

Abstract
Using a microelectrode technique, acetylcholine (ACh)-induced membrane potential changes were characterized using various types of inhibitors of K+ and Cl− channels in rabbit aortic valve endothelial cells (RAVEC).
ACh produced transient then sustained membrane hyperpolarizations. Withdrawal of ACh evoked a transient depolarization.
High K+ blocked and low K+ potentiated the two ACh-induced hyperpolarizations. Charybdotoxin (ChTX) attenuated the ACh-induced transient and sustained hyperpolarizations; apamin inhibited only the sustained hyperpolarization. In the combined presence of ChTX and apamin, ACh produced a depolarization.
In Ca2+-free solution or in the presence of Co2+ or Ni2+, ACh produced a transient hyperpolarization followed by a depolarization. In BAPTA-AM-treated cells, ACh produced only a depolarization.
A low concentration of A23187 attenuated the ACh-induced transient, but not the sustained, hyperpolarization. In the presence of cyclopiazonic acid, the hyperpolarization induced by ACh was maintained after ACh removal; this maintained hyperpolarization was blocked by Co2+.
Both NPPB and hypertonic solution inhibited the membrane depolarization seen after ACh washout. Bumetanide also attenuated this depolarization.
It is concluded that in RAVEC, ACh produces a two-component hyperpolarization followed by a depolarization. It is suggested that ACh-induced Ca2+ release from the storage sites causes a transient hyperpolarization due to activation of ChTX-sensitive K+ channels and that ACh-activated Ca2+ influx causes a sustained hyperpolarization by activating both ChTX- and apamin-sensitive K+ channels. Both volume-sensitive Cl− channels and the Na+-K+-Cl− cotransporter probably contribute to the ACh-induced depolarization.
Keywords: endothelial cell, aortic valve, acetylcholine, potassium channel, chloride channel
Introduction
The vascular endothelium plays an important role in the regulation of vascular tone through the release of vasorelaxing factors such as prostacyclin, endothelium-derived nitric oxide and endothelium-derived hyperpolarizing factor (Chen et al., 1988). Acetylcholine (ACh) is a well-known stimulant that releases these vasorelaxing factors via an increase in the cellular concentration of Ca2+ ([Ca2+]i) (Lückhoff & Busse, 1990b). In endothelial cells, ACh activates Ca2+-activated K+ (KCa) channels through an increase in [Ca2+]i (Himmel et al., 1994), thus causing a membrane hyperpolarization (Brunet & Bény, 1989; Mehrke & Daut, 1990; Chen & Cheung, 1992; Marchenko & Sage, 1993; Carter & Ogden, 1994; Sharma & Davis, 1994; Frieden & Bény, 1995). It has been suggested that this membrane hyperpolarization in endothelial cells in turn increases Ca2+ influx due to an increase in the electrical driving force for Ca2+ (Lückhoff & Busse, 1990b). Several types of K+ channels have been found to be expressed in endothelial cells (Chen & Cheung, 1992; Marchenko & Sage, 1996; Wang et al., 1996; Nilius et al., 1997c). However, exactly which K+ channels contribute to the ACh-induced hyperpolarization in intact endothelial cells under physiological conditions is not entirely clear.
It has been reported that ACh produces not only hyperpolarization but also depolarization in endothelial cells (Marchenko & Sage, 1993). Vascular endothelial cells express both Cl− transporters (Klein & O'Neill, 1990, Perry & O'Neill, 1993) and Cl− channels (Nilius et al., 1997b), and it is known that Cl− plays an important role in the regulation of the membrane potential and cell volume in endothelial cells (Nilius et al., 1997c). However, the roles played by Cl− channels and Cl− transporters in the ACh-induced membrane potential changes have not yet been clarified.
It has been reported that membrane potential changes evoked in smooth muscle cells can propagate into endothelial cells since these two types of cells are electronically coupled in a unidirectional way (Weid & Bény 1993; Bény & Pacicca, 1994; Marchenko & Sage, 1994b; Bény, 1997). This indicates the difficulty of recording electrical activity that truly arises from endothelial cells when intact vascular tissues are used. An alternative strategy might be to use cultured or isolated endothelial cells, but some alterations in receptors, ion channels and agonist-activated intracellular signal transduction systems have been noted in such cells (Sturek et al., 1991). It has been found that the endothelial cells of canine aortic valves can release endothelium-derived relaxing factors (Ku et al., 1990), suggesting that aortic valvular endothelial cells may be functionally similar to vascular endothelial cells. It is also known that the intact rabbit aortic valve is relatively thin and has no smooth muscle cells underlying the surface endothelial monolayer. Thus, the endothelium of the rabbit aortic valve is a preparation that allows us to circumvent the above problems.
The aim of the present study was to elucidate the characteristic features of the membrane potential changes induced by ACh in rabbit aortic valve endothelial cells (RAVEC). The ionic mechanisms underlying the ACh-induced electrical responses were pharmacologically assessed using a range of inhibitors of K+ channels, Cl− channels and Cl− transporters.
Methods
Male Japanese White albino rabbits (supplied by Kitayama Labes Co. Ltd., Japan), weighing 1.9–2.3 kg, were anaesthetized by injection of pentobarbitone sodium (40 mg kg−1, i.v.) and then exsanguinated. The protocols used conformed with guidelines on the conduct of animal experiments issued by Nagoya City University Medical School and by the Japanese government (Law no. 105; Notification no. 6), and were approved by The Committee on the Ethics of Animal Experiments in Nagoya City University Medical School. The heart was rapidly excised and placed in a chamber filled with Krebs solution. The aorta was opened by a longitudinal incision at the attachment to the left ventricle. The aortic valve was dissected out under a binocular microscope and transferred to a chamber of 0.5 ml volume set up on an inverted-microscope (Diaphoto TMD, Nikon, Japan). The chamber was superfused with oxygenated Krebs solution at a rate of 4 ml min−1.
Recording of membrane potential
The membrane potential was measured using conventional glass microelectrodes filled with 1 M KCl. The resistance of the electrode was 120–180 MΩ. To impale an endothelial cell, the electrode was advanced gently towards the surface of the aortic valve using a micromanipulator (model MHW-3; Narishige International, Tokyo, Japan) until the sudden appearance of a negative potential (Weid & Bény, 1992). The experimental protocol was started when the membrane potential had been stable for over 5 min after impalement. Successful recordings were usually obtainable for from 20 min to 5 h. Membrane potentials were recorded using an Axoclamp-2B amplifier (Axon Instruments, Foster, CA, U.S.A.) and were displayed on a cathode-ray oscilloscope (model VC-6020; Hitachi, Tokyo, Japan). The data were stored at an acquisition rate of 20 Hz using an AxoScope 1.1.1/Digidata 1200 data acquisition system (Axon Instruments, Foster, CA, U.S.A.) on an IBM/AT compatible PC.
To observe the concentration-dependent effects of ACh on membrane potential, ACh (0.01–10 μM) was applied for 3 min at 25 min intervals in an ascending order in the same preparation. Maximum responses were obtained with 3 μM ACh. The effects of inhibitors of K+ channels and Cl− channels were observed on the membrane potential changes induced by 3 μM ACh. To this end, the ACh-induced response was first recorded, followed by a 25 min intermission. Next, one of a number of inhibitors was applied so that it was added 10 min before the 2nd application of ACh and was present throughout the period for which ACh was applied. The concentrations of K+ channel inhibitors were chosen so as to retain their selectivity. To study the effect of ACh in Ca2+-free solution, nominal Ca2+-free solution was first applied for 10 min, then ACh was repetitively applied to the same cell for 3 min at 25 min intervals in Ca2+-free solution. To study the effect of the acetoxymethyl ester of bis-(aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA-AM) on the ACh-induced response, a control response was first recorded, and then BAPTA-AM (30 μM) was applied for 40 min, followed by a 10 min washout of BAPTA-AM. Finally, ACh was again applied. Ni2+ (5 mM) or Co2+ (2 mM) was added 10 min before ACh and was present throughout the application of ACh. A23187 (0.1 μM) was also added 10 min before ACh and was present throughout the application of ACh.
Solutions
The composition of the Krebs solution was as follows (mM): NaCl, 122; KCl, 4.7; MgCl2, 1.2; CaCl2, 2.6; NaHCO3, 15.5; KH2PO4, 1.2; glucose, 11.5. The solutions were bubbled with 95% O2 and 5% CO2 and their pH was maintained at 7.3–7.4. High K+ solution was made by isotonic replacement of NaCl with KCl. Nominal Ca2+-free solution was made by substituting an equimolar concentration of MgCl2 for CaCl2 (no EGTA added). To make hypertonic solutions, 20 mM sucrose or 20 mM D-mannitol was added to the Krebs solution.
Chemicals
Drugs used were acetylcholine hydrochloride (Daiichi Pharmaceutical Co. Ltd., Tokyo, Japan), A23187 and cyclopiazonic acid (CPA) (Calbiochem, La Jolla, CA, U.S.A.), BAPTA-AM (Dojin, Kumamoto, Japan), 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) (Research Biochemicals International, Natick, MA, U.S.A.), bumetanide, niflumic acid and glibenclamide (Sigma, St. Louis, MO, U.S.A.), sucrose and D-mannitol, (Wako Pure Chemical, Osaka, Japan), apamin and charybdotoxin (ChTX) (Peptide Institute, Osaka, Japan).
A23187, CPA, BAPTA-AM and glibenclamide were dissolved in dimethyl sulphoxide (DMSO, Dojin, Kumamoto, Japan). The final concentration of DMSO when diluted in Krebs solution was 0.06% at its maximum. This concentration of DMSO had no effect on the ACh-induced electrical responses. NPPB was dissolved in ethanol (stock, 10 mM). Further dilutions were made using Krebs solution. The powdered forms of bumetanide, niflumic acid, sucrose and mannitol were directly dissolved in Krebs solution.
Statistics
All values are expressed as means±s.e.mean; n denotes the number of valves. Statistical significance was determined using Student's paired and unpaired t-tests. Differences were considered to be significant at P<0.05.
Results
ACh-induced membrane potential changes
The resting membrane potential of RAVEC was −49.6±1.2 mV (n=29). ACh (0.01–3 μM) concentration-dependently produced a transient, followed by a sustained membrane hyperpolarization (Figure 1Aa). In the presence of 3 μM ACh, the sustained hyperpolarization could be maintained for over 10 min, but with a progressive reduction in its amplitude (Figure 1Ab). Following the removal of ACh, a transient membrane depolarization appeared. The depolarization reached its peak within 2 min and the membrane then repolarized to the original level within 15 min. The amplitude of this depolarization was dependent on the concentration of the pre-applied ACh (up to 1 μM) (Figure 1Aa and Ab). These responses to ACh were reproducible when ACh was applied at 25 min intervals.
Figure 1.
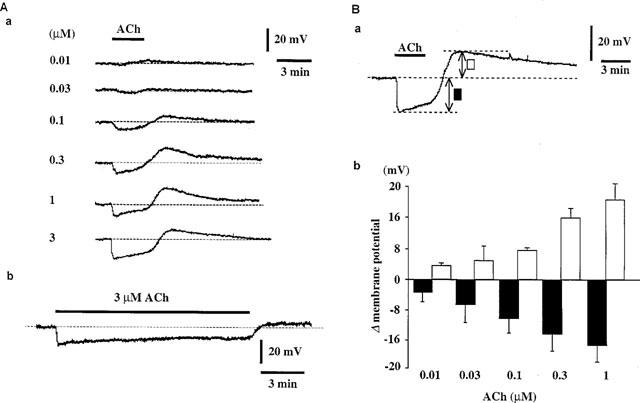
Effects of ACh on membrane potential in RAVEC. (Aa) Concentration-dependent effects of ACh on membrane potential. ACh (0.01–3 μM) was applied for 3 min. Recordings were all from the same preparation. (Ab) Hyperpolarization was maintained throughout a 10-min application of 3 μM ACh. (Ba) and (Bb) Summary of the effects of ACh on membrane potential. Bb shows the maximum hyperpolarization and depolarization elicited by each concentration of ACh (mean of data from three preparations, with s.e. shown by vertical lines). Ba indicates how these maxima were obtained.
High K+ (50 mM) depolarized the membrane from −54.5±1.7 to −23.5±0.2 mV (n=3), and ACh (3 μM) did not modify the membrane potential in the presence of high K+ (−23.5±0.2 mV, n=3). Low K+ (1.2 mM) did not alter the resting membrane potential (−49.3±2.9 to −51.8±2.9 mV, P>0.05, n=4) but it did enhance the ACh-induced transient (18.7±1.7 to 38.6±2.4 mV, P<0.01, n=4) and sustained (17.5±3.0 to 37.6±2.8 mV, P<0.01, n=4) hyperpolarizations.
Effects of K+-channel inhibitors on ACh-induced electrical responses (Figure 2)
Figure 2.
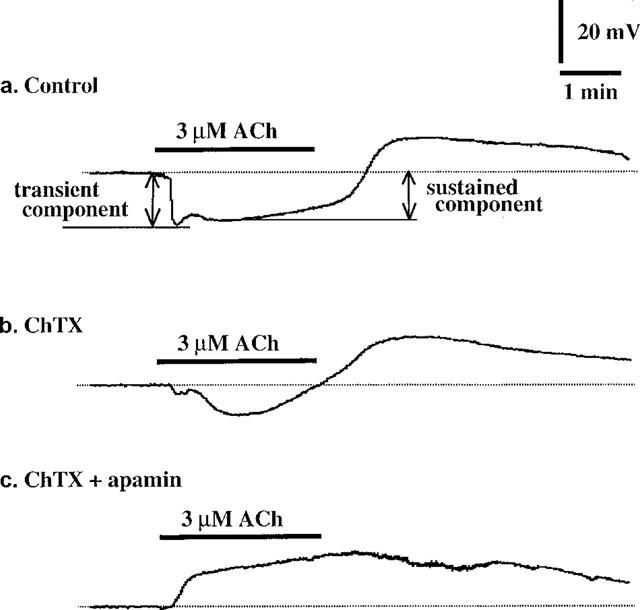
Effects of ChTX and apamin on ACh-induced membrane potential changes. Actual tracings of the effects of ChTX and apamin. (a) Control; (b) in the presence of ChTX (50 nM); (c) in the presence of both ChTX (50 nM) and apamin (0.1 μM). Recordings were all obtained from the same cell.
Apamin (0.1 μM) did not modify the resting membrane potential (−51.5±1.3 to −53.2−1.0 mV, P>0.05, n=6) but it significantly attenuated the ACh-induced sustained hyperpolarization (15.3±1.2 to 11.1±1.5 mV, P<0.05, n=6) with no effect on the ACh-induced transient hyperpolarization (17.1±1.3 to 16.4±1.1 mV, P>0.05, n=6). Apamin significantly enhanced the depolarization seen after washout of ACh (8.8±0.9 to 10.8±1.5 mV, P<0.05, n=6). ChTX (50 nM) did not modify the resting membrane potential (−49.7±1.2 to −49.4±0.9 mV, P>0.05, n=5) but it significantly attenuated the ACh-induced transient (16.5±2.1 to 2.8±2.8 mV, P<0.01, n=5) and sustained (16.6±2.2 to 8.7±1.1 mV, P<0.05, n=5) hyperpolarizations (Figure 2b). ChTX significantly enhanced the membrane depolarization seen after washout of ACh (8.4±1.3 to 13.0±1.2 mV, P<0.05, n=5). In the presence of ChTX (50 nM), application of apamin (0.1 μM) did not modify the resting membrane potential (−51.2±4.3 to −49.4±4.7 mV, P>0.05, n=3). However, under these conditions, ACh produced a sustained depolarization, not a hyperpolarization, with a relatively slow onset (Figure 2c).
Glibenclamide (10 μM) modified neither the resting membrane potential nor the ACh-induced hyperpolarization (n=3). Ba2+ (0.1 mM) significantly depolarized the membrane by 10.4±0.5 mV (n=3, P<0.01) but did not modify the ACh-induced hyperpolarization.
Effects of Ca2+-free solution and BAPTA-AM on ACh-induced responses
At the end of a 10 min application of nominal Ca2+-free solution, the membrane was depolarized by 9.3±3.2 mV (n=4). Repetitive application of ACh in Ca2+-free solution at 25 min intervals resulted in a progressive alteration in the ACh-induced hyperpolarization: it changed from a sustained hyperpolarization (with a depolarization after removal of ACh) to a transient hyperpolarization followed, while the ACh was still present, by a depolarization (n=4) (Figure 3a).
Figure 3.

Effects of Ca2+-free solution (a) and BAPTA-AM (b) on ACh-induced membrane potential changes. (a) ACh (3 μM) was first applied in normal Krebs solution (as control). The preparation was then exposed to Ca2+-free solution for 10 min, and ACh was subsequently applied at 225 min intervals. The traces were obtained from the same cell. Similar observations were made in two other cells. (b) BAPTA-AM (30 μM) was pretreated for 40 min followed by a 10 min washout, then ACh (3 μM) was applied. Similar observations were made in two other cells.
When RAVEC were pre-treated with 30 μM BAPTA-AM for 40 min, the membrane potential was not significantly changed (−50.4±0.2 to −53.2±1.1 mV, P>0.05, n=4). In such BAPTA-AM-treated cells, ACh produced only a sustained depolarization (3.2±1.8 mV, n=4, Figure 3b).
Effects of Co2+ and Ni2+ on ACh-induced responses
Ni2+ (5 mM) and Co2+ (2 mM) depolarized the membrane by 6.4±1.5 mV (n=5) and 5.4±2.5 mV (n=3), respectively. In the presence of Ni2+ or Co2+, ACh produced a transient hyperpolarization while the ACh was still present (Figure 4a and b). When Ni2+ was applied during the ACh-induced sustained hyperpolarization, the membrane response was converted to a depolarization (n=4) (Figure 4c).
Figure 4.
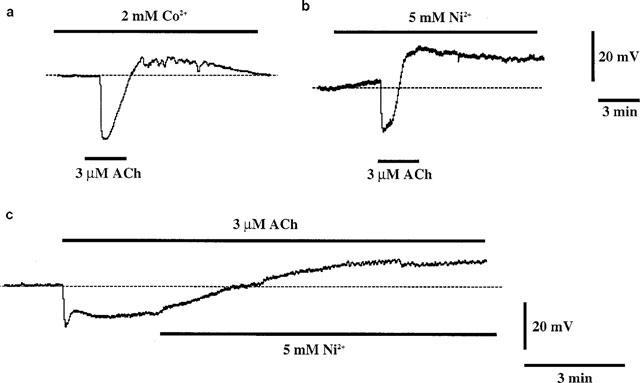
Effects of 2 mM Co2+ (a) and 5 mM Ni2+ (b and c) on ACh-induced membrane potential changes. ACh (3 μM) was applied for 3 min as indicated by bars (a and b). Co2+ and Ni2+ were added 10 min before ACh and were present throughout the application of ACh. (c) Ni2+ (5 mM) was applied during the sustained component of the hyperpolarization induced by 3 μM Ach.
Effects of CPA and A-23187 on ACh-induced responses
CPA (3 μM) itself produced a small hyperpolarization (3.4±1.4 mV, P<0.05, n=3). In the presence of CPA, ACh produced a hyperpolarization which lasted long after the washout of ACh (Figure 5a). When ACh was again applied after a 30 min interval (in the continued presence of CPA), it produced only a small hyperpolarization. The membrane potential level reached as a result of the second application of ACh (−67.0±3.0 mV, P>0.05, n=3) was similar to that produced by the first. Under these conditions, Co2+ converted the membrane response to a depolarization (n=3) (Figure 5b).
Figure 5.
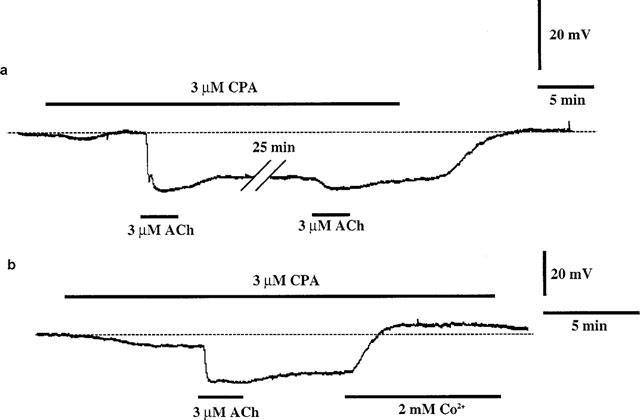
Effect of CPA (3 μM) on ACh-induced membrane potential changes. (a) In the presence of CPA (3 μM), the recovery from the ACh-induced hyperpolarization that occurred after washout of ACh was largely prevented. When ACh was again applied after an interval of 25 min (in the continued presence of CPA), ACh evoked only a small hyperpolarization. The hyperpolarization was maintained for as long as CPA was present. (b) In the presence of CPA, ACh produced a hyperpolarization that was maintained after the removal of ACh. When Co2+ (2 mM) was applied during this maintained hyperpolarization, the membrane response was converted to a depolarization.
A23187 (0.1 μM) had no significant effect on the resting membrane potential but it did significantly attenuate the ACh-induced transient hyperpolarization (17.3±1.7 to 6.5±2.0 mV, P<0.01) with no change in the amplitude of the sustained component (though its duration was increased) (n=4, Figure 6).
Figure 6.
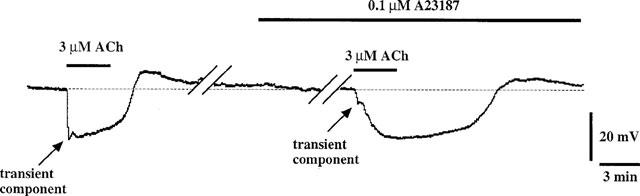
Effect of A23187 (0.1 μM) on ACh-induced membrane potential changes. The transient component of the ACh-induced hyperpolarization was attenuated in the presence of A23187.
Effects of inhibitors of Cl− channels and Cl− transporters on ACh-induced responses
Neither NPPB (20 nM) nor bumetanide (10 μM) modified the resting membrane potential or the ACh-induced maximum hyperpolarization (n=4). However, these agents did significantly attenuate the depolarization seen after the washout of ACh. The depolarizations were by 7.8±0.9 and 4.4±0.3 mV in the absence and presence of NPPB, respectively (P<0.05, n=4, Figure 7a), and by 6.9±1.0 and 2.1±0.9 mV in the absence and presence of bumetanide, respectively (P<0.01, n=5, Figure 7b). Niflumic acid (0.5 mM) hyperpolarized the membrane by 14.5±2.5 mV (n=4) but it had no effect on the depolarization seen after the removal of ACh. The peak amplitude of the depolarization was 10.2±2.2 and 11.2±0.1 mV in the absence and presence of niflumic acid, respectively (P>0.05, n=3).
Figure 7.
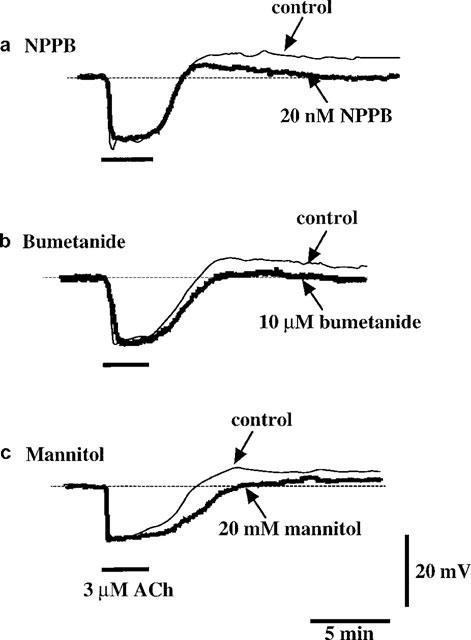
Effects of NPPB, bumetanide and mannitol on ACh-induced membrane potential changes. ACh-induced responses obtained before and after application of inhibitors [NPPB (20 nM, a), bumetanide (10 μM, b) and mannitol (20 mM, c)] in the same cell.
Hypertonic solution containing 20 mM D-mannitol did not significantly modify either the resting membrane potential (−53.5±1.1 to −54.3±1.7 mV, P>0.05, n=9) or the ACh-induced maximum hyperpolarization (14.6±1.6 to 13.1±1.2 mV, P>0.05, n=7). This hypertonic solution, however, did significantly attenuate the depolarization seen after the removal of ACh (5.9±1.1 to 1.9±0.6 mV, P<0.05, n=8, Figure 7c). Similar results were obtained when the hypertonic solution used was made by the addition of 20 mM sucrose (n=4, data not shown).
Discussion
ACh produces a transient, followed by a sustained hyperpolarization in the endothelial cells of the porcine coronary artery (Chen & Cheung, 1992) and in those of the rabbit aorta (Wang et al., 1996). In the endothelial cells of the intact rat aorta, ACh also produces a biphasic response: a transient hyperpolarization followed in that case by a depolarization (Marchenko & Sage, 1993; 1994a). In the present experiments, ACh produced a transient, then a sustained hyperpolarization (which could be maintained for at least 10 min) followed by a transient depolarization that occurred after the removal of the ACh. Thus, the ACh-induced membrane potential changes in endothelial cells may vary by species and by region.
ACh-induced hyperpolarization
In the present experiments, ACh produced a transient, followed by a sustained hyperpolarization. These two ACh-induced responses were both abolished in high K+ (50 mM) solution and enhanced in low K+ (1.2 mM) solution. Furthermore, the ACh-induced hyperpolarization was abolished in BAPTA-AM-treated cells. These results indicate that in RAVEC, the ACh-induced hyperpolarization is due to an activation of those K+ channels that are regulated by [Ca2+]i.
The ACh-induced transient hyperpolarization was preserved in Ca2+-free solution and it was resistant to both Ni2+ and Co2+. Furthermore, a low concentration of A23287 (0.1 μM), which is thought to act selectively on the Ca2+ storage sites (Itoh et al., 1985), greatly attenuated the ACh-induced transient hyperpolarization but not the sustained hyperpolarization. These results suggest that the transient hyperpolarization is due to a release of Ca2+ from the storage sites. In contrast, the ACh-induced sustained hyperpolarization was abolished in Ca2+-free solution and by Co2+ or Ni2+. Furthermore, in the presence of CPA (an inhibitor of Ca2+-ATPase in the Ca2+ storage sites: Li & van Breemen, 1996; Sasajima et al., 1997), the ACh-induced hyperpolarization was maintained long after the washout of this agonist. This result is consistent with a previous finding in the endothelial cells of the guinea-pig coronary artery (Cheung & Chen, 1992). The sustained ACh-induced hyperpolarization seen in the presence of CPA was blocked by Co2+. These results suggest that ACh activates Co2+-sensitive Ca2+ influx, thus causing a sustained membrane hyperpolarization.
Several types of K+ channels have been identified in endothelial cells (Lückhoff & Busse, 1990a; Sakai, 1990; Chen & Cheung, 1992; Sharma & Davis, 1994; Song & Davis, 1994; Nilius et al., 1997c). It has been found that ChTX and tetraethylammonium, inhibitors of KCa channels, both inhibit the ACh-induced hyperpolarization seen in the endothelial cells of the guinea-pig coronary artery (Chen & Cheung, 1992) and in those of the rabbit aorta (Wang et al., 1996). Recently, it has been reported that an apamin-sensitive K+ channel is functionally expressed in the endothelial cells of the rat aorta (Marchenko & Sage, 1996) and in those of the human umbilical vein (Muraki et al., 1997). In the present experiments, the transient component of the ACh-induced hyperpolarization was markedly attenuated by ChTX (but not by apamin). In contrast, the sustained component of the ACh-induced hyperpolarization was inhibited by ChTX and by apamin. Furthermore, a combined application of ChTX and apamin completely blocked both components of the ACh-induced hyperpolarization. Neither glibenclamide (an inhibitor of ATP-sensitive K+ channels) nor Ba2+ (an inhibitor of inwardly rectifying K+ channels) affected the ACh-induced hyperpolarization. These results suggest that the ACh-induced transient hyperpolarization is probably due to an activation of ChTX-sensitive KCa channels, while the sustained hyperpolarization is due to an activation of both ChTX-sensitive and apamin-sensitive KCa channels.
In the present experiments, CPA (3 μM) and A23187 (0.1 μM) by themselves produced only minor hyperpolarizations. In preliminary experiments, however, we found that 0.3 μM A23187 produced a profound hyperpolarization (∼12–15 mV), suggesting that the small hyperpolarization induced by either agent in the experiments reported here might have been due to them being in a concentration barely sufficient to produce hyperpolarization. In line with this hypothesis, it was reported a few years ago that in pig coronary artery endothelial cells, A23187 produces a hyperpolarization at 0.5 μM (Weid & Bény, 1992).
ACh-induced depolarization
In the present experiments, a transient membrane depolarization was seen after the removal of ACh. Although the membrane hyperpolarization could be sustained as long as ACh was present (at least up to 10 min), application of Ni2+ or Co2+ converted this response to a depolarization. Furthermore, in Ca2+-free solution, the transient hyperpolarization was followed by a depolarization while the ACh was still present. Moreover, in the combined presence of ChTX and apamin, ACh evoked only a sustained depolarization of slow onset. The sustained component of the hyperpolarization diminished in amplitude as a function of time despite continuous stimulation by ACh, so these results, taken together, suggest that the depolarization phase was being generated while ACh was still present and that it was being masked by the hyperpolarization. This idea is consistent with previous findings by Marchenko & Sage (1993, 1994a).
In the present experiments, an ACh-induced depolarization was observed in BAPTA-AM-treated cells. Furthermore, niflumic acid (an inhibitor of Ca2+-activated Cl− channels, Nilius et al., 1997a) failed to inhibit the ACh-induced depolarization, suggesting that [Ca2+]i does not play an important role in the production of the ACh-induced depolarization. It has been suggested that endothelial cell volume is increased by vasoactive agonists (bradykinin, angiotensin II, vasopressin) through an activation of Na+-K+-Cl− co-transport (O'Donnell, 1993). Furthermore, it has been found that the volume-sensitive chloride current is cross-correlated with changes in cell volume in endothelial cells of the bovine pulmonary artery (Nilius et al., 1996; 1997b; Voets et al., 1998). In the present experiments, the ACh-induced depolarization was markedly attenuated by bumetanide (an inhibitor of the Na+-K+-Cl− transporter) (Cabantchik & Greger, 1992), by NPPB (a non-selective Cl− channel inhibitor) (Cabantchik & Greger, 1992) and by the presence of hypertonic conditions. It is thought that Na+-K+-Cl− co-transport could be electrically neutral (Haas, 1994). Thus, we speculate that the final pathway for the ACh-induced depolarization may involve an activation of volume-sensitive Cl− (ClVol) channels. Taken together, these results could be taken to suggest that ACh increases cell volume through an activation of the Na+-K+-Cl− co-transporter, thus leading to ClVol-channel activation. However, it remains to be clarified how ClVol channels might be activated during stimulation with ACh.
In the present study, neither NPPB nor hypertonic solution changed the resting membrane potential. In accordance with the findings of Nilius et al. (1996) in the endothelial cells of the bovine pulmonary artery, this result suggests that ClVol channels may not be activated under resting conditions.
In conclusion, in RAVEC, ACh produces a transient, followed by a sustained membrane hyperpolarization due to a co-activation of ChTX-sensitive and apamin-sensitive K+ channels. It is also suggested that volume-sensitive Cl− channels most likely contribute to the ACh-induced membrane depolarization (a phase of the response that is largely concealed by the ACh-induced hyperpolarization under physiological conditions).
Acknowledgments
We thank Professor Hikaru Suzuki (Department of Physiology, Nagoya City University Medical School) for critical comments and Dr R.J. Timms for a critical reading of the English. This work was partly supported by a Grant-In-Aid from the Ministry of Education of Japan.
Abbreviations
- ACh
acetylcholine
- BAPTA-AM
acetoxymethyl ester of bis-(aminophenoxy)ethane-N, N, N′, N′-tetraacetic acid
- ChTX
charybdotoxin
- ClVol channel
volume-sensitive chloride channel
- CPA
cyclopiazonic acid
- DMSO
dimethyl sulphoxide
- NPPB
5-nitro-2-(3-phenylpropylamino) benzoic acid
- RAVEC
rabbit aortic valve endothelial cells
References
- BÉNY J.-L., PACICCA C. Bidirectional electrical communication between smooth muscle and endothelial cells in the pig coronary artery. Am. J. Physiol. 1994;266:H1465–H1472. doi: 10.1152/ajpheart.1994.266.4.H1465. [DOI] [PubMed] [Google Scholar]
- BÉNY J.-L. Electrical coupling between smooth muscle cells and endothelial cells in pig coronary arteries. Pflügers Arch. 1997;433:364–367. doi: 10.1007/s004240050289. [DOI] [PubMed] [Google Scholar]
- BRUNET P.C., BÉNY J.-L. Substance P and bradykinin hyperpolarize pig coronary artery endothelial cells in primary culture. Blood Vessels. 1989;26:228–234. doi: 10.1159/000158770. [DOI] [PubMed] [Google Scholar]
- CABANTCHIK Z.I., GREGER R. Chemical probes for anion transporters of mammalian cell membranes. Am. J. Physiol. 1992;262:C803–C827. doi: 10.1152/ajpcell.1992.262.4.C803. [DOI] [PubMed] [Google Scholar]
- CARTER T.D., OGDEN D. Acetylcholine-stimulated changes of membrane potential and intracellular Ca2+ concentration recorded in endothelial cells in situ in the isolated rat aorta. Pflügers Arch. 1994;428:476–484. doi: 10.1007/BF00374568. [DOI] [PubMed] [Google Scholar]
- CHEN G., SUZUKI H., WESTON A.H. Acetylcholine releases endothelium-derived hyperpolarizing factor and EDRF from rat blood vessels. Br. J. Pharmacol. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN G., CHEUNG D.W. Characterization of acetylcholine-induced membrane hyperpolarization in endothelial cells. Circ. Res. 1992;70:257–263. doi: 10.1161/01.res.70.2.257. [DOI] [PubMed] [Google Scholar]
- CHEUNG D.W., CHEN G. Calcium activation of hyperpolarization response to acetylcholine in coronary endothelial cells. J. Cardiovasc. Pharmacol. 1992;20 suppl. 12:S120–S123. doi: 10.1097/00005344-199204002-00034. [DOI] [PubMed] [Google Scholar]
- FRIEDEN M., BÉNY J.-L. Effect of 5-hydroxytryptamine on the membrane potential of endothelial and smooth muscle cells in pig coronary artery. Br. J. Pharmacol. 1995;115:95–100. doi: 10.1111/j.1476-5381.1995.tb16325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAAS M. The Na-K-Cl cotransporters. Am. J. Physiol. 1994;267:C869–C885. doi: 10.1152/ajpcell.1994.267.4.C869. [DOI] [PubMed] [Google Scholar]
- HIMMEL H.M., RASMUSSON R.L., STRAUSS H.C. Agonist-induced changes of [Ca2+]i and membrane currents in single bovine aortic endothelial cells. Am. J. Physiol. 1994;267:C1338–C1350. doi: 10.1152/ajpcell.1994.267.5.C1338. [DOI] [PubMed] [Google Scholar]
- ITOH T., KANMURA Y., KURIYAMA H. A23187 increases calcium permeability of store sites more than of surface membranes in the rabbit mesenteric artery. J. Physiol. 1985;359:467–484. doi: 10.1113/jphysiol.1985.sp015597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLEIN J.D., O'NEILL W.C. Effect of bradykinin on Na-K-2Cl cotransport and bumetanide binding in aortic endothelial cells. J. Biol. Chem. 1990;265:22238–22242. [PubMed] [Google Scholar]
- KU D.D., NELSON J.M., CAULFIELD J.B., WINN M.J. Release of endothelium-derived relaxing factors from canine cardiac valves. J. Cardiovasc. Pharmacol. 1990;16:212–218. doi: 10.1097/00005344-199008000-00006. [DOI] [PubMed] [Google Scholar]
- LI L., VAN BREEMEN C. Agonist- and CPA-induced elevation of cytoplasmic free Ca2+ in intact valvular endothelium from rabbits. Am. J. Physiol. 1996;270:H837–H848. doi: 10.1152/ajpheart.1996.270.3.H837. [DOI] [PubMed] [Google Scholar]
- LÜCKHOFF A., BUSSE R. Activators of potassium channels enhance calcium influx into endothelial cells as a consequence of potassium currents. Naunyn-Schmiedeberg's Arch. Pharmacol. 1990a;342:94–99. doi: 10.1007/BF00178979. [DOI] [PubMed] [Google Scholar]
- LÜCKHOFF A., BUSSE R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflügers Arch. 1990b;416:305–311. doi: 10.1007/BF00392067. [DOI] [PubMed] [Google Scholar]
- MARCHENKO S.M., SAGE S.O. Electrical properties of resting and acetylcholine-stimulated endothelium in intact rat aorta. J. Physiol. 1993;462:735–751. doi: 10.1113/jphysiol.1993.sp019579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARCHENKO S.M., SAGE S.O. Mechanism of acetylcholine action on membrane potential of endothelium of intact rat aorta. Am. J. Physiol. 1994a;266:H2388–H2395. doi: 10.1152/ajpheart.1994.266.6.H2388. [DOI] [PubMed] [Google Scholar]
- MARCHENKO S.M., SAGE S.O. Smooth muscle cells affect endothelial membrane potential in rat aorta. Am. J. Physiol. 1994b;267:H804–H811. doi: 10.1152/ajpheart.1994.267.2.H804. [DOI] [PubMed] [Google Scholar]
- MARCHENKO S.M., SAGE S.O. Calcium-activated potassium channels in the endothelium of intact rat aorta. J. Physiol. 1996;492:53–60. doi: 10.1113/jphysiol.1996.sp021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEHRKE G., DAUT J. The electrical response of cultured guinea-pig coronary endothelial cells to endothelium-dependent vasodilators. J. Physiol. 1990;430:251–272. doi: 10.1113/jphysiol.1990.sp018290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURAKI K., IMAIZUMI Y., OHYA S., SATO K., TAKII T., ONOZAKI K., WATANABE M. Apamin-sensitive Ca2+-dependent K+ current and hyperpolarization in human endothelial cells. Biochem. Biophys. Res. Commun. 1997;236:340–343. doi: 10.1006/bbrc.1997.6949. [DOI] [PubMed] [Google Scholar]
- NILIUS B., EGGERMONT J., VOETS T., DROOGMANS G. Volume-activated Cl− channels. Gen. Pharmacol. 1996;27:1131–1140. doi: 10.1016/s0306-3623(96)00061-4. [DOI] [PubMed] [Google Scholar]
- NILIUS B., PRENEN J., SZÜCS G., WEI L., TANZI F., VOETS T., DROOGMANS G. Calcium-activated chloride channels in bovine pulmonary artery endothelial cells. J. Physiol. 1997a;498:381–396. doi: 10.1113/jphysiol.1997.sp021865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NILIUS B., SZÜCS G., HEINKE S., VOETS T., DROOGMANS G. Multiple types of chloride channels in bovine pulmonary artery endothelial cells. J. Vasc. Res. 1997b;34:220–228. doi: 10.1159/000159226. [DOI] [PubMed] [Google Scholar]
- NILIUS B., VIANA F., DROOGMANS G. Ion channels in vascular endothelium. Annu. Rev. Physiol. 1997c;59:145–170. doi: 10.1146/annurev.physiol.59.1.145. [DOI] [PubMed] [Google Scholar]
- O'DONNELL M.E. Role of Na-K-Cl cotransport in vascular endothelial cell volume regulation. Am. J. Physiol. 1993;264:C1316–C1326. doi: 10.1152/ajpcell.1993.264.5.C1316. [DOI] [PubMed] [Google Scholar]
- PERRY P.B., O'NEILL W.C. Swelling-activated K fluxes in vascular endothelial cells: volume regulation via K-Cl cotransport and K channels. Am. J. Physiol. 1993;265:C763–C769. doi: 10.1152/ajpcell.1993.265.3.C763. [DOI] [PubMed] [Google Scholar]
- SAKAI T. Acetylcholine induces Ca-dependent K currents in rabbit endothelial cells. Jpn. J. Pharmacol. 1990;53:235–246. doi: 10.1254/jjp.53.235. [DOI] [PubMed] [Google Scholar]
- SASAJIMA H., WANG X., VAN BREEMEN C. Fractional Ca2+ release from the endoplasmic reticulum activates Ca2+ entry in freshly isolated rabbit aortic endothelial cells. Biochem. Biophys. Res. Comm. 1997;241:471–475. doi: 10.1006/bbrc.1997.7844. [DOI] [PubMed] [Google Scholar]
- SHARMA N.R., DAVIS M.J. Mechanism of substance P-induced hyperpolarization of porcine coronary artery endothelial cells. Am. J. Physiol. 1994;266:H156–H164. doi: 10.1152/ajpheart.1994.266.1.H156. [DOI] [PubMed] [Google Scholar]
- SONG J., DAVIS M.J. Chloride and cation currents activated by bradykinin in coronary venular endothelial cells. Am. J. Physiol. 1994;267:H2508–H2515. doi: 10.1152/ajpheart.1994.267.6.H2508. [DOI] [PubMed] [Google Scholar]
- STUREK M., SMITH P., STEHNO-BITTEL L.In vitro models of vascular endothelial cell calcium regulation Ion channels of vascular smooth muscle cells and endothelial cells 1991New York: Elsevier; 349–364.ed. Sperelakis, N. & Kuriyama, H. pp [Google Scholar]
- VOETS T., MANOLOPOULOS V., EGGERMONT J., ELLORY C., DROOGMANS G., NILIUS B. Regulation of a swelling-activated chloride current in bovine endothelium by protein tyrosine phosphorylation and G proteins. J. Physiol. 1998;506:341–352. doi: 10.1111/j.1469-7793.1998.341bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG X., CHU W., VAN BREEMEN C. Potentiation of acetylcholine-induced responses in freshly isolated rabbit aortic endothelial cells. J. Vasc. Res. 1996;33:414–424. doi: 10.1159/000159170. [DOI] [PubMed] [Google Scholar]
- WEID P.-Y., BÉNY J.-L. Effect of Ca2+ ionophores on membrane potential of pig coronary artery endothelial cells. Am. J. Physiol. 1992;262:H1823–H1831. doi: 10.1152/ajpheart.1992.262.6.H1823. [DOI] [PubMed] [Google Scholar]
- WEID P.-Y., BÉNY J.-L. Simultaneous oscillations in the membrane potential of pig coronary artery endothelial and smooth muscle cells. J. Physiol. 1993;471:13–24. doi: 10.1113/jphysiol.1993.sp019888. [DOI] [PMC free article] [PubMed] [Google Scholar]