibody selectively immunoprecipitates RII binding activity exclusively at 80 kDa. Moreover, immunoprecipitation of MAP2D with anti-MAP2 antibody (Fig. 2C, lane 2) depletes extracts of AKAP 80 (Fig. 2C, lane 1). Taken together, these results confirm the identity of AKAP 80 as MAP2D.
Fig. 2. In vitro expression of MAP2D.
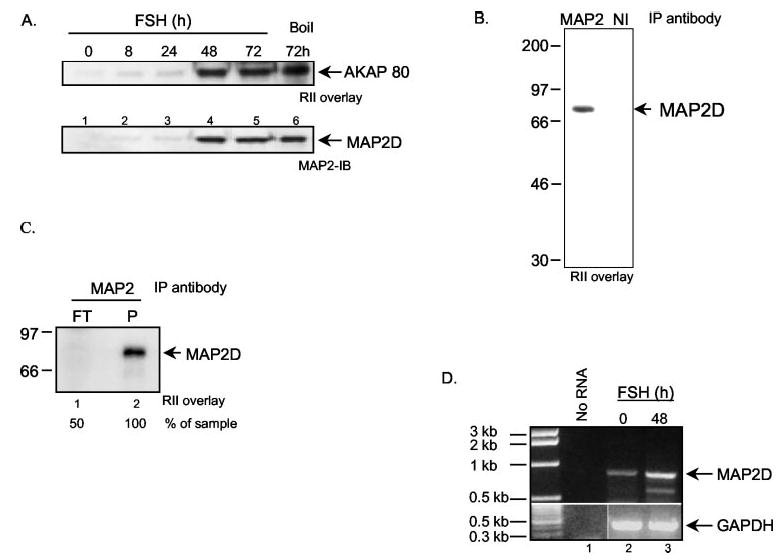
A, granulosa cells from estrogen-primed rats were treated with 50 ng/ml FSH for 0, 8, 24, 48, or 72 h. Total cell extracts were boiled in SDS-PAGE sample buffer and subjected to either RII overlay assay (top) or Western blotting using a mouse monoclonal MAP2 antibody (bottom). Lane 6, cells were harvested in buffer A, boiled for 10 min, and centrifuged at 10,000 × g for 10 min at 4 °C, and the supernatant fraction was loaded onto the gel. Results are representative of three separate experiments. B, detergent-soluble ovarian extracts from PMSG-treated rats (500 μg of protein) were subjected to immunoprecipitation using either the mouse monoclonal anti-MAP2 antibody (10 μl) or control NI mouse anti-HA antibody (10 μl) in the presence of protein A+G-agarose. The agarose pellets were washed in buffer D, and proteins were separated by SDS-PAGE, blotted to Immobilon, and subjected to an RII overlay assay as described under “Experimental Procedures.” Results are representative of two separate experiments. C, detergent-soluble ovarian extracts from PMSG-treated rats (800 μg of protein) were subjected to immunoprecipitation using either the mouse monoclonal anti-MAP2 antibody (10 μl) or control NI mouse anti-HA antibody (10 μl) in the presence of protein A+G-agarose in a total volume of 200 μl. The flow-through (FT) collected upon pelleting the agarose was boiled, and 50% was subjected to SDS-PAGE. P, washed agarose pellet. Following SDS-PAGE, proteins were subjected to an RII overlay assay. Results are representative of two separate experiments. D, cells were treated with 50 ng/ml FSH for either 0 or 48 h. Cells were harvested in 1 ml of Trizol® reagent, and RNA was isolated by phenol/chloroform extraction and isopropyl alcohol precipitation. 500 ng of RNA was DNase-treated and subjected to reverse transcriptase-PCR using the primers and conditions described under “Experimental Procedures.”
In order to show that FSH increases the gene expression of MAP2D, reverse transcriptase-PCR was performed. Results showed that whereas there is detectable MAP2D mRNA expression in vehicle-treated cells, FSH up-regulates the expression of MAP2D mRNA by 48 h post-treatment (Fig. 2D, lanes 2 and 3). Glyceraldehyde-3-phosphate dehydrogenase was used as an internal loading control.
MAP2D Is the Only MAP2 Isoform Expressed in the Ovary and Is Expressed Only in Granulosa Cells of PO Follicles and the Corpus Luteum
Our identification of AKAP 80 as MAP2D is the first report of the expression of a member of the MAP2 protein family in the ovary. Since the MAP2 family of proteins was previously thought to be localized primarily to neural cells (40, 41, 60, 61), we sought to compare the expression of MAP2 in ovarian extracts with that of brain extracts. Rats were not injected or were injected with PMSG for 48 h to induce maturation of PA follicles to a PO phenotype or were injected with PMSG for 48 h followed by hCG for 1 h. The ovaries as well as the brains from PMSG-treated rats were harvested, homogenized in detergent-enriched buffer A, and subjected to immunoprecipitation with the MAP2 antibody. A Western blot using the MAP2 antibody revealed that whereas all four isoforms of MAP2 are present in the brain (Fig. 3, lane 4), only the MAP2D isoform is present in the ovary (Fig. 3, lane 4 compared with lanes 2 and 3). These results also demonstrate that MAP2D is only present in ovarian extracts enriched in PO follicles (Fig. 3, lanes 2 and 3, compared with lane 1).
To further explore the expression of MAP2D in the ovary, whole ovarian extracts were analyzed for MAP2D protein during both the follicular and luteal phases of maturation. For Fig. 4A, rats were injected with 25 IU of PMSG to induce the maturation of PA to PO follicles. Some rats were also injected first with 25 IU of PMSG and 48 h later with 25 IU of hCG to induce ovulation (~12 h post-hCG injection) and corpus luteum formation. The ovaries were harvested at the indicated time points, and ovarian extracts were subjected to both an RII overlay assay and Western blot with the MAP2 antibody. Blots were also probed with an antibody to cyclin D2 as a loading control. Consistent with in vitro granulosa cell results seen in Fig. 2A, the expression of MAP2D in vivo is induced by PMSG by 48 h (Fig. 4A, lane 4). Treatment with hCG did not substantially alter the expression of MAP2D; MAP2D expression is maintained throughout the luteal phase (Fig. 4A, lanes 5–10). These results suggest that MAP2D may play a role not only in the PO follicle but also in the corpus luteum. To determine whether MAP2D was restricted to granulosa cells or was expressed in other ovarian cells, we performed immunohistochemistry on paraffin-embedded sections of whole rat ovaries. Rats were injected with PMSG for 48 h or injected with PMSG for 48 h and then with hCG for 48 h to stimulate ovulation and corpus luteum formation. Paraffin-embedded sections were incubated with antibodies to either MAP2 or, as a positive control, inhibin-α. Inhibin-α is induced by PMSG in granulosa cells of PO follicles and is absent from the corpus luteum (33, 62). Protein expression of both MAP2D and inhibin-α was low in PA follicles (Fig. 4B, a and e). PMSG induces the expression of both inhibin-α and MAP2D in the PO follicle (Fig. 4B, b and f). Like inhibin-α expression, the expression of MAP2D is localized to the granulosa cell layer of the PO follicle, with minimal expression in the thecal, stromal, and surface epithelial cells of the ovary (Fig. 4B, c and g). In contrast to inhibin-α expression, which is down-regulated in response to the preovulatory LH surge (62), MAP2D expression is maintained after ovulation and in the cells of the corpus luteum (Fig. 4B, d and h).
Fig. 4. Expression of MAP2D during ovarian differentiation.
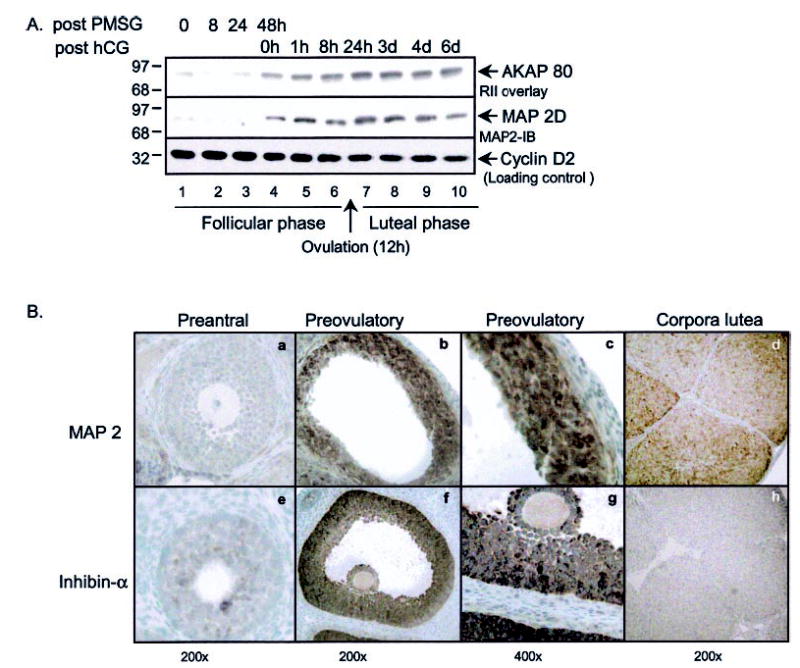
A, rats were not treated or were injected with either 25 IU of PMSG or with 25 IU of PMSG followed by 25 IU of hCG. Ovaries were harvested at the time points indicated after injections. Soluble extracts were made by homogenizing whole ovaries in buffer A, as described under “Experimental Procedures.” Equal concentrations of protein (60 μg) were loaded. Following SDS-PAGE and transfer, blots were subjected to RII overlay assay (top) or Western blotting using anti-MAP2D antibody or, as a loading control, anti-cyclin D2 antibody. Results are representative of two separate experiments. B, rats were injected with either 25 IU of PMSG or with 25 IU of PMSG followed by 25 IU of hCG. Ovaries were harvested 48 h post-PMSG or 48 h post-hCG into 4% paraformaldehyde. Ovarian tissue was sectioned onto glass slides and subjected to immunohistochemistry. -Fold magnification is indicated at the bottom. Results are representative of two separate experiments.
MAP2D Is Phosphorylated in Granulosa Cells
Phosphorylation of the MAP2 family of proteins has been extensively studied in the brain (43, 63). MAP2 phosphorylation has been shown to regulate its association with other proteins as well as the microtubule network (43, 63, 64). At least 15 of the 46 phosphorylation sites present in the high molecular weight MAP2 isoforms are conserved in MAP2D (43). We first sought to determine whether or not an acute treatment of PO granulosa cells with hCG stimulated MAP2 phosphorylation. To this end, granulosa cells from PMSG-primed rats were cultured overnight in the presence of 32Pi and treated the next day for 10 min with either vehicle or hCG. Cell lysates were then subjected to immunoprecipitation using the MAP2 antibody or a mouse monoclonal NI control. MAP2D was already phosphorylated in vehicle-treated cells (Fig. 5A, lane 1), and acute treatment with hCG did not further increase the phosphorylation of MAP2D (Fig. 5A, lane 2). 32P-Labeled MAP2D protein was absent from NI control immunoprecipitations (Fig. 5A, lanes 3 and 4). Since MAP2D was already phosphorylated in vehicle-treated cells, we sought to determine the time point at which MAP2D was phosphorylated relative to its expression using an antibody that recognizes MAP2 phosphorylation on Thr256 and/or Thr259 in the proline-rich domain (MAP2 305 antibody) (43). Granulosa cells from PA follicles were treated with FSH for 0, 24, 48, and 72 h, and total cell lysates were subjected to Western blotting. MAP2 phosphorylation on Thr256 and/or Thr259 matched the expression of MAP2D in response to FSH treatment (Fig. 5B, compare top with middle and lower panels). These results suggest that MAP2D is phosphorylated either coincidentally with or soon after its synthesis at least on Thr256 and/or Thr259.
Fig. 5. Phosphorylation of MAP2D in granulosa cells.
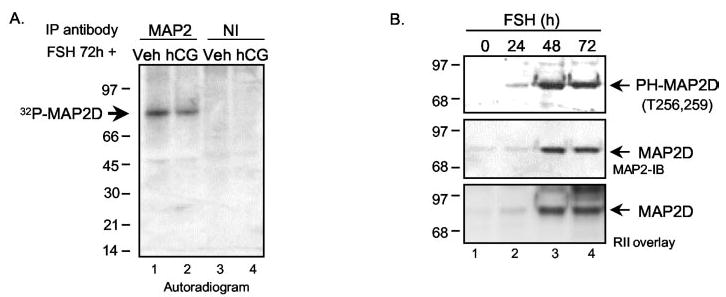
A, PO granulosa cells obtained from rats 48 h after injection of 10 IU of PMSG were incubated overnight with 0.5 mCi of 32Pi to label cellular ATP pools. Granulosa cells were then treated for 10 min with vehicle or 1 IU/ml hCG. Cell extracts were prepared in buffer A and then subjected to immunoprecipitation with indicated antibodies, as detailed under “Experimental Procedures.” Following SDS-PAGE, gel was dried and exposed to film. Results are representative of three separate experiments. B, granulosa cells were treated with 50 ng/ml FSH for the times indicated, and total cell extracts were subjected to Western blotting with either MAP2 antibody or anti-phospho-MAP2 (antibody 305) or to RII overlay. Results are representative of two separate experiments.
MAP2D Associates with the R Subunits of PKA in Granulosa Cells of PO Follicles
We previously showed that AKAP 80 preferentially binds RIIα over RIIβ in an in vitro solid phase overlay assay (37). However, we also showed, following separation of PKA holoenzymes in ovarian extracts by DEAE-cellulose chromatography, that AKAP 80 copurified with DEAE fractions containing PKA IIβ holoenzyme as well as catalytic subunit-free RI and not with PKA IIα holoenzyme or the prominent peak of catalytic subunit-free RII β (8). We therefore sought to ascertain whether MAP2D additionally anchored RI as well as RII subunits in granulosa cells. Most known AKAPs predominately anchor RII and exhibit at least a 100-fold lower affinity for RI (9). A growing number of “dual” AKAPs readily bind RI (12–15, 18), yet these AKAPs still exhibit a 10 –25-fold preference for PKA II (65). However, some recently described AKAPs appear to preferentially bind RI (16 –19).
To determine whether MAP2D additionally bound to RI, we performed immunoprecipitations from ovarian extracts. Under standard immunoprecipitation conditions, neither RI nor RII antibodies immunoprecipitated detectable levels of MAP2D (not shown). However, upon cross-linking of proteins with the reversible cross-linker DSP, RI antibodies immunoprecipitated primarily a slower migrating form of MAP2D from ovarian extracts, whereas detectable levels of MAP2D were never immunoprecipitated with RII antibodies (Fig. 6A). The reverse immunoprecipitations were unsuccessful, because we could not distinguish RI and RII from the prominent IgG bands in our immunoprecipitates. These results suggested that MAP2D could be an RI-preferential AKAP in ovarian extracts. If this is true, then MAP2D and RI should copurify.
Fig. 6. DEAE-cellulose and cAMP-agarose affinity chromatography of PO ovarian extracts.
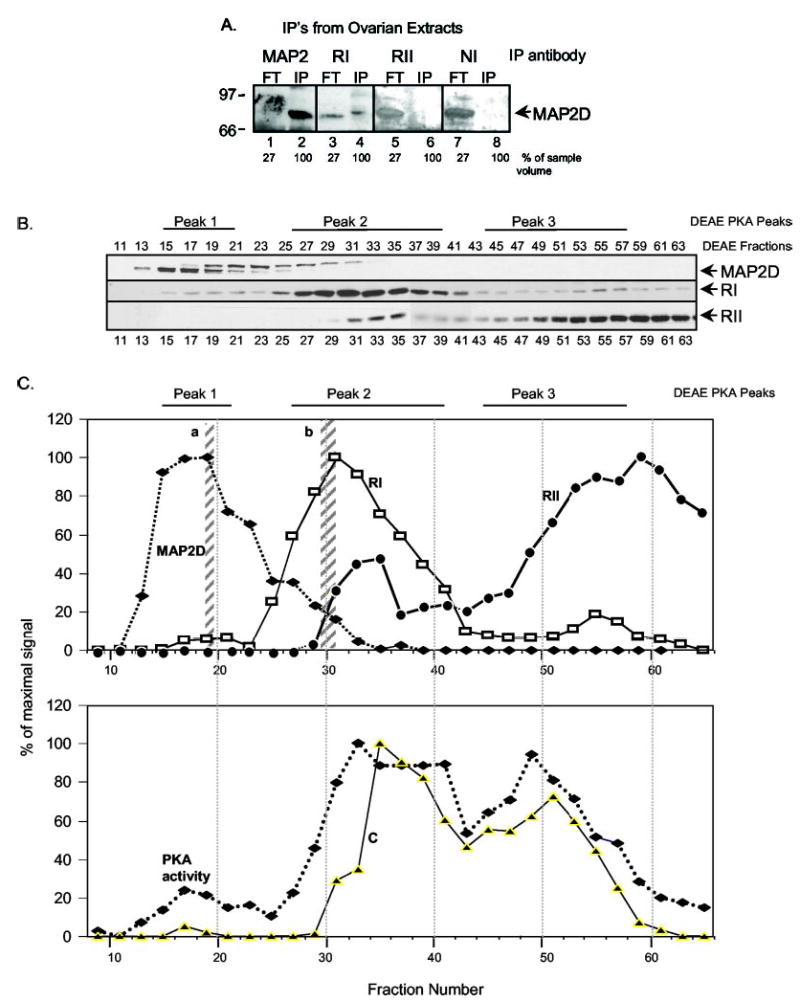
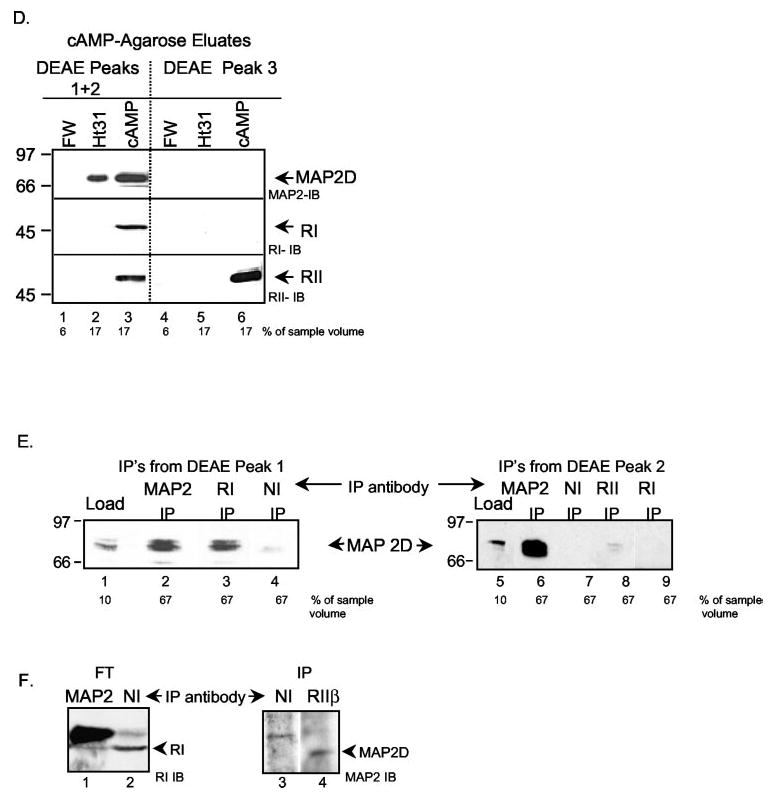
A, detergent-soluble ovarian extracts (800 μg of protein in 300 μl) were prepared (without DTT), subjected to cross-linking by incubating with 1 mM DSP for 15 min at room temperature, and then subjected to immunoprecipitation using the indicated antibodies (anti-MAP2 (Sigma), anti-RI (BD Biosciences), and anti-RII (Upstate Biotechnology)). Flow-through (FT) represents 27% of the extract that was not pulled down by the antibody-agarose complex. IP, immunoprecipitated complex. Samples were subjected to SDS-PAGE and blotted with anti-MAP2 antibody. Results for RII and NI immunoprecipitations are overexposed to confirm the absence of signal in the immunoprecipitation complex lanes. Results are representative of three separate experiments. B, ovaries from 15 rats were harvested 48 h post-PMSG injection and homogenized in buffer E, and a soluble extract was prepared by centrifuging at 105,000 × g for 15 min and loading onto a DEAE-cellulose column. Proteins were eluted with a linear salt gradient, collecting fractions in buffer B but without DTT. Aliquots of odd-numbered fractions were mixed with SDS-PAGE sample buffer, boiled, and subjected to SDS-PAGE and Western blotting with the indicated antibodies. Results are representative of four separate experiments. C, a graphic representation of the Western data presented in B but now normalized to percentage of maximal signal. Also shown is the cAMP-stimulated PKA activity, also normalized to percentage of maximal signal (lower portion of C). In D, fractions from the indicated DEAE-cellulose peak fractions (shown in C) were pooled, concentrated, and incubated with cAMP agarose. The agarose was washed with low and high salt buffers to remove nonspecifically bound proteins, and the final wash (FW) was collected. Specifically bound AKAPs were eluted first with 5 μM Ht31 (Ht31) and then with 75 mM cAMP. The samples were mixed with SDS-PAGE sample buffer and boiled, and aliquots were then subjected to SDS-PAGE and Western blotting using the antibodies indicated. Results are representative of four separate experiments. E, fractions from indicated regions of the DEAE-cellulose column (shown in C) were pooled, 1-ml aliquots were incubated with the protein cross-linker (DSP; 1 μM) for 15 min at room temperature, and samples were subjected to immunoprecipitation with the indicated antibodies (see A). A 100-μl aliquot of the 1-ml starting material was boiled and applied to the gel (Load). Agarose pellets were washed with buffer D, and proteins were eluted from the protein A+G-agarose with 150 μl of Immunopure® elution buffer, pH 2.8, mixed with SDS-PAGE sample buffer, and boiled. 67% of the total eluate (IP) was loaded onto the gel for SDS-PAGE. Results are representative of two separate experiments. F, immunoprecipitations from pooled and concentrated DEAE peak 1 and 2 fractions (lanes 1 and 2) or from PKA IIβ holoenzyme in these fractions that was sedimented by sucrose density gradient centrifugation (lanes 3 and 4) were conducted with Sigma anti-MAP2 antibody, anti-HA (NI), or BD Biosciences anti-RIIβ antibodies. Lanes 1 and 2, immunodepletion results for proteins that were not imunoprecipitated. Lanes 3 and 4, immunoprecipitation results. Results are representative of two independent experiments.
We therefore sought to compare the DEAE-cellulose elution positions of MAP2D, RI, and RII. Ovarian PKAs are separated by DEAE-cellulose chromatography into three peaks of kinase activity (8, 52) (see Fig. 6C). We have previously established that peak 1 consists of the PKA I holoenzyme; peak 2 consists of catalytic subunit-free RI and a PKA IIβ holoenzyme; and peak 3 consists of a second PKA IIβ holoenzyme, a relatively minor PKA IIα holoenzyme, and a prominent peak of catalytic subunit-free RIIβ (8, 52). The elution positions of MAP2D, RI, RII, PKA activity, and the PKA C subunit are shown in Fig. 6, B and C. Notably, the highest concentrations of RI are detected in DEAE peak 2 (fractions 25–39), consistent with the more acidic elution of this prominent peak of catalytic subunit-free RI relative to PKA I holoenzyme (52, 66, 67). The majority of RII elutes in peak 3. The peak of MAP2D co-elutes with the PKA I holoenzyme in peak 1, although a portion of the MAP2D also elutes in the same fractions as RI and RII in peak 2. However, MAP2D does not coelute with the bulk of the RII protein in peak 3.
We then bound PKA R subunits in pooled fractions from DEAE peaks 1 plus 2 versus peak 3 to cAMP-agarose. We compared the elution of MAP2D and PKA R subunits using Ht31, a synthetic peptide derived from the amphipathic helical region of the human thyroid Ht31 AKAP (10), to compete with AKAPs for binding to PKA RII subunits (69), thus competing off RII-AKAPs. cAMP was used to compete off R subunits and remaining RI AKAPs bound to R subunits. Results showed that a portion of the MAP2D in DEAE peaks 1 and 2 was eluted with the RII AKAP Ht31 (Fig. 6D, lane 2), and as expected, none of the R subunits were eluted. cAMP effectively eluted RI and RII as well as a significant fraction of the MAP2D, possibly bound to RI. MAP2D was not detected in DEAE peak 3 cAMP-agarose eluates (Fig. 6D, lanes 4 –6), where the majority of RII protein was present. These results confirm that MAP2D is bound to R subunits in DEAE fractions that contain RI and RII (pooled from DEAE peak 1 and 2 fractions) but is not bound to R subunits in fractions that contain predominately RII (pooled peak 3 fractions). Moreover, these results showing differential elution of MAP2D with the RII AKAP Ht31 versus cAMP suggest that MAP2D is dual AKAP.
To explore more directly the association of MAP2D with RI and RII, we performed coimmunoprecipitation experiments from DEAE column fractions containing MAP2D, RI, and RII. Fractions were incubated with the cross-linking reagent DSP and then subjected to immunoprecipitation with antibodies to MAP2, RI, RII, or NI controls. An aliquot of the starting material representing 10% of the total volume was stopped in SDS-PAGE sample buffer and loaded onto the gel (Fig. 6E, lanes 1 and 5). Bound proteins were eluted with an acidic elution buffer (and are labeled IP). Immunoprecipitation from DEAE peak 1 fractions (region a in Fig. 6C) with either MAP2 or RI antibody pulled down MAP2D protein (Fig. 6E, lanes 2 and 3). However, when immunoprecipitations were conducted with DEAE peak 2 fractions (region b in Fig. 6C), MAP2 antibody and to a much lesser extent RII antibody pulled down MAP2D, whereas MAP2D was undetectable in anti-RI antibody immunoprecipitations (Fig. 6E, lanes 5–9).
These results suggest that MAP2D may preferentially associate with RI over RII but that not all of the RI is bound to MAP2D. Indeed, upon sucrose density gradient centrifugation (52) of DEAE peak 2, the majority of MAP2D and catalytic subunit-free RIα co-sediment with hemoglobin at 4.6 S (not shown), consistent with both RI and MAP2D existing in an unbound state. However, MAP2D also co-sediments with both PKA I and II from DEAE peaks 1 and 2, respectively (not shown), consistent with the formation of a larger complex.
We also evaluated the associations of MAP2D and R subunits by coimmunoprecipitation from DEAE-cellulose fractions corresponding to peaks 1 and 2 that were batch-eluted and then concentrated. Consistent with results shown in Fig. 6E, immunoprecipitation with MAP2D but not with NI antibodies depleted the extract of RI (Fig. 6F, lanes 1 and 2). RII was obscured by the prominent immunoglobulin band in MAP2 immunoprecipitation. Immunoprecipitation with RII but not with NI antibodies diminished RII from sucrose density gradient fractions containing PKA IIβ peak (not shown) and immunoprecipitated MAP2 (Fig. 6F, lanes 3 and 4). These results show that whereas MAP2D appears to strongly associate with RI (see Fig. 6, E and F), it also binds RIIβ in ovarian extracts. We conclude that MAP2D is a dual RI/RII AKAP.
RI and RII Colocalize with MAP2D in the Golgi Apparatus of PO Granulosa Cells
We next sought to determine whether or not an association between MAP2D and RI and RII was evident in PO granulosa cells as detected by immunofluorescence. Granulosa cells from PA follicles were plated on glass cover-slips and treated for 72 h with either vehicle or FSH. The coverslips were fixed, blocked, and incubated with the MAP2D antibody overnight and subsequently with a secondary antibody conjugated to rhodamine or fluorescein labels, as indicated, and then visualized using confocal microscopy. The induction of MAP2D in FSH-treated granulosa cells is readily detected (Fig. 7A, compare a and c). The arrows (Fig. 7A, c and d) mark the location of concentrated amounts of MAP2D in a prominent, phase-dense circular structure of the cell located adjacent to the nucleus, which is not detected in vehicle-treated cells (Fig. 7A, b). Both RI and RII co-localized to this phase-dense structure in PO granulosa cells (Fig. 7B, e–h and i–l), although both RI and RII are also detected throughout the cytoplasm of PO cells.
Fig. 7. Cellular location of MAP2D.
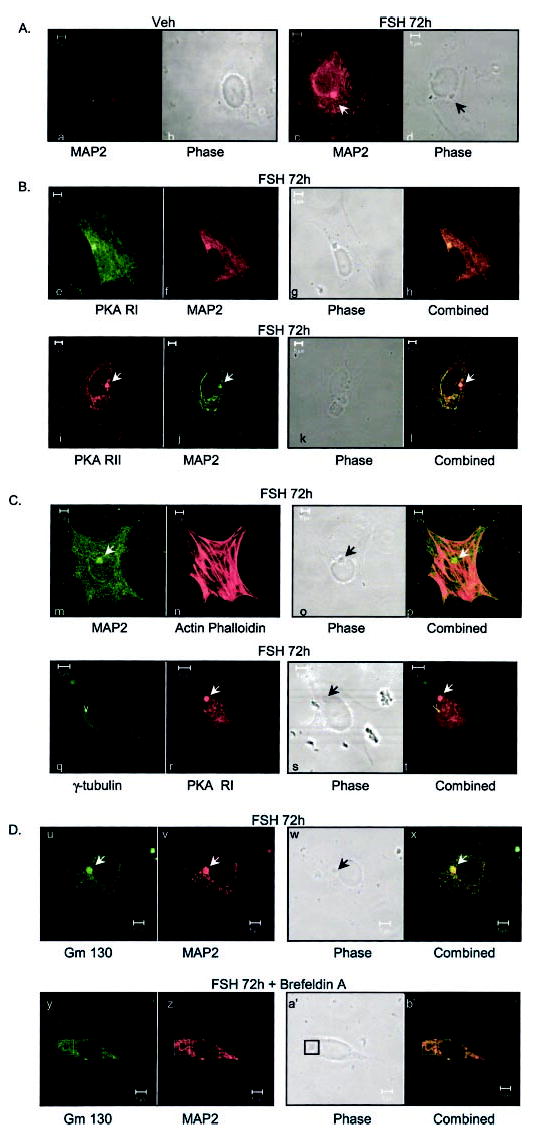
Cells were plated on glass coverslips coated with fibronectin and treated for 72 h with vehicle (Veh) or 50 ng/ml FSH, as indicated. The cells were fixed in 4% paraformaldehyde plus Triton X-100, blocked in 1% bovine serum albumin, and incubated with the indicated primary antibodies overnight at 4 °C at a dilution of 1:200, with the exception of anti-PKA RII and MAP2 in B, in which primary antibodies were at a 1:1000 dilution. Secondary antibodies were added at a dilution of 1:100, with the exception of those for anti-PKA RII and MAP2 in B, in which secondary antibodies were at a 1:200 dilution. Slides were analyzed by confocal microscopy using a Zeiss LSM510 laser-scanning microscope. A phase-contrast image of the cells is also shown. No fluorescent signal was detected in the presence of primary or secondary antibodies alone. Primary and corresponding secondary antibodies used were as follows. A, mouse anti-MAP2 (Sigma) and goat anti-mouse rhodamine; B, e–h, rabbit anti-RI (82) and goat anti-rabbit fluorescein, mouse anti-MAP2, and goat anti-mouse rhodamine; i–l, goat anti-RII (Upstate Biotechnology) and donkey anti-goat rhodamine, mouse anti-MAP2 (Sigma), and donkey anti-mouse fluorescein; C, m–p, mouse anti-MAP2, goat anti-mouse fluorescein, and actin phalloidin; q–t, mouse anti-γ-tubulin (Sigma) and goat anti-mouse fluorescein, rabbit anti-RI, and goat anti-rabbit rhodamine; D, mouse anti-Gm 130 conjugated to fluorescein, mouse anti-MAP2, and goat anti-mouse rhodamine. D, cells were treated for 72 h with FSH and then for 1 h with 10 μM brefeldin A and fixed as described above. Results are representative of at least three separate experiments.
In order to identify this phase-dense structure in PO granulosa cells that appears to concentrate MAP2D, we determined whether this structure reacted with antibodies that mark the centrosome, the Golgi apparatus, or the actin cytoskeleton. Results show that this circular structure does not correspond to the actin cytoskeleton (Fig. 7C, m–p) or the centrosome (Fig. 7C, q–t), based on the inability of actin phalloidin or an antibody to γ-tubulin, respectively, to localize at this spot. However, incubation of coverslips with an antibody to the Golgi matrix protein Gm 130 conjugated to fluorescein together with the MAP2 antibody shows that both MAP2D and Gm 130 localized to this phase-dense, circular structure (Fig. 7D, u–x). Treatment of granulosa cells, cultured on glass coverslips with FSH for 72 h, with 10 μg/ml brefeldin A for 1 h, which has been shown to disrupt the Golgi apparatus as well as to disrupt the Golgi localization of anchor proteins such as AKAP 350 (70), caused a redistribution of MAP2D and Gm 130 out of the circular structure and into the cytosol (Fig. 7D, y–b′). This result provides evidence that MAP2D localizes to the Golgi apparatus in PO granulosa cells. Taken together, these results indicate that MAP2D is localized in granulosa cells at a large circular structure of the Golgi apparatus and suggest that this MAP2D may anchor a portion of the cellular PKA RI and perhaps RII in granulosa cells of PO follicles through its association with the Golgi apparatus.
MAP2D Plays a Functional Role in LH Receptor Signaling Events
To begin to ascertain the physiological role of MAP2D in granulosa cells of PO follicles, we explored the effect of antisense oligonucleotides against MAP2D on LH receptor signaling responses. Granulosa cells from estrogen-primed rats were treated with vehicle or FSH for 72 h and treated with either scrambled or antisense MAP2 oligonucleotides that have been shown to inhibit MAP2 expression in neuronal cells (41). In the presence of the scrambled, control MAP2 oligonucleotide, FSH induced MAP2D expression over vehicle (Fig. 8, compare lanes 1 and 2). In the presence of the antisense MAP2 oligonucleotide, FSH-induced MAP2D expression is decreased by 35 ± 5% (mean ± range, n = 2) relative to total ERK protein compared with MAP2D protein expression in control FSH-treated cells (Fig. 8, compare lanes 2 and 5). The effect of decreased MAP2D expression on CREB phosphorylation stimulated by the LH receptor agonist hCG was examined by treating the cells for 72 h with FSH and subsequently treating with hCG for 10 min in the presence of either the scrambled or the antisense MAP2 oligonucleotide. Under control conditions, hCG stimulated CREB phosphorylation (Fig. 8, compare lanes 2 and 3). When MAP2D expression was decreased by MAP2D antisense oligonucleotides, hCG-stimulated CREB phosphorylation was inhibited 62 ± 12% (mean ± range, n = 2) relative to total ERK protein (Fig. 8, compare lanes 3 and 6). However, hCG-stimulated histone H3 phosphorylation was not reduced when MAP2D protein levels were reduced (not shown). These results suggest that MAP2D may play a crucial role in acute select PKA-mediated hCG-stimulated signaling events in granulosa cells.
Fig. 8. Effect of reduced MAP2D expression on LH receptor signaling to CREB.
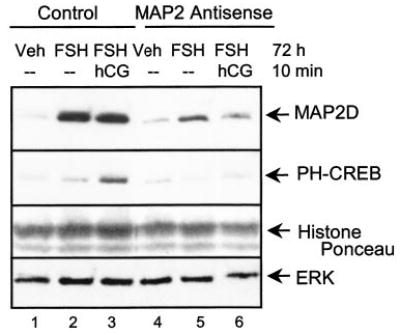
Granulosa cells from estrogen-primed rats were plated on tissue culture plates coated with 100 ng/ml poly-L-lysine and treated with vehicle (Veh) or FSH for 72 h or with FSH for 72 h followed by 1 IU/ml hCG (hCG) for 10 min in the presence of either scrambled (Control) or antisense (Antisense) oligonucleotides against MAP2 as described under “Experimental Procedures.” Oligonucleotides were added at a concentration of 10 μg/ml every 12 h during the 72-h incubation period. Cells were harvested in SDS-PAGE sample buffer and subjected to Western blotting with antibodies against MAP2 (A, top) or phospho-CREB (PH-CREB) (middle). Protein load was determined using an antibody that detects total ERK and by Ponceau S staining (bottom). Results are representative of two independent experiments.
DISCUSSION
Our results demonstrate that the 80-kDa AKAP induced by FSH as ovarian granulosa cells mature from a PA to a PO phenotype corresponds to the neuronal protein MAP2D. MAP2 has been extensively studied in brain tissue not only as a microtubule-binding protein necessary for neurite outgrowth (38) but also as an RII-binding protein (47, 48). MAP2D is a low molecular weight splice variant of the MAP2 protein that is expressed predominately in adult brain (44) and functions to stabilize microtubules and especially microfilaments (42). Our results show that only the MAP2D isoform, and not the other MAP2 family members, is expressed in the ovary and that its expression is induced by FSH and restricted to PO granulosa cells. Moreover, whereas MAP2D is distributed throughout the granulosa cell, it is concentrated at a distinct, phase-dense circular region of the Golgi apparatus, as evidenced by its ability to co-localize with the Golgi marker Gm 130 and the ability of brefeldin A to redistribute MAP2D out of this circular structure. MAP2D does not localize to the actin cytoskeleton or the centrosome (the microtubule-organizing center), as evidenced by its inability to co-localize with actin phalloidin or γ-tubulin. These results suggest that, in contrast to its localization in neurons, the portion of MAP2D that is concentrated at the Golgi in PO granulosa cells does not appear to be associated with the microfilament or microtubular networks present in granulosa cells (71, 72). However, since microtubules are important for the cellular position and organization of the Golgi (73, 74), additional studies are required to determine whether the MAP2D in granulosa cells is associated with the microtubules that are specifically associated with the Golgi apparatus. Additional studies are also required to determine whether the remaining MAP2D not associated with the Golgi is associated with the microtubule network in granulosa cells.
Neuronal MAP2 contains a large number of phosphorylation sites that can be phosphorylated by a variety of kinases including PKA, ERK, glycogen synthase kinase 3β, casein kinase II, cyclin-dependent kinases, and protein kinase C (43). It is generally believed that the phosphorylation of MAP2 reduces its association with microtubules (43). At least 15 of the 46 phosphorylation sites present in MAP2A/2B are conserved in MAP2D (43). MAP2C, which is identical to MAP2D except that it is missing the fourth microtubule binding domain present in MAP2D (45), is phosphorylated by glycogen synthase kinase 3β in the proline-rich domain, resulting in an inhibition of microtubule bundling (64), and by PKA at KXGS motifs, resulting in a disruption of MAP2C-microtubule interaction and increased localization of MAP2C with actin at membrane ruffles (63). In granulosa cells from PO follicles, we have shown, upon labeling of intracellular ATP pools with 32Pi, that MAP2D is phosphorylated in the absence of acute hormone stimulation. Acute LH receptor signaling in response to the LH receptor agonist hCG has no detectable effect on the amount of phosphorylated MAP2D protein; how-ever, it is possible that hCG stimulates a rearrangement of phosphorylated sites on MAP2D to modulate its association with cellular organelles or other proteins. MAP2D phosphorylation on Thr256/Thr259 in the proline-rich domain, which immediately precedes the four microtubule binding domains, appears to be coincident with MAP2D expression in response to FSH treatment; however, the consequence of this phosphorylation has not yet been determined. Since it is well documented that MAP2 phosphorylation reduces its affinity for microtubules (43, 63), our results suggest that the majority of MAP2D in PO granulosa cells might not be associated with the microtubule network that extends from the centrosome to the cells’ periphery. It is possible, however, that the phosphorylation of MAP2D on Thr256/Thr259 or on other sites regulates its association with the microtubule network selectively associated with the Golgi apparatus in PO granulosa cells.
Most of the AKAPs identified have been shown to bind RII subunits with at least a 100-fold higher affinity than RI (9). Even the dual specificity AKAPs, such as sAKAP-84 and DAKAP 1 and 2, bind RII with as much as a 25-fold higher affinity over RI (65) and appear to contain overlapping RI/RII binding domains (76). The sperm fibrous sheath protein FSC1 contains two separate RI binding domains, one of which also binds RII, and this protein preferentially pulls down RI from testis extracts (18). Similarly, the neurofibromatosis 2 tumor suppressor protein merlin (16) as well as the peripheral type benzodiazepine receptor-associated protein PAP7 (19) preferentially binds RI. MAP2 contains a well characterized RII binding domain in its N-terminal region (48) that in vitro preferentially binds RIIα over RIIβ (78). Using a solid phase in vitro RII overlay binding assay, we showed that MAP2D also preferentially binds RIIα over RIIβ (37). These results establish that MAP2D is an RII AKAP. Although Therakauf et al. (47) showed that purified brain microtubules bound equal amounts of RIIβ and RIIβ but did not bind RI, using a cAMP photoaffinity ligand to identify RI and RII subunits, fractionation and immunoprecipitation data from granulosa cells suggest that MAP2D is a dual AKAP that appears to preferentially associate with RI. This result suggests a unique role for MAP2D as a selective RI-binding AKAP in granulosa cells of the PO follicle. However, the PKA I holoenzyme comprises a very small fraction (~5%) of the PKA holoenzyme activity in PO granulosa cells (8) (see Fig. 6C). Although the predominant PKA holoenzyme contains RIIβ subunits, the RIIα knock-out mouse is fertile (79), and RIIα is compensated for by an increase in RI, at least in adipose tissue (80). Our results show that whereas MAP2D and RI coelute upon ion exchange chromatography of PO ovarian extracts (see Fig. 6C) and coimmunoprecipitate (see Fig. 6E), MAP2D also coelutes and coimmunoprecipitates with RII. However, not all of the MAP2D in granulosa cells is bound to R subunits. Likewise, most of the RIIβ and RIα do not appear to be associated with MAP2D, based on their distinct elution positions from DEAE-cellulose (see Fig. 6C). Both RI and RII colocalize with MAP2D in the Golgi matrix, as evidenced by confocal microscopy. However, there are other AKAPs associated with the Golgi apparatus that appear to preferentially bind either RII, such as MTG (81), an unidentified 85-kDa AKAP (57), and AKAP 350 (70) or RI, such as PAP7 (19). Thus, RI and RII localized to the Golgi are not exclusively bound to MAP2D. Taken together, these results suggest that MAP2D binds RI and RII; that the association of MAP2D with RI and RII is tightly regulated, since significant fractions of MAP2D, RI, and RII are not associated; and that the binding of RI and RII to MAP2D probably reflects distinct cellular localizations of these proteins.
Inhibition of MAP2 expression using antisense oligonucleotides in cultured rat cortical neurons inhibits elongation of neurites as well as causing destabilization of microtubules (41). Inhibition of MAP2D expression in PO granulosa cells inhibited the ability of hCG to effectively stimulate the PKA-mediated phosphorylation of the signaling intermediate CREB. This result suggests that MAP2D may play a functional role in acute LH receptor signaling events, possibly by acting as a molecular scaffold that coordinates the actions of PKA to phosphorylate select substrates. Perhaps MAP2D is a key component of the mechanism that distinguishes PKA signaling in granulosa cells of PA follicles compared with that of PO follicles.
In summary, we have shown that the neuronal protein MAP2D is also expressed in ovarian granulosa cells in response to the hormone FSH. We identify MAP2D as an apparent dual RI/RII AKAP based on its ability to coimmunoprecipitate with RI and RII. MAP2D in granulosa cells is predominately localized to the Golgi and not to the microtubule-organizing center and appears to be important for LH receptor signaling. Thus, the function of MAP2D in granulosa cells is expected to be quite novel and distinct from its established neuronal roles.
Footnotes
This work was funded by National Institutes of Health (NIH) Grant P01 HD21921 (to M. H. D.), a Veterans Affairs Merit Grant and NIH Grants HD36408 (to D. W. C.), and NIH Grant DK48239 (to J. D. S.).
The abbreviations used are: PA, preantral; AKAP, A-kinase anchoring protein; C subunit, catalytic subunit of PKA; CREB, cAMP-response element-binding protein; DEX, dexamethasone; DSP, dithiobis[succin-imidylpropionate]; ERK, extracellular signal-regulated kinase; FSH, follicle-stimulating hormone; hCG, human chorionic gonadotropin; LH, luteinizing hormone; MAP2, microtubule-associated protein 2; NI, non-immune; PKA, protein kinase A; PO, preovulatory; PMSG, pregnant mares serum gonadotropin; R subunit, PKA regulatory subunit; HA, hemagglutinin; DTT, dithiothreitol; PBS, phosphate-buffered saline.
References
- 1.McGee EA, Hsueh AJ. Endocr Rev. 2000;21:200 –214. doi: 10.1210/edrv.21.2.0394. [DOI] [PubMed] [Google Scholar]
- 2.Richards JS. Physiol Rev. 1980;60:51–89. doi: 10.1152/physrev.1980.60.1.51. [DOI] [PubMed] [Google Scholar]
- 3.Robinson-White A, Stratakis CA. Ann N Y Acad Sci. 2002;968:256 –270. doi: 10.1111/j.1749-6632.2002.tb04340.x. [DOI] [PubMed] [Google Scholar]
- 4.Taylor SS. J Biol Chem. 1989;264:8443–8446. [PubMed] [Google Scholar]
- 5.Dell’Acqua ML, Scott JD. J Biol Chem. 1997;272:12881–12884. doi: 10.1074/jbc.272.20.12881. [DOI] [PubMed] [Google Scholar]
- 6.Hunzicker-Dunn M, Lorenzini NA, Lynch LL, West DE. J Biol Chem. 1985;260:13360 –13369. [PubMed] [Google Scholar]
- 7.Hunzicker-Dunn M, Maizels ET, Kern LC, Ekstrom RC, Constantinou AI. Mol Endocrinol. 1989;3:780 –789. doi: 10.1210/mend-3-5-780. [DOI] [PubMed] [Google Scholar]
- 8.Carr DW, Cutler RE, Jr, Cottom JE, Salvador LM, Fraser IDC, Scott JD, Hunzicker-Dunn M. Biochem J. 1999;344:613–623. [PMC free article] [PubMed] [Google Scholar]
- 9.Michel JJ, Scott JD. Annu Rev Pharmacol Toxicol. 2002;42:235–257. doi: 10.1146/annurev.pharmtox.42.083101.135801. [DOI] [PubMed] [Google Scholar]
- 10.Carr DW, Hausken ZE, Fraser IDC, Stofko-Hahn RE, Scott JD. J Biol Chem. 1992;267:13376 –13382. [PubMed] [Google Scholar]
- 11.Carr DW, Stofko-Hahn RE, Fraser IDC, Bishop SM, Acott TS, Brennan RG, Scott JD. J Biol Chem. 1991;266:14188 –14192. [PubMed] [Google Scholar]
- 12.Reinton N, Collas P, Haugen TB, Skalhegg BS, Hansson V, Jahnsen T, Tasken K. Dev Biol. 2000;223:194 –204. doi: 10.1006/dbio.2000.9725. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Adamik R, Pacheco-Rodriguez G, Moss J, Vaughan M. Proc Natl Acad Sci U S A. 2003;100:1627–1632. doi: 10.1073/pnas.0337678100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang LJ, Wang L, Ma Y, Durick K, Perkins G, Deerinck TJ, Ellisman MH, Taylor SS. J Cell Biol. 1999;145:951–959. doi: 10.1083/jcb.145.5.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. Proc Natl Acad Sci U S A. 1997;94:11184 –11189. doi: 10.1073/pnas.94.21.11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gronholm M, Vossebein L, Carlson CR, Kuja-Panula J, Teesalu T, Alfthan K, Vaheri A, Rauvala H, Herberg FW, Tasken K, Carpen O. J Biol Chem. 2003;278:41167–41172. doi: 10.1074/jbc.M306149200. [DOI] [PubMed] [Google Scholar]
- 17.Angelo R, Rubin CS. J Biol Chem. 1998;273:14633–14643. doi: 10.1074/jbc.273.23.14633. [DOI] [PubMed] [Google Scholar]
- 18.Miki K, Eddy EM. J Biol Chem. 1998;273:34384 –34390. doi: 10.1074/jbc.273.51.34384. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Degenhardt B, Tobin D, Yao ZX, Tasken K, Papadopoulos V. Mol Endocrinol. 2001;15:2211–2228. doi: 10.1210/mend.15.12.0736. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Ndubuka C, Rubin CS. J Biol Chem. 1996;271:16862–16869. doi: 10.1074/jbc.271.28.16862. [DOI] [PubMed] [Google Scholar]
- 21.Dong F, Feldmesser M, Casadevall A, Rubin CS. J Biol Chem. 1998;273:6533–6541. doi: 10.1074/jbc.273.11.6533. [DOI] [PubMed] [Google Scholar]
- 22.Dodge K, Scott JD. FEBS Lett. 2000;476:58 –61. doi: 10.1016/s0014-5793(00)01671-9. [DOI] [PubMed] [Google Scholar]
- 23.Lin RY, Moss SR, Rubin CS. J Biol Chem. 1995;270:27804 –27811. doi: 10.1074/jbc.270.46.27804. [DOI] [PubMed] [Google Scholar]
- 24.Ginsberg MD, Feliciello A, Jones JK, Avvedimento EV, Gottesman ME. J Mol Biol. 2003;327:885–897. doi: 10.1016/s0022-2836(03)00173-6. [DOI] [PubMed] [Google Scholar]
- 25.Keryer G, Rios RM, Landmark BF, Skalhegg B, Lohmann SM, Bornens M. Exp Cell Res. 1993;204:230 –240. doi: 10.1006/excr.1993.1029. [DOI] [PubMed] [Google Scholar]
- 26.Diviani D, Langeberg LK, Doxsey SJ, Scott JD. Curr Biol. 2000;10:417–420. doi: 10.1016/s0960-9822(00)00422-x. [DOI] [PubMed] [Google Scholar]
- 27.Steen RL, Beullens M, Landsverk HB, Bollen M, Collas P. J Cell Sci. 2003;116:2237–2246. doi: 10.1242/jcs.00432. [DOI] [PubMed] [Google Scholar]
- 28.Pawson T, Scott JD. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 29.DeManno DA, Cottom JE, Kline MP, Peters CA, Maizels ET, Hunzicker-Dunn M. Mol Endocr. 1999;13:91–105. doi: 10.1210/mend.13.1.0222. [DOI] [PubMed] [Google Scholar]
- 30.Mukherjee A, Park-Sarge OK, Mayo KE. Endocrinology. 1996;137:3234 –3245. doi: 10.1210/endo.137.8.8754745. [DOI] [PubMed] [Google Scholar]
- 31.Pei L, Dodson R, Schoderbek WE, Maurer RA, Mayo KE. Mol Endocr. 1991;5:521–534. doi: 10.1210/mend-5-4-521. [DOI] [PubMed] [Google Scholar]
- 32.Cottom J, Salvador LM, Maizels ET, Reierstad S, Park Y, Carr DW, Davare MA, Hell JW, Palmer SS, Dent P, Kawakatsu H, Ogata M, Hunzicker-Dunn M. J Biol Chem. 2003;278:7167–7179. doi: 10.1074/jbc.M203901200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richards JS. Endocr Rev. 1994;15:725–751. doi: 10.1210/edrv-15-6-725. [DOI] [PubMed] [Google Scholar]
- 34.Hsueh AJW, Adashi EY, Jones PBC, Welsh TH., Jr Endocr Rev. 1984;5:76 –110. doi: 10.1210/edrv-5-1-76. [DOI] [PubMed] [Google Scholar]
- 35.Richards JS, Fitzpatrick SL, Clemens JW, Morris JK, Alliston T, Sirois J. Rec Prog Horm Res. 1995;50:223–254. doi: 10.1016/b978-0-12-571150-0.50014-7. [DOI] [PubMed] [Google Scholar]
- 36.Salvador LM, Maizels E, Hales DB, Miyamoto E, Yamamoto H, Hunzicker-Dunn M. Endocrinol. 2002;143:2986 –2994. doi: 10.1210/endo.143.8.8976. [DOI] [PubMed] [Google Scholar]
- 37.Carr DW, DeManno DA, Atwood A, Hunzicker-Dunn M, Scott JD. J Biol Chem. 1993;268:20729 –20732. [PubMed] [Google Scholar]
- 38.Matus A. J Cell Sci (Suppl) 1991;15:61–67. doi: 10.1242/jcs.1991.supplement_15.9. [DOI] [PubMed] [Google Scholar]
- 39.Doll T, Meichsner M, Riederer BM, Honegger P, Matus A. J Cell Sci. 1993;106:633–639. doi: 10.1242/jcs.106.2.633. [DOI] [PubMed] [Google Scholar]
- 40.Lim RW, Halpain S. J Biol Chem. 2000;275:20578 –20587. doi: 10.1074/jbc.M001887200. [DOI] [PubMed] [Google Scholar]
- 41.Sharma N, Kress Y, Shafit-Zagardo B. Cell Motil Cytoskeleton. 1994;27:234 –247. doi: 10.1002/cm.970270305. [DOI] [PubMed] [Google Scholar]
- 42.Ferhat L, Represa A, Bernard A, Ben-Ari Y, Khrestchatisky M. J Cell Sci. 1996;109:1095–1103. doi: 10.1242/jcs.109.5.1095. [DOI] [PubMed] [Google Scholar]
- 43.Sanchez C, Diaz-Nido J, Avila J. Prog Neurobiol. 2000;61:133–168. doi: 10.1016/s0301-0082(99)00046-5. [DOI] [PubMed] [Google Scholar]
- 44.Ferhat L, Represa A, Ferhat W, Ben-Ari Y, Khrestchatisky M. Eur J Neurosci. 1998;10:161–171. doi: 10.1046/j.1460-9568.1998.00044.x. [DOI] [PubMed] [Google Scholar]
- 45.Ferhat L, Bernard A, Ribas dP, Ben-Ari Y, Khrestchatisky M. Neurochem Int. 1994;25:327–338. doi: 10.1016/0197-0186(94)90139-2. [DOI] [PubMed] [Google Scholar]
- 46.Ferhat L, Ben-Ari Y, Khrestchatisky M. Comptes Rendus l’Academie Sciences Ser 3 Sci Vie. 1994;317:304 –309. [PubMed] [Google Scholar]
- 47.Theurkauf WE, Vallee RB. J Biol Chem. 1982;257:3284 –3290. [PubMed] [Google Scholar]
- 48.Rubino HM, Dammerman M, Shafit-Zagardo B, Erlichman J. Neuron. 1989;3:631–638. doi: 10.1016/0896-6273(89)90273-0. [DOI] [PubMed] [Google Scholar]
- 49.Lowry OW, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 50.Hunzicker-Dunn M. J Biol Chem. 1981;256:12185–12193. [PubMed] [Google Scholar]
- 51.Rudolph SA, Krueger BK. Adv Cyclic Nucleotide Res. 1979;10:107–133. [PubMed] [Google Scholar]
- 52.Hunzicker-Dunn M, Cutler RE, Jr, Maizels ET, DeManno DA, Lamm MLG, Erlichman J, Sanwal BD, LaBarbera AR. J Biol Chem. 1991;266:7166 –7175. [PubMed] [Google Scholar]
- 53.Scott JD, Stofko RE, McDonald JR, Comer JD, Vitalis EA, Mangili JA. J Biol Chem. 1990;265:21561–21566. [PubMed] [Google Scholar]
- 54.Coghlan VM, Langeberg LK, Fernandex A, Lamb NJC, Scott JD. J Biol Chem. 1994;269:7658 –7665. [PubMed] [Google Scholar]
- 55.Park Y, Freedman BF, Lee EJ, Jameson LJ. Diabetologia. 2003;46:365–377. doi: 10.1007/s00125-003-1037-4. [DOI] [PubMed] [Google Scholar]
- 56.Yu RN, Ito M, Jameson JL. Mol Endocrinol. 1998;12:1010 –1022. doi: 10.1210/mend.12.7.0131. [DOI] [PubMed] [Google Scholar]
- 57.Rios RM, Celati C, Lohmann SM, Borens M, Keryer G. EMBO J. 1992;11:1723–1731. doi: 10.1002/j.1460-2075.1992.tb05224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hunzicker-Dunn M, Scott JD, Carr DW. Biol Repro. 1998;58:1496 –1502. doi: 10.1095/biolreprod58.6.1496. [DOI] [PubMed] [Google Scholar]
- 59.Hernandez MA, Avila J, Andreu JM. Eur J Biochem. 1986;154:41–48. doi: 10.1111/j.1432-1033.1986.tb09356.x. [DOI] [PubMed] [Google Scholar]
- 60.Matsunaga W, Miyata S, Itoh M, Kiyohara T, Maekawa S. Neuroscience. 2002;111:151–162. doi: 10.1016/s0306-4522(01)00607-8. [DOI] [PubMed] [Google Scholar]
- 61.Tucker RP. Brain Res Brain Res Rev. 1990;15:101–120. doi: 10.1016/0165-0173(90)90013-e. [DOI] [PubMed] [Google Scholar]
- 62.Woodruff TK, D’Agostino JB, Schwartz NB, Mayo KE. Endocrinology. 1989;124:2193–2199. doi: 10.1210/endo-124-5-2193. [DOI] [PubMed] [Google Scholar]
- 63.Ozer RS, Halpain S. Mol Biol Cell. 2000;11:3573–3587. doi: 10.1091/mbc.11.10.3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanchez C, Perez M, Avila J. Eur J Cel Biol. 2000;79:252–260. doi: 10.1078/s0171-9335(04)70028-x. [DOI] [PubMed] [Google Scholar]
- 65.Diviani D, Scott JD. J Cell Sci. 2001;114:1431–1437. doi: 10.1242/jcs.114.8.1431. [DOI] [PubMed] [Google Scholar]
- 66.Malkinson AM, Butley MS. Cancer Res. 1981;41:1334 –1341. [PubMed] [Google Scholar]
- 67.Corbin JD, Keely SL, Park CR. J Biol Chem. 1975;250:218 –225. [PubMed] [Google Scholar]
- 68.Deleted in proof
- 69.Herberg FW, Maleszka A, Eide T, Vossebein L, Tasken K. J Mol Biol. 2000;298:329 –339. doi: 10.1006/jmbi.2000.3662. [DOI] [PubMed] [Google Scholar]
- 70.Shanks RA, Steadman BT, Schmidt PH, Goldenring JR. J Biol Chem. 2002;277:40967–40972. doi: 10.1074/jbc.M203307200. [DOI] [PubMed] [Google Scholar]
- 71.Herman B, Albertini DF. Cell Motil. 1982;2:583–597. doi: 10.1002/cm.970020607. [DOI] [PubMed] [Google Scholar]
- 72.Albertini DF, Clark JI. Cell Biol Int Rep. 1981;5:387–397. doi: 10.1016/0309-1651(81)90009-6. [DOI] [PubMed] [Google Scholar]
- 73.Allan VJ, Thompson HM, McNiven MA. Nat Cell Biol. 2002;4:E236 –E242. doi: 10.1038/ncb1002-e236. [DOI] [PubMed] [Google Scholar]
- 74.Thyberg J, Moskalewski S. Exp Cell Res. 1999;246:263–279. doi: 10.1006/excr.1998.4326. [DOI] [PubMed] [Google Scholar]
- 75.Deleted in proof
- 76.Huang LJ, Durick K, Weiner JA, Chun J, Taylor SS. J Biol Chem. 1997;272:8057–8064. doi: 10.1074/jbc.272.12.8057. [DOI] [PubMed] [Google Scholar]
- 77.Deleted in proof
- 78.Valle RB, DiBartolomeis MJ, Theurkauf WE. J Cell Biol. 1981;90:568 –576. doi: 10.1083/jcb.90.3.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cummings DE, Brandon EP, Planas JV, Motamed K, Idzerda RL, McKnight GS. Nature. 1996;382:622–626. doi: 10.1038/382622a0. [DOI] [PubMed] [Google Scholar]
- 80.Planas JV, Cummings DE, Idzerda RL, Knight GS. J Biol Chem. 1999;274:36281–36287. doi: 10.1074/jbc.274.51.36281. [DOI] [PubMed] [Google Scholar]
- 81.Schillace RV, Andrews SF, Liberty GA, Davey MP, Carr DW. J Immunol. 2002;168:1590 –1599. doi: 10.4049/jimmunol.168.4.1590. [DOI] [PubMed] [Google Scholar]
- 82.Vijayaraghavan S, Olson GE, Nag Das S, Winfrey VP, Carr DW. Biol Reprod. 1997;57:1517–1523. doi: 10.1095/biolreprod57.6.1517. [DOI] [PubMed] [Google Scholar]