

Abstract
The mechanisms of the sustained vasodilator actions of corticotrophin-releasing factor (CRF) and sauvagine (SVG) were studied using rings of endothelium de-nuded rat thoracic aorta (RTA) and the isolated perfused rat superior mesenteric arterial vasculature (SMA).
SVG was ≈amp;50 fold more potent than CRF on RTA (EC40: 0.9±0.2 and 44±9 nM respectively, P<0.05), and ≈amp;10 fold more active in the perfused SMA (ED40: 0.05±0.02 and 0.6±0.1 nmol respectively, P<0.05). Single bolus injections of CRF (100 pmol) or SVG (15 pmol) in the perfused SMA caused reductions in perfusion pressure of 23±1 and 24±2% that lasted more than 20 min.
Removal of the endothelium in the perfused SMA with deoxycholic acid attenuated the vasodilatation and revealed two phases to the response; a short lasting direct action, and a sustained phase which was fully inhibited.
Inhibition of nitric oxide synthase with L-NAME (100 μM) L-NMMA (100 μM) or 2-ethyl-2-thiopseudourea (ETPU, 100 μM) had similar effects on the vasodilator responses to CRF as removal of the endothelium, suggesting a pivotal role for nitric oxide. However the selective guanylate cyclase inhibitor 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 10 μM) did not affect the response to CRF.
High potassium (60 mM) completely inhibited the vasodilator response to CRF in the perfused SMA, indicating a role for K+ channels in this response.
Compared to other vasodilator agents acting via the release of NO, the actions of CRF and SVG are strikingly long-lasting, suggesting a novel mechanism of prolonged activation of nitric oxide synthase.
Keywords: Corticotrophin-releasing factor, sauvagine, nitric oxide, endothelium, adenylate cyclase, guanylate cyclase, K+ channels, vasodilatation, CRF receptor
Introduction
Corticotrophin-releasing factor (CRF) was originally identified as the principle hypothalamic regulator of corticotrophin (ACTH) secretion from the anterior pituitary gland (Vale et al., 1981). During the same period two further peptides with structural homology and similar biological actions to CRF were isolated and characterized – namely, sauvagine (SVG), from the skin of the South American frog Phyllomedusa Sauvagei (Montecucchi et al., 1980), and urotensin I (UI) from the urophysis of the teleost fish Catostomus commersoni (Lederis & Medakovic, 1974; Lederis et al., 1982). Subsequent studies have shown both amphibians and teleost fish to possess their own CRF molecules (Morley et al., 1991; Stenzel-Poore et al., 1992). The cloning from rat brain of urocortin, a peptide with greater structural homology to UI than to CRF (Vaughan et al., 1995), has shown that mammals also have more than one CRF-like peptide. This indicates that some physiological functions ascribed to CRF may involve other endogenous members of this family of peptides.
Current evidence suggests important peripheral actions of CRF or a structurally homologous peptide from this family. Intravenous injection of CRF results in an increase in blood glucose levels (Brown et al., 1982a,1982b), and inhibition of both gastric emptying and intestinal transit, whilst increasing colonic transport (Sheldon et al., 1990; Williams et al., 1987). Systemic administration of CRF also causes a pronounced reduction in blood pressure with a transient increase in heart rate in a number of species including rat, dog and primates (Gardiner et al., 1988; Corder et al., 1992; Lenz et al., 1985; MacCannell et al., 1982; Schurmeyer et al., 1985; Udelsman et al., 1986). Two distinct CRF receptors have been cloned, one of which is found in the heart and skeletal muscle and displays higher affinity for SVG and UI (Chang et al., 1993; Kishimoto et al., 1995). This is consistent with the greater potency of these peptides, compared to CRF, at decreasing blood pressure and dilating the mesenteric vasculature (MacCannell et al., 1982; Lenz et al., 1985).
The hypotensive action of these peptides is primarily due to widespread vasodilatation, including the mesenteric vasculature and hindquarters (Gardiner et al., 1988; MacCannell et al., 1982), and is thought to be a direct action of CRF on blood vessels. Studies with mesenteric resistance arteries (Lei et al., 1993) and the isolated heart (Grunt et al., 1993) support this, as does work showing the binding of [125I]-Tyr-CRF to rabbit aorta (Dashwood et al., 1987). However the exact mechanism of action remains unclear, with evidence for a direct action on vascular smooth muscle mediated by increased cyclic AMP levels (Lei et al., 1993; Gerritsen & Lederis, 1979), or an indirect action for instance in the the coronary circulation where the response is partly endothelium-dependent and attenuated by L-NG-monomethylarginine (L-NMMA) and indomethacin (Grunt et al., 1993). Consistent with our initial investigations (Barker & Corder, 1995), recent studies in the rat aorta have reported a partial inhibition of the response by removal of the endothelium, L-NG-nitroarginine methyl ester (L-NAME), or LY83583, an inhibitor of guanylate cyclase (Jain et al., 1997).
We have investigated the vasodilator response to CRF using the isolated perfused superior mesenteric arterial vasculature (SMA) of the rat, and compared its actions with those of SVG. The role of the endothelium in the vasodilator action was determined by its removal with the detergent deoxycholic acid, selective inhibition of nitric oxide synthase (NOS) or inhibition of guanylate cyclase with 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, Moro et al., 1996). The involvement of potassium channels in the vasodilator mechanism was evaluated using high extracellular potassium chloride, the non-selective potassium channel blocker tetraethylammonium (TEA), and the ATP-sensitive potassium channel blocker glibenclamide (Langton et al., 1991; Edwards & Weston, 1993). Direct vasodilator effects of CRF and SVG were compared using rings of rat thoracic aorta (RTA) where the endothelium had been removed.
Methods
Isolated perfused rat superior mesenteric arterial vasculature
Male Wistar rats (A. Tuck & Son, Battlebridge, U.K.) weighing 200–400 g were anaesthetized with pentobarbitone (60 mg kg−1, i.p.). A midline incision was made and the superior mesenteric artery was located, cleared of surrounding tissue, cannulated and perfused with heparinized Krebs solution (100 U ml−1) at 2.5 ml min−1. The superior mesenteric arterial vascular bed was separated from the intestines and transferred to the perfusion apparatus where it was perfused at 5 ml min−1 with Krebs solution (mM); NaCl 118, NaHCO3 25, KCl 4.7, MgSO4 1.2, KH2PO4 0.7, CaCl2 2.5 and glucose 5.6, warmed to 37°C and gassed with 95%O2 : 5%CO2. Perfusion pressure was recorded on a Gould RS3400 recording system, via Gould P23XL pressure transducers. A 15 min stabilization period was allowed, after which an infusion of methoxamine (final concentration 50 μM) was started. A further stabilization period was allowed before testing the endothelium-dependent and endothelium-independent vasodilator responses by administration of single bolus doses of acetylcholine (ACh, 100 pmol), sodium nitroprusside (SNP, 1 nmol), and isoprenaline (1 nmol, for evaluating the effects of detergent and L-NAME only).
Dose response curves to CRF or SVG were constructed using doses in the range 10 pmol–1 nmol. For subsequent investigations single bolus doses of CRF (100 pmol) or SVG (15 pmol) were used, as these gave comparable reductions in perfusion pressure (PP). The vasodilator responses to single bolus doses of CRF and SVG were measured for 20 min. Due to the sustained action of CRF and SVG, for studies evaluating the mechanism of the vasodilatation each SMA preparation was used to measure only one response.
The role of the endothelium
The effect of removal of the endothelium was studied using the detergent deoxycholic acid. After the initial verification of vasodilator responses with ACh, SNP and isoprenaline, methoxamine infusion was stopped and deoxycholic acid (5 mM) was infused for 20 s. After a period of recovery (approximately 30 min) the methoxamine infusion was restarted, and once perfusion pressure was stable, the responses to ACh, SNP and isoprenaline were re-tested. This was followed by a single bolus dose of CRF (100 pmol) or SVG (15 pmol).
The role of nitric oxide
The role of endothelial NOS (eNOS) derived nitric oxide (NO) was investigated in seperate experiments, using the NOS inhibitors L-NAME (100 μM), L-NMMA (100 μM) or 2-ethyl-2-thiopseudourea (ETPU, 100 μM; Southan et al., 1995), or the inactive isomer D-NAME (100 μM). The effect of inhibiting guanylate cyclase was studied using ODQ (10 μM).
The initial doses of ACh and SNP were followed by an infusion of one of the above compounds. The perfusion pressure was allowed to stabilize, after which the doses of ACh and SNP were repeated. A single bolus dose of CRF (100 pmol) or SVG (15 pmol; L-NAME experiments only) was then administered and the response recorded as above.
The role of NO was also investigated using L-NAME to reverse the vasodilator response to CRF. The endothelium-dependent and -independent responses were verified with ACh and SNP as above. One of three treatments was employed in each set of tissues, with all responses being measured for 35 min. In the control group basal pressure was recorded for 20 min, followed by an infusion of 100 μM L-NAME for 15 min. A second group was given a single bolus of 500 pmol CRF, followed 20 min later by an infusion of L-NAME (100 μM). The third group received an infusion of isoprenaline (8 nmol min−1; 1.6 μM), which gave a vasodilatation of similar magnitude to the CRF. After 20 min L-NAME (100 μM) was also infused, and the response recorded for a further 15 min.
The role of prostaglandins
The influence of products of cyclo-oxygenase on the vasodilator response was investigated by infusion of indomethacin (3 μM) after the initial doses of ACh and SNP. Further doses of ACh and SNP were then given followed by a single dose of CRF (100 pmol).
The role of potassium channels
The involvement of potassium channels in the vasodilator response to CRF was investigated by constricting the SMA with a high concentration of potassium. After the initial doses of ACh and SNP, the infusion of methoxamine was terminated and an infusion of potassium chloride (60 mM) was commenced. Once the perfusion pressure had stabilized, further doses of ACh and SNP were given followed by CRF (100 pmol).
The role of specific potassium channels in the response to CRF was studied using the non-selective potassium channel blocker, TEA, and the selective blocker of ATP-sensitive potassium channels, glibenclamide. After the initial doses of ACh and SNP an infusion of TEA (3 or 10 mM) or gli-benclamide (10 μM) was started. Further doses of ACh and SNP were given followed by a single dose of CRF (100 pmol).
Rings of rat thoracic aorta
Male Wistar rats were killed with an overdose of anaesthetic (sodium pentobarbitone, 120 mg kg−1, i.p.). The thoracic aorta was removed, cleared of surrounding tissue and cut into rings 3–4 mm in length. The endothelium was removed by gently scraping the inside of each ring with the tip of a pair of forceps. Rings were mounted in organ baths containing Krebs solution (formula as above) warmed to 37°C and gassed with 95%O2 : 5%CO2, and gradually placed under 2.5 g tension. Changes in tension were measured on an isometric Siegestab K30 force transducer, and recorded on a Graphtec Linearcorder Mark VI. Tissues were contracted with 50 μM methoxamine, and the response to ACh (10−7–10−5 M) was evaluated to ensure the absence of endothelium-dependent vasodilator responses. Any tissues that responded to ACh were discarded. After washing the tissues and reapplication of methoxamine, concentration response curves to either CRF or SVG (10−10–10−7 M) were constructed. Due to the long lasting effect of CRF and SVG each RTA preparation was used for only one concentration response curve.
Materials
SVG was obtained from Bachem, U.K. Rat CRF was a gift from Professor N. Ling, Neurocrine (La Jolla, CA, U.S.A.). Peptides were dissolved in 10 mM sodium bicarbonate and dilutions were made in Krebs solution containing 0.1% bovine serum albumin. ODQ was obtained from Calbiochem. All other compounds were purchased from Sigma. Compounds were dissolved and diluted in Krebs solution except for indomethacin which was dissolved in 0.6 M sodium bicarbonate, and ODQ and glibenclamide which were dissolved in DMSO. For each a dilution of 1 : 1000 into Krebs was followed by a further 1 : 30 dilution on entry into the perfusion system, where appropriate vehicle controls were carried out.
Data analysis
Results shown as means±s.e.mean. were compared by analysis of variance or Student's t-test as appropriate, with statistically significant differences accepted at P<0.05. Basal perfusion pressures (PP, mmHg) are given in figure legends. To facilitate comparison of vasodilator responses during different treatments, values were expressed as a percentage of PP prior to administration of each agent. Comparison of relative potencies was made using EC40 (the concentration causing a 40% reduction in tension) and ED40 (the dose causing a 40% reduction in PP). Changes in perfusion pressure are indicated by ΔPP expressed as a percentage.
Results
In the perfused SMA SVG was 10 fold more potent than CRF (Figure 1a), with ED40 values of 0.05±0.02 nmol (n=8) and 0.6±0.1 nmol (n=7) respectively (P<0.05). On the RTA SVG was also the most potent of the two peptides, and ≈amp;50 fold more active than CRF. The threshold concentration for vasodilatation of the RTA was 0.3 nM for SVG (n=8) and 10 nM for CRF (n=7), and EC40 values for these peptides were 0.9±0.2 and 44±9 nM respectively (Figure 1b).
Figure 1.
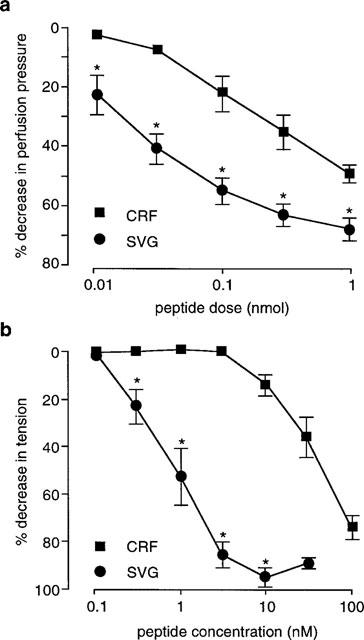
Dose response curves to CRF and SVG in (a) perfused SMA (CRF, n=7, basal PP 116±9 mmHg; SVG, n=8; basal PP, 88±9 mmHg), and (b) endothelium de-nuded RTA (CRF, n=5; SVG, n=6). *P<0.05 compared to CRF.
A single bolus injection of either CRF or SVG produced a long lasting vasodilatation in the SMA (Figure 2). CRF (100 pmol, n=10) produced a maximum fall in perfusion pressure of 23±2% at 3 min. This response was sustained with perfusion pressure still reduced by 15±2% at 20 min. Similarly, SVG (15 pmol, n=8) caused a maximum fall in perfusion pressure of 24±2% at 4 min, with a reduction in perfusion pressure of 17±4% still present at 20 min.
Figure 2.
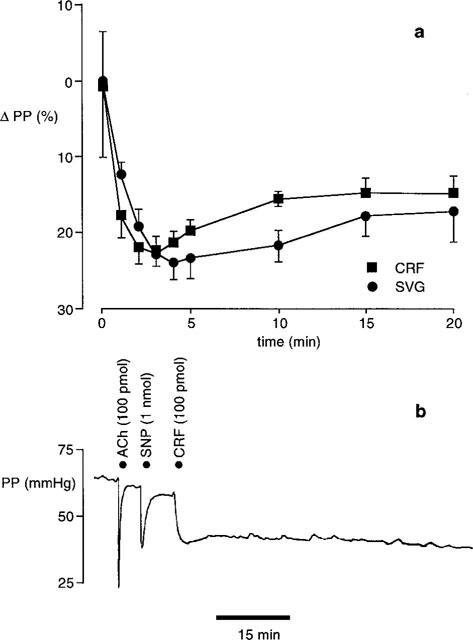
Effect of a single bolus dose of CRF or SVG in the perfused SMA. (a) Vasodilator responses to 100 pmol CRF (n=10, basal PP 79±7 mmHg) and 15 pmol SVG (n=7, basal PP 104±10 mmHg). (b) Typical vasodilator response to ACh (100 pmol), SNP (1 nmol) and CRF (100 pmol).
Deoxycholic acid
Infusion of detergent inhibited endothelium-dependent but not endothelium-independent responses. There was no vasodilator response to ACh (100 pmol), compared with a pre-deoxycholic acid fall in perfusion pressure of 48±1%. The magnitude of the SNP induced vasodilatation was increased markedly (ΔPP −18±5%, control; ΔPP −34±5%, detergent treated; P<0.05), but the response to isoprenaline was unchanged (ΔPP −15±4%, control; ΔPP −13±3%, detergent treated). The response to a single bolus dose of CRF (100 pmol) after deoxycholic acid infusion (n=6) was reduced in both size and duration (Figure 3a). The initial fall in perfusion pressure was only 9±2%, compared to a fall of 23±2% in the control preparations (P<0.05). In contrast to the sustained response in control tissues, after removal of the endothelium perfusion pressure returned to pre-CRF levels within 5 min. Deoxycholic acid also attenuated the response to SVG (Table 1). Perfusion pressure in detergent treated tissues (n=6) fell by 9±3%, compared to a decrease of 24±2% in control tissues (P<0.05). In comparison with CRF, a small vasodilator response to SVG persisted for 10 min but it was substantially less than the response in control preparations.
Figure 3.
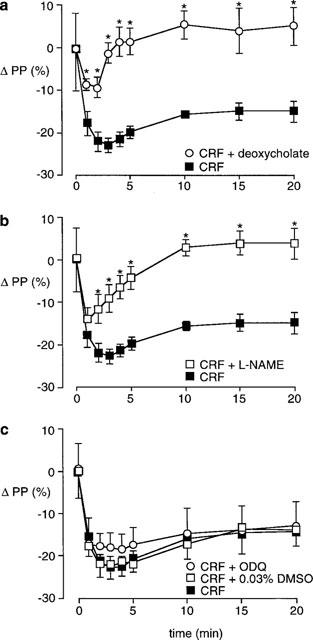
Effect of different treatments on the vasodilator response to CRF (n=10, basal PP 79±7 mmHg) in the perfused SMA. (a) 5 mM deoxycholic acid for 20 s (n=6, basal PP 83±9 mmHg); (b) 100 μM L-NAME (n=6, basal PP 165±14 mmHg); (c) 10 μM ODQ (n=8, basal PP 138±10 mmHg), or 0.03% DMSO control (n=7, basal PP 87±7 mmHg). *P<0.05 compared to control response.
Table 1.
Effect of deoxycholic acid (5 mM for 20 s) and L-NAME (100 μM) on SVG (15 pmol)-induced vasodilatation of the perfused SMA

NOS inhibition
Infusion of L-NAME reduced the ACh-induced vasodilatation (ΔPP −48±1%, control; ΔPP −25±4%, L-NAME; P<0.05), but increased the response to SNP (ΔPP −18±5%, control; ΔPP −39±5%, L-NAME; P<0.05). The response to isoprenaline was unaffected (ΔPP −15±4%, control; ΔPP −16±3%, L-NAME; P>0.05).
L-NAME had a similar effect on the CRF induced vasodilatation in the SMA (Figure 3b) to the removal of the endothelium with detergent. The initial fall in perfusion pressure of 14±3% (n=6) was significantly less than the control response (P<0.05). At 5 min the perfusion pressure in the L-NAME treated preparations had almost returned to pre-CRF levels, and by 10 min the CRF response was completely inhibited. The response to SVG was also attenuated by L-NAME (Table 1). The initial vasodilatation (ΔPP −12±3%) was smaller during L-NAME infusion (n=6) compared to the control tissues (P<0.05), and perfusion pressure had returned to baseline 10 min after SVG administration.
L-NMMA attenuated the fall in perfusion pressure to ACh (ΔPP −48±1%, control; ΔPP −38±4%, L-NMMA; P<0.05) but increased the size of the response to SNP (ΔPP −18±5, control; ΔPP −37±4%, L-NMMA; P<0.05). Although the magnitude of the initial response to CRF was not decreased, L-NMMA (n=6) attenuated the response to CRF with significant inhibition of the response at 5 min (P<0.05), and a return to pre-CRF perfusion pressure by 10 min (Table 2).
Table 2.
Effect of L-NMMA, ETPU and D-NAME on CRF (100 -pmol)-induced vasodilatation of the perfused SMA
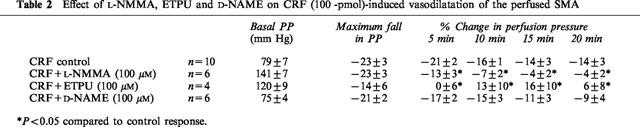
In common with L-NAME and L-NMMA, ETPU significantly reduced the ACh-induced fall in perfusion pressure (ΔPP −48±1%, control; ΔPP −23±5%, ETPU; P<0.05) whilst increasing the SNP-induced vasodilatation (ΔPP −18±5%, control; ΔPP −43±7%, ETPU; P<0.05). ETPU (n=4) also inhibited the response to CRF reducing the maximum fall in perfusion pressure and the duration of the response (Table 2).
In contrast D-NAME had no significant effect on either the ACh-induced fall in perfusion pressure (ΔPP −48±1%, control; ΔPP −46±5%, D-NAME) or the reduction in perfusion pressure to SNP (ΔPP −18±5%, control; ΔPP −18±2%, D-NAME). There was also no significant effect on either the magnitude or the length of the CRF-induced vasodilator response (n=6, Table 2).
DMSO vehicle (0.03%) had no effect on responses to ACh (ΔPP −48±1%, control; ΔPP −51±2%, DMSO), SNP (ΔPP −18±5%, control; ΔPP −20±1%, DMSO), or CRF (n=7, Figure 3c). Infusion of ODQ (10 μM) significantly reduced the fall in perfusion pressure to ACh (ΔPP −48±1%, control; ΔPP −33±3%, ODQ) and completely abolished the SNP response. However ODQ (n=6) did not inhibit either the size of the response to CRF or its duration of action (Figure 3c).
In order to evaluate the effect of eNOS inhibition on already established vasodilator responses a second set of experiments with L-NAME was performed (Figure 4). Both CRF and isoprenaline showed large vasodilator effects, with the maximum fall in perfusion pressure (ΔPP −62±5%) to the bolus of 500 pmol CRF being recorded at 2 min (n=6). The largest reduction in perfusion pressure during the isoprenaline infusion was 57±6% at 20 min (n=6). In control tissues the perfusion pressure remained stable throughout the initial 20 min (n=6), and L-NAME infusion increased perfusion pressure by 141±33% above the basal level. In tissues where CRF was administered, perfusion pressure was still 26±10% below the basal level 20 min after bolus administration. L-NAME infusion reversed the effect of CRF and perfusion pressure increased to 128±28% above the baseline level. This increase was not significantly different from the response to L-NAME in control preparations. In isoprenaline treated tissues, L-NAME infusion increased perfusion pressure to 16±16% above the baseline level which was significantly less than the L-NAME-induced increase in control and CRF-treated preparations (P<0.05).
Figure 4.
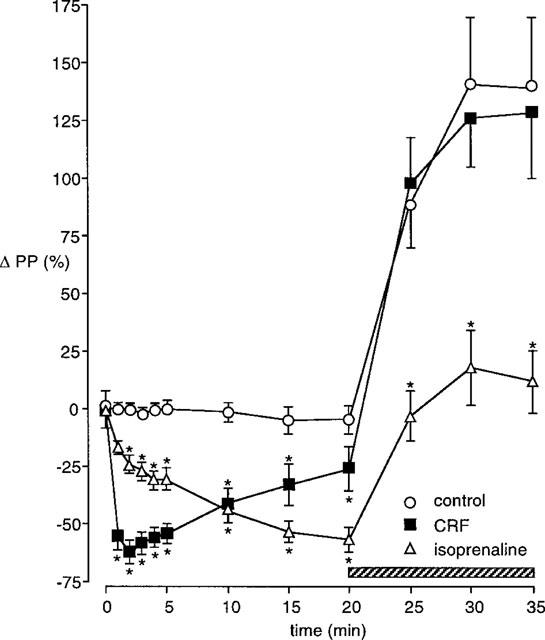
Effect of infusing L-NAME (100 μM) during the vasodilator response to CRF (500 pmol) or isoprenaline (8 nmol min−1) in the perfused SMA. The hatched bar shows the period of L-NAME infusion. Control L-NAME infusion, basal PP 46±7 mmHg; L-NAME during the CRF response, basal PP 48±8 mmHg; L-NAME during isoprenaline infusion, basal PP 51±6 mmHg. n=6 for each, *P<0.05 compared to control L-NAME response.
The role of prostaglandins
Indomethacin infusion (3 μM) did not significantly affect the fall in perfusion pressure to ACh (ΔPP −48±2%, control ΔPP −39±5%, indomethacin) or SNP (ΔPP −28±2%, control; ΔPP −23±3%, indomethacin). The initial fall in perfusion pressure to CRF was slightly but significantly increased (ΔPP −25±3%, control; −34±5%, indomethacin; n=6, P<0.05), but there were no significant differences at any other time.
Potassium channel antagonists
Infusion of 60 mM KCl inhibited the fall in perfusion pressure to ACh (ΔPP −48±1%, control; ΔPP −17±3%, 60 mM KCl; P<0.05), but did not significantly alter the SNP-induced fall in perfusion pressure (ΔPP −18±5, control; ΔPP −11±1%, 60 mM KCl). The response to CRF was completely inhibited at all time points (n=6, Figure 5a).
Figure 5.
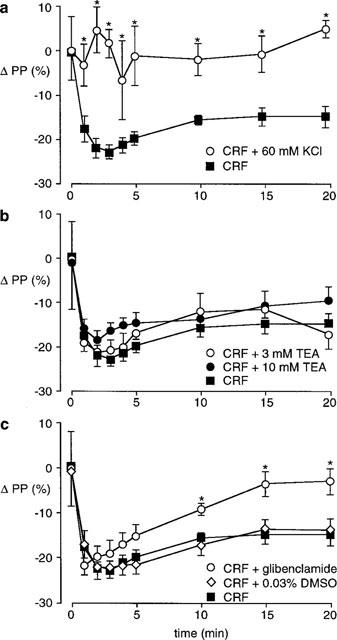
Effect of inhibiting potassium channel function on the vasodilator response to 100 pmol CRF in the perfused SMA (CRF n=10, basal PP 79±7 mmHg). (a) 60 mM KCl (n=4, basal PP 81±6 mmHg); (b) 3 mM TEA (n=4, basal PP 68±8 mmHg), 10 mM TEA (n=6, basal PP 90±7 mmHg); (c) 10 μM glibenclamide (n=9, basal PP 81±7 mmHg), 0.03% DMSO (n=7, basal PP 84±7 mmHg). *P<0.05 compared to control response.
The non-selective potassium channel antagonist TEA (3 and 10 mM) significantly inhibited ACh-induced decreases in perfusion pressure (ΔPP −48±1%, control; ΔPP −20±6%, 3 mM TEA; ΔPP −20±3%, 10 mM TEA; P<0.05) but had no effect on the fall in perfusion pressure to SNP (ΔPP −18±5%, control; ΔPP −23±1%, 3 mM TEA; ΔPP −26±4%, 10 mM TEA). However neither 3 mM (n=4) nor 10 mM (n=6) TEA had any effect on the vasodilator response to CRF (Figure 5b).
In contrast to TEA, glibenclamide, a selective blocker of ATP-sensitive potassium channels did not inhibit either the fall in perfusion pressure to ACh (ΔPP −48±1%, control; ΔPP −54±3%, glibenclamide) or SNP (ΔPP −18±5%, control; ΔPP −20±1%, glibenclamide), but significantly reduced the response to CRF (Figure 5c). Although the initial response was not affected (ΔPP −22±2%), 10 min after CRF the vasodilator response was partially inhibited and by 15 min perfusion pressure had returned to basal levels.
Discussion
CRF and SVG produced dose-dependent vasodilatations of endothelium de-nuded RTA and the perfused SMA. A single bolus dose of CRF or SVG causes a long lasting vasodilatation of the perfused SMA. In agreement with in vivo studies that have compared the two peptides, SVG was more potent than CRF in both vascular preparations (Brown et al., 1982a; Lenz et al., 1985). Earlier studies of the vasodilator action of CRF have reported the effect to be either a direct vascular smooth muscle effect (Lei et al., 1993), or partly endothelium-dependent (Grunt et al., 1993; Jain et al., 1997). Here using the RTA from which the endothelium had been removed CRF and SVG produced concentration-dependent relaxation of vascular smooth muscle by a direct action. But of greater interest, and of more relevance to the effect of systemic administration of these peptides on blood pressure, studies of the perfused SMA showed these peptides to have two components to their vasodilator action; a short direct action on vascular smooth muscle, followed by a sustained endothelium-dependent vasodilatation. This was demonstrated firstly by the removal of the endothelium with deoxycholic acid and secondly using three different eNOS inhibitors. From these results it can be concluded that the long-lasting endothelium-dependent vasodilator response is due to the sustained activation of eNOS and a prolonged release of NO.
The role of NO in the prolonged vasodilator response to CRF was also demonstrated by infusing L-NAME during the prolonged phase of CRF-induced vasodilatation. This increased the perfusion pressure to a level that was not significantly different to L-NAME infusion alone. Hence the vasodilatation at the point when L-NAME is infused is due solely to NO, because any direct effect on vascular smooth muscle, or release of other endothelial vasodilator molecules would be expected to attenuate the rise in perfusion pressure. This is clearly seen with the infusion of isoprenaline, where there is a small increase in perfusion pressure, due to the inhibition of basal NO synthesis, but it remains below that seen during L-NAME infusion in control- and CRF-treated preparations. Some studies indicate a role for NO in the vasodilator actions of isoprenaline (Graves & Poston, 1993), but here the response to a single bolus dose of isoprenaline was unaffected by either deoxycholic acid or L-NAME, indicating a vasodilator effect which is largely independent of any action on the endothelium. Inhibition by indomethacin of the vasodilator response to CRF in the isolated heart (Grunt et al., 1993) suggests that prostaglandins play a role in the endothelium-dependent vasodilatation, but no evidence for CRF-induced synthesis of vasodilator prostanoids was observed in the perfused SMA.
In agreement with the pivotal role of the endothelium in the vasodilator responses to CRF or SVG, autoradiographic studies of rabbit aorta have shown the endothelium as well as the underlying vascular smooth muscle to bind [125]-Tyr-CRF (Dashwood et al., 1987). Direct effects of this family of peptides on vascular smooth muscle have been observed through measurement of increased cyclic AMP in response to UI (Gerritsen & Lederis, 1979). Consistent with the results described here, the effect of UI on cyclic AMP levels in vascular smooth muscle is short lasting (Gerritsen & Lederis, 1979). A peripheral CRF receptor (CRF-R2β) has been cloned which is G-protein coupled to adenylate cyclase (Kishimoto et al., 1995). This receptor has been identified in blood vessels (Lovenberg et al., 1995), but its localization to endothelial or vascular smooth muscle cells has yet to be confirmed. In previous studies of the vasodilator effect of CRF in intact rat mesenteric arteries it was suggested that the CRF-R2β receptor was involved in the response (Rohde et al., 1996). In the studies of direct vasodilator effects on the endothelium de-nuded RTA described here, the EC40 values for SVG and CRF, and the 50 fold difference in potency of these peptides are very similar to values reported for the induction of cyclic AMP synthesis by the human CRF-R2β receptor (Liaw et al., 1997). Hence, the CRF-R2β receptor may well mediate the direct vasodilator effects of CRF on vascular smooth muscle, but it is unclear whether the endothelium-dependent effects are also via this receptor subtype.
A surprising aspect of these studies was the mechanism of the CRF-induced NO-dependent vasodilatation. Although it is generally accepted that NO produces relaxation of vascular smooth muscle primarily by activation of guanylate cyclase and an increase in cyclic GMP, the response to CRF in the perfused SMA was not altered by the selective guanylate cyclase inhibitor ODQ at concentrations which inhibited completely SNP-induced vasodilatation. This contrasts with results obtained in studies of CRF on the rat thoracic aorta using LY83583 which is also described as a guanylate cyclase inhibitor (Jain et al., 1997). However, LY83583 also inhibits NOS, and generates free radicals that scavenge NO, thus preventing the actions of NO independently of any inhibitory effect on guanylate cyclase (Kontos & Wei, 1993; Kannan & Johnson, 1995; Luo et al., 1995). When compared with the lack of effect of ODQ described here, it seems likely that LY83583 inhibited CRF-induced vasodilatation by a mechanism unrelated to guanylate cyclase inhibition.
An alternative mechanism of action for NO has been described, involving the opening of potassium channels in vascular smooth muscle to produce vasodilatation (Bolotina et al., 1994). The inhibition of both CRF and ACh-induced vasodilator effects by high extracellular potassium indicates a role for potassium channels in their vasodilator responses. However, comparison of the effects of TEA and glibenclamide on the actions of ACh and CRF suggest the involvement of different potassium channels in their mechanisms. TEA attenuated the ACh response at a concentration (3 mM) which shows selectivity for inhibition of calcium-activated potassium channels (Langton et al., 1991), without altering the response to CRF. Conversely the NO-dependent phase of the response to CRF was attenuated by glibenclamide, a selective inhibitor of ATP-sensitive potassium channels (Edwards & Weston, 1993), even though it did not alter the response to ACh. High concentrations of TEA (10 mM) are reported to cause a non-selective blockade of potassium channels (Langton et al., 1991), but no effect on the vasodilator action of CRF was observed.
It has been proposed that endothelium-derived hyperpolarizing factor (EDHF) is more important than NO in mediating the endothelium-dependent response to ACh in the small resistance vessels of the mesenteric vasculature (Garland & McPherson, 1992; Parsons et al., 1994). However the complete inhibition of the sustained phase of CRF vasodilatation with inhibitors of eNOS indicates that EDHF is not involved. In the mesenteric vasculature and isolated perfused kidney of the rat, EDHF appears to act via calcium-activated potassium channels (McPherson & Angus 1991; Rapacon et al., 1996), but the hyperpolarizing response to NO is mediated by ATP-sensitive channels (Garland & McPherson, 1992). The precise role of ATP-sensitive potassium channels in the NO-dependent vasodilatation induced by CRF is not evident at this time.
The ability of a single bolus of CRF or SVG to produce a long lasting, NO-mediated vasodilatation is of particular interest given that other vasodilator responses involving the release of NO tend to be of short duration. For example ACh produces a large endothelium-dependent vasodilatation lasting <1 min. In some respects the cascade superfusion technique using cultured endothelial cells (Gryglewski et al., 1986) is similar to that used here as the agents applied are rapidly washed through. This has shown that bradykinin, another peptide mediator causing endothelium-dependent vasodilatation induces NO release for <5 min. In comparison, the calcium ionophore, A23187, can cause a longer lasting output of NO (Gryglewski et al., 1986), indicating that a lengthy influx of Ca2+ causes a sustained activation of eNOS. Hence, a prolonged receptor mediated influx of Ca2+ could play a role in the endothelium-dependent vasodilator effect of CRF.
However, physiologically, the most notable factor causing a sustained NO-dependent vasodilatation is shear stress (Frangos et al., 1996). Hence, the mechanism underlying shear stress-induced NO synthesis may be of more relevance to the response to CRF. NO release stimulated by constant shear involves both calcium-dependent and ATP-sensitive potassium channels (Hutcheson & Griffith, 1994). Indeed, the inhibitory effect of glibenclamide on the response to CRF may well be due to an effect on NO release (Hutcheson & Griffith, 1994), thus indicating similarities in the mechanisms of shear stress and CRF-induced NO release. Interestingly, heat shock protein 90 (Hsp90) has recently been shown to modulate the activity of eNOS during stimulation with agonists or shear stress (García-Cardeña et al., 1998). Based on these findings, the sustained NO-dependent vasodilator effect of CRF could involve the induction of eNOS-Hsp90 heterocomplexes with a resultant sensitization of the endothelium to shear stress induced NO release in the perfused SMA. In conclusion, further work is required for a full appreciation of this unusual and potentially important mechanism of eNOS activation by the CRF-family of peptides.
Acknowledgments
This work was supported by a grant from the Joint Research Board of St. Bartholomew's Hospital.
Abbreviations
- CRF
corticotrophin-releasing factor
- EPTU
2 ethyl-2-thiopseudourea
- RTA
rat thoracic aorta
- SMA
superior mesenteric artery vasculature
- SVG
sauvagine
- UI
urotensin I
References
- BARKER D.M., CORDER R. The long-lasting vasodilator action of corticotrophin releasing hormone is endothelium-dependent in the rat perfused mesentery. Br. J. Pharmacol. 1995;115:42P. [Google Scholar]
- BOLOTINA V.M., NAJIBI S., PALACINO J.J., PAGANO P.J., COHEN R.A. Nitric oxide directly activates the calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–851. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- BROWN M.R., FISHER L.A., SPIESS J., RIVIER J., RIVIER C., VALE W. Comparison of the biologic actions of corticotrophin releasing factor and sauvagine. Regul. Pept. 1982a;4:107–114. doi: 10.1016/0167-0115(82)90101-x. [DOI] [PubMed] [Google Scholar]
- BROWN M.R., FISHER L.A., SPIESS J., RIVIER C., RIVIER J., VALE W. Corticotrophin releasing factor: Actions on the sympathetic nervous system and metabolism. Endocrinology. 1982b;111:928–931. doi: 10.1210/endo-111-3-928. [DOI] [PubMed] [Google Scholar]
- CHANG C.P., PEARSE R.V., II, O'CONNELL S., ROSENFELD M.G. Identification of a seven transmembrane helix receptor for corticotrophin releasing factor and sauvagine in mammalian brain. Neuron. 1993;11:1187–1195. doi: 10.1016/0896-6273(93)90230-o. [DOI] [PubMed] [Google Scholar]
- CORDER R., TURNILL D., LING N., GAILLARD R.C. Attenuation of corticotrophin releasing factor-induced hypertension in anesthetised rats with the CRF antagonist, α-helical CRF9–41; comparison with effect on ACTH release. Peptides. 1992;13:1–6. doi: 10.1016/0196-9781(92)90132-m. [DOI] [PubMed] [Google Scholar]
- DASHWOOD M.R., ANDREWS H.E., WEI E.T. Binding of [125I]Tyr-corticotrophin releasing factor to rabbit aorta is reduced by removal of the endothelium. Eur. J. Pharmacol. 1987;135:111–112. doi: 10.1016/0014-2999(87)90766-7. [DOI] [PubMed] [Google Scholar]
- EDWARDS G., WESTON A.H. The pharmacology of ATP-sensitive potassium channels. Ann. Rev. Pharmacol. Toxicol. 1993;33:597–637. doi: 10.1146/annurev.pa.33.040193.003121. [DOI] [PubMed] [Google Scholar]
- FRANGOS J.A., HUANG T.Y., CLARK C.B. Steady shear and step changes in shear stimulate endothelium via independent mechanisms – superposition of transient and sustained nitric oxide production. Biochem. Biophys. Res. Commun. 1996;224:660–665. doi: 10.1006/bbrc.1996.1081. [DOI] [PubMed] [Google Scholar]
- GARCIA-CARDENA G., FAN R., SHAH V., SORRENTINO R., CIRINO G., PAPAPETROPOULOS A., SESSA W.C. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- GARDINER S.M., COMPTON A.M., BENNETT T. Regional haemodynamic effects of depressor neuropeptides in conscious unrestrained Long Evans and Brattleboro rats. Br. J. Pharmacol. 1988;95:197–208. doi: 10.1111/j.1476-5381.1988.tb16565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARLAND C.J., MCPHERSON G.A. Evidence that nitric oxide does not mediate the hyperpolarisation and relaxation to acetylcholine in the rat small mesentery. Br. J. Pharmacol. 1992;105:429–435. doi: 10.1111/j.1476-5381.1992.tb14270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERRITSEN M.E., LEDERIS K. Urotensin I effects on intracellular content of cyclic AMP in the rat tail artery. Eur. J. Pharmacol. 1979;60:211–220. doi: 10.1016/0014-2999(79)90220-6. [DOI] [PubMed] [Google Scholar]
- GRAVES J., POSTON L. β-adrenoceptor agonist mediated relaxation of rat isolated resistance arteries: a role for the endothelium and nitric oxide. Br. J. Pharmacol. 1993;108:631–637. doi: 10.1111/j.1476-5381.1993.tb12853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRUNT M., GLASER J., SCHMIDHUBER H., PAUSCHINGER P., BORN J. Effects of corticotrophin releasing factor on isolated heart activity. Am. J. Physiol. 1993;264:H1124–H1129. doi: 10.1152/ajpheart.1993.264.4.H1124. [DOI] [PubMed] [Google Scholar]
- GRYGLEWSKI R.J., MONCADA S., PALMER R.M.J. Bioassay of prostacyclin and endothelium-derived relaxing factor (EDRF) from porcine aortic endothelial cells. Br. J. Pharmac. 1986;87:685–694. doi: 10.1111/j.1476-5381.1986.tb14586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUTCHESON I.R., GRIFFITH T.M. Heterogenous populations of K+ channels mediate EDRF release to flow but not agonists in rabbit aorta. Am. J. Physiol. 1994;266:H590–H596. doi: 10.1152/ajpheart.1994.266.2.H590. [DOI] [PubMed] [Google Scholar]
- JAIN V., VENDERNIKOV Y.P., SAADE G.R., CHWALISZ K., GARFIELD R.E. The relaxation responses to corticotropin-releasing factor in rat aorta are endothelium dependent and gestationally regulated. Am. J. Obstet. Gynecol. 1997;176:234–240. doi: 10.1016/s0002-9378(97)80042-7. [DOI] [PubMed] [Google Scholar]
- KANNAN M.S., JOHNSON D.E. Modulation of nitric oxide-dependent relaxation of pig tracheal smooth muscle by inhibitors of guanylyl cyclase and calcium activated potassium channels. Life Sci. 1995;56:2229–2238. doi: 10.1016/0024-3205(95)00212-o. [DOI] [PubMed] [Google Scholar]
- KISHIMOTO T., PEARSE R.V., II, LIN C.R., ROSENFELD M.G. A sauvagine/corticotrophin releasing factor receptor expressed in heart and skeletal muscle. Proc. Natl. Acad. Sci. USA. 1995;92:1108–1112. doi: 10.1073/pnas.92.4.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KONTOS H.A., WEI E.P. Hydroxyl radical-dependent inactivation of guanylate cyclase in cerebral arterioles by methylene blue and by LY83583. Stroke. 1993;24:427–434. doi: 10.1161/01.str.24.3.427. [DOI] [PubMed] [Google Scholar]
- LANGTON P.D., NELSON M.T., HUANG Y., STANDEN N.B. Block of calcium-activated potassium channels in arterial myocytes by tetraethylammonium ions. Am. J. Physiol. 1991;260:H927–H934. doi: 10.1152/ajpheart.1991.260.3.H927. [DOI] [PubMed] [Google Scholar]
- LEDERIS K., LETTER A., MCMASTER D., MOORE G., SCHLESINGER D. Complete amino acid sequence of urotensin I, a hypotensive and corticotrophin releasing neuropeptide from Catastomus. Science. 1982;218:162–164. doi: 10.1126/science.6981844. [DOI] [PubMed] [Google Scholar]
- LEDERIS K., MEDAKOVIC M. Pharmacological observations on the hypotensive actions of extracts of teleost fish urophyses (urotensin I) in the rat. Br. J. Pharmocol. 1974;51:315–324. doi: 10.1111/j.1476-5381.1974.tb10665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEI S., RICHTER R., BIENERT M., MULVANY M.J. Relaxing actions of corticotrophin releasing factor on rat resistance arteries. Br. J. Pharmacol. 1993;108:941–947. doi: 10.1111/j.1476-5381.1993.tb13490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LENZ H.J., FISHER L.A., VALE W., BROWN M.R. Corticotrophin releasing factor, sauvagine and urotensin I: Effects on blood flow. Am. J. Physiol. 1985;249:R85–R90. doi: 10.1152/ajpregu.1985.249.1.R85. [DOI] [PubMed] [Google Scholar]
- LIAW C.W., GRIGORIADIS D.E., LORANG M.T., DE SOUZA E.B., MAKI R.A. Localization of agonist- and antagonist-binding domains of human corticotrophin-releasing factor receptors. Mol. Endocrinol. 1997;11:2048–2053. doi: 10.1210/mend.11.13.0034. [DOI] [PubMed] [Google Scholar]
- LOVENBERG T.W., CHALMERS D.T., LIU C., DE SOUZA E.B. CRF2α and CRF2β receptor mRNAs are differently distributed between the rat central nervous system and peripheral tissues. Endocrinology. 1995;136:4139–4142. doi: 10.1210/endo.136.9.7544278. [DOI] [PubMed] [Google Scholar]
- LUO D., DAS S., VINCENT S.R. Effects of methylene blue and LY83583 on neuronal nitric oxide synthase and NADPH-diaphorase. Eur. J. Pharmacol. 1995;290:247–51. doi: 10.1016/0922-4106(95)00084-4. [DOI] [PubMed] [Google Scholar]
- MACCANNELL K.L., LEDERIS K., HAMILTON P., RIVIER J. Amunine (ovine CRF), urotensin I and sauvagine, three structurally related peptides, produce selective dilation of the mesenteric circulation. Pharmacology. 1982;25:116–120. doi: 10.1159/000137732. [DOI] [PubMed] [Google Scholar]
- MCPHERSON G.A., ANGUS J.A. Evidence that acetylcholine-mediated hyperpolarisation of the rat small mesenteric artery does not involve the K+ channel opened by cromakalim. Br. J. Pharmacol. 1991;103:1184–1190. doi: 10.1111/j.1476-5381.1991.tb12321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONTECUCCHI P.C., ANASTASI A., DE CASTIGLIONE R., ERSPAMER V. Isolation and amino acid composition of Sauvagine. Int. J. Peptide and Protein Res. 1980;16:191–199. [PubMed] [Google Scholar]
- MORLEY S.D., SCHÖNROCK C., RICHTER D., OKAWARA Y., LEDERIS K. Corticotrophin (CRF) gene family in the brain of the teleost fish Catostomus commersoni (white sucker): molecular analysis predicts distinct precursors for two CRFs and one urotensin I peptide. Mol. Marine Biol. and Biotech. 1991;1:48–57. [PubMed] [Google Scholar]
- MORO M.A., RUSSELL R.J., CELLEK S., LIZASOAIN I., SU Y., DARLEY-USMAR V.M., RADOMSKI M.W., MONCADA S. cGMP mediates the vascular and platelet actions of nitric oxide: conformation using an inhibitor of the soluble guanylyl cyclase. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1480–1485. doi: 10.1073/pnas.93.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARSONS S.J.W., HILL A., WALDRON G.J., PLANE F., GARLAND C.J. The relative importance of nitric oxide and nitric oxide-independent mechanisms in acetylcholine-evoked dilatation of the rat mesenteric bed. Br. J. Pharmacol. 1994;113:1275–1280. doi: 10.1111/j.1476-5381.1994.tb17136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAPACON M., MIEYAL P., MCGIFF J.C., FULTON D., QUILLEY J. Contribution of calcium-activated potassium channels to the vasodilator effect of bradykinin in the isolated, perfused kidney of the rat. Br. J. Pharmacol. 1996;118:1504–1508. doi: 10.1111/j.1476-5381.1996.tb15566.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROHDE E., FURKERT J., FECHNER K., BEYERMANN M., MULVANY M.J., RICHTER R.M., DENEF C., BIENERT M., BERGER H. Corticotrophin-releasing hormone (CRH) receptors in the mesenteric small arteries of rats resemble the (2)-subtype. Biochem. Pharmacol. 1996;52:829–833. doi: 10.1016/0006-2952(96)00300-0. [DOI] [PubMed] [Google Scholar]
- SCHÜRMEYER T.H., GOLD P.W., GALLUCI W.T., TOMAI T.P., CUTTLER G.B. , JR, LORIAUX D.L., CHROUSOS G.P. Effects and pharmokinetic properties of the rat/human corticotrophin releasing factor in rhesus monkeys. Endocrinology. 1985;117:300–306. doi: 10.1210/endo-117-1-300. [DOI] [PubMed] [Google Scholar]
- SHELDON R.J., JIANG Q., PORRECA F., FISHER L.A. Gastrointestinal motor effects of corticotrophin releasing factor in mice. Regul. Pept. 1990;28:137–151. doi: 10.1016/0167-0115(90)90013-m. [DOI] [PubMed] [Google Scholar]
- SOUTHAN G.J., SZABÓ C., THIEMERMANN C. Isothioureas: potent inhibitors of nitric oxide synthases with variable isoform selectivity. Br. J. Pharmacol. 1995;114:510–516. doi: 10.1111/j.1476-5381.1995.tb13256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STENZEL-POORE M.P., HELDWEIN K.A., STENZEL P., LEE S., VALE W. Characterisation of the genomic corticotrophin releasing factor (CRF) gene from Xenopus laevis: Two members of the CRF family exist in amphibians. Mol. Endocrin. 1992;6:1716–1724. doi: 10.1210/mend.6.10.1448118. [DOI] [PubMed] [Google Scholar]
- UDELSMAN R., GALLUCCI W.T., BACHER J., LORIAUX D.L., CHROUSOS G.P. Haemodynamic effects of corticotrophin releasing factor in the anaesthetised cynomolus monkey. Peptides. 1986;7:465–471. doi: 10.1016/0196-9781(86)90016-1. [DOI] [PubMed] [Google Scholar]
- VALE W., SPIESS J., RIVIER C., RIVIER J. Characterisation of a 41 residue ovine hypothalamic peptide that stimulates secretion of corticotrophin and β-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- VAUGHAN J., DONALDSON C., BITTENCOURT J., PERRIN M.H., LEWIS K., SUTTON S., CHAN R., TURNBULL A.V., LOVEJOY D., RIVIER C., RIVIER J., SAWCHENKO P.E., VALE W. Urocortin, a mammalian neuropeptide related to fish urotensin I and corticotrophin releasing factor. Nature. 1995;378:287–292. doi: 10.1038/378287a0. [DOI] [PubMed] [Google Scholar]
- WILLIAMS C.L., PETERSON J.M., VILLAR R.G., BURKS T.F. Corticotrophin releasing factor directly mediates colonic responses to stress. Am. J. Physiol. 1987;253:G582–G586. doi: 10.1152/ajpgi.1987.253.4.G582. [DOI] [PubMed] [Google Scholar]