

Abstract
The effect of IL-4 on responses to intraplantar (i.pl.) carrageenin, bradykinin, TNFα, IL-1β, IL-8 and PGE2 was investigated in a model of mechanical hyperalgesia in rats. Also, the cellular source of the IL-4 was investigated.
IL-4, 30 min before the stimulus, inhibited responses to carrageenin, bradykinin, and TNFα, but not responses to IL-1β, IL-8 and PGE2.
IL-4, 2 h before the injection of IL-1β, did not affect the response to IL-1β, whereas IL-4, 12 or 12+2 h before the IL-1β, inhibited the hyperalgesia (−30%, −74%, respectively).
In murine peritoneal macrophages, murine IL-4 for 2 h before stimulation with LPS, inhibited (−40%) the production of IL-1β but not PGE2. Murine IL-4 (for 16 h before stimulation with LPS) inhibited LPS-stimulated PGE2 but not IL-1β.
Anti-murine IL-4 antibodies potentiated responses to carrageenin, bradykinin and TNFα, but not IL-1β and IL-8, as well as responses to bradykinin in athymic rats but not in rats depleted of mast cells with compound 40/80.
These data suggest that IL-4 released by mast cells limits inflammatory hyperalgesia. During the early phase of the inflammatory response the mode of action of the IL-4 appears to be inhibition of the production TNFα, IL-1β and IL-8. In the later phase of the response, in addition to inhibiting the production of pro-inflammatory cytokines, IL-4 also may inhibit the release of PGs.
Keywords: Inflammatory hyperalgesia, interleukin-4, bradykinin, tumour necrosis factor alpha, interleukin-1β, interleukin-8, prostaglandin E2
Introduction
Cytokines constitute a link between cellular injury and recognition of non-self and the development of local and systemic signs and symptoms of inflammation, such as cell migration, oedema, fever and hyperalgesia (Dinarello et al., 1986; Ferreira et al., 1988, Faccioli et al., 1990, Dinarello, 1991). In this context, it was shown, in a model of mechanical hyperalgesia, that carrageenin-evoked hyperalgesia results from the combined effects of the release of cyclo-oxygenase products and sympathomimetic amines (Nakamura & Ferreira, 1987). A cascade of cytokine release preceded the generation of these mediators (Cunha et al., 1992). Carrageenin and LPS caused the release of bradykinin, which stimulated the release of TNFα. TNFα induced the release of IL-1β and IL-6, which stimulated the production of cyclo-oxygenase products, as well as IL-8, which stimulated production of sympathomimetic mediators (Cunha et al., 1992; Ferreira et al., 1993). In a different model, the ‘tail-flick' method, LPS-induced hyperalgesia was also mediated by TNFα and IL-1, and the effect of TNFα was dependent upon IL-1, although prostaglandins did not appear to be involved in the responses (Watkins et al., 1994, 1995). In addition, TNFα has been suggested to be a mediator of inflammatory pain in man since a monoclonal antibody (MAb) neutralizing human TNFα diminished the pain associated with rheumatoid arthritis (Rankin et al., 1995).
In the last decade, cytokines generally regarded as ‘anti-inflammatory' have been described which inhibit the production of cytokines such as IL-1β, IL-6, IL-8 and TNFα, which are generally regarded as ‘pro-inflammatory'. The class of anti-inflammatory cytokines includes IL-4, IL-10, IL-13 and TGFβ (Elias et al., 1991; Callard et al., 1996; Hart et al., 1989; Standiford et al., 1990; Fiorentino et al., 1991; Cassatella et al., 1993; Lord & Lamb, 1996). Recently, it was shown that IL-10 (probably originating from local macrophages) inhibited the inflammatory hyperalgesic responses to carrageenin and bradykinin by two mechanisms: inhibition of hyperalgesic cytokine release and blockade of induction of cyclo-oxygenase-2 (COX-2, Poole et al., 1995). Another inhibitory cytokine, IL-4, produced by Th2 lymphocytes and mast cells, is known to limit production of the cytokines IL-1β, IL-6, IL-8 and TNFα (Lord & Lamb, 1996; Tunon de Lara et al., 1994). Also, IL-4 can suppress the delayed type hypersensitivity responses in experimental animals and in man, (Rocken et al., 1996), possibly because of its capacity to induce Th2 T cell responses. In addition to inhibition of the production of inflammatory cytokines, IL-4 has been reported to inhibit the induction of COX-2, with a consequent reduction in the production of PGs (Seitz et al., 1994; Endo et al., 1994).
Given the capacity of IL-4 to inhibit the production of pro-inflammatory cytokines, such as TNFα, IL-1β, and IL-8, we have tested the possibility that IL-4 limits inflammatory hyperalgesia by diminishing the cytokine mediated hyperalgesic responses to carrageenin, bradykinin, and pro-inflammatory cytokines. In addition, the source of the IL-4 released in inflammatory hyperalgesia was investigated.
Methods
Nociceptive test
A constant pressure of 20 mmHg was applied to the hind-paws of rats and discontinued when they presented a typical freezing reaction (reaction time). This reaction was characterized by a reduction of escape movements: animals usually made several attempts to escape from the position imposed by the experimental situation. These were followed by alterations in respiratory frequency with the onset of a typical shivering reaction. The intensity of hyperalgesia was quantified as the variation in reaction time (delta reaction time) obtained by subtracting values measured 3 h after administration of hyperalgesic substances from (control) reaction times measured before injection at time zero (Ferreira et al., 1978). Reaction times were typically 32–34 s (with s.e.means of 0.5–1.0 s) before injection and 2–4 s after stimulation with hyperalgesic agents. Multiple paw treatments did not alter basal reaction times.
Experimental protocol
In vivo measurements
Hyperalgesia was measured 3 h after injections (in 100 μl, i.pl.) of carrageenin (10 or 100 μg), bradykinin (50 or 500 ng), TNFα (0.25 or 2.5 pg), IL-1β (0.05 or 0.5 pg), IL-8 (10 or 100 pg), and PGE2 (10 or 100 ng) into the hind-paws of rats. [The doses of hyperalgesic agents were the smallest that evoked maximum responses and the time interval of 3 h was a time at which responses to the chosen doses of hyperalgesic agents were all at or close to their peak (Ferreira et al., 1988, 1993; Cunha et al., 1991, 1992; Poole et al., 1995). The order of potency of the cytokines was IL-1β>TNFα>>IL-8 for these human sequence cytokines in rats (Cunha et al., 1992). Whether this order of potency reflects the order of potency of the endogenous cytokines of the rat is unknown. The rat sequence cytokines were not available for this study and certain cytokines exhibit species preference or specificity (Lumpkin, 1987; Morstyn & Burgess, 1988; Stefferl et al., 1996) and the native cytokines remain to be tested.] IL-4 (2.5–10 ng in 50 μl, i.pl.) or saline (50 μl) was injected in the (same) hind-paws, 30 min, 2, 12, or 12+2 h before hyperalgesic agents. MAbs to murine IL-4 (BVDG or 11B11, 50 μg in 50 μl, i.pl) were injected into hind-paws to be injected with hyperalgesic agents, 30 min before carrageenin, bradykinin, TNFα, IL-1β, IL-8, and dextran (200 μg, 100 μl). BVDG (50 μg in 50 μl, i.pl) was injected into the hind-paws of athymic rats and, 30 min later, bradykinin (50 ng, 100 μl) was injected into the (same) hind-paws. Hyperalgesia was assessed after a further 3 h. Also, hyperalgesia was measured 3 h after injection of bradykinin into hind-paws of rats depleted of mast cells by chronic administration (in 50 μl, i.pl.) of compound 48/80 (day 1: 1 μg, day 2: 3 μg, days 3 and 4: 10 μg). Bradykinin (50 ng, 100 μl, i.pl.) was injected on day 5.
In vitro measurements
Murine peritoneal macrophages, harvested from the peritoneal cavities of mice treated, 3 days earlier, with sterile thioglycollate (2 ml of a 3% w/v solution), were allowed to adhere to 24-well plastic tissue culture plates. The macrophages were seeded at 106 cells ml−1 well−1 in RPMI with 10% foetal calf serum, penicillin (100 U ml−1) and streptomycin (100 mg ml−1), for 1 h at 37°C, in an atmosphere of 5% CO2 in air. The monolayers that formed were washed three times with phosphate buffered saline (PBS), pH 7.4, and cultured at 37°C with the following: (a) RPMI medium, (b) LPS (3 μg ml−1), (c) murine IL-4 (1.6 and 16 ng ml−1), (d) murine IL-4 (1.6 and 16 ng ml−1) and, 12 h later, LPS (3 μg ml−1), (e) murine IL-4 (16 ng ml−1) and, 2 h later, LPS (3 μg ml−1). After culture for a further 16 h, the concentrations of IL-1β and PGE2 in the supernatants were measured as described previously (TNFα: Cunha et al., 1993; PGE2: Amersham kit, Poole et al., 1995).
Results are presented as means with s.e.means of groups of five animals for the in vivo measurements and of triplicate wells for in vitro assays. Two independent in vitro assays were performed, each using macrophages from different mice. Differences between responses were evaluated by ANOVA, followed by Bonferroni's' t-test.
Materials
Drugs
The following materials were obtained from the sources indicated. Recombinant human IL-1β, IL-4, IL-8, TNFα, murine IL-1β and murine IL-4 (NIBSC preparations coded 86/680, 88/656, 89/520, 87/650, 93/668 and 91/656. The specific activities of these materials are IL-1β: 100,000 international units (IU) 1 μg−1 ampoule−1, IL-4: 1,000 IU 0.1 μg−1 ampoule−1, IL-8: 1,000 IU 1 μg−1 ampoule−1, TNFα: 40,000 IU 1 μg−1 ampoule−1, murine IL-1β: 100,000 IU 0.1 μg−1 ampoule−1, murine IL-4: 10,000 IU 1 μg−1 ampoule−1). Carrageenin (FMC Corporation, Philadelphia, U.S.A.). PGE2, bradykinin, dextran, concanavalin A (Con-A) and compound 48/80 (Sigma, St. Louis, U.S.A.). Bacterial endotoxin from E. coli 055 : B5 (referred to here as lipopolysaccharide, LPS) and thioglycollate (Difco Laboratories Ltd, West Molsey, Surrey, U.K.). PGE2 assay reagents and [3H]-thymidine (Amersham International plc, U.K.). Monoclonal IgG antibodies (MAbs) to murine IL-4: BVDG and 11B11 (Professor F. Liew, University of Glasgow, U.K.). Control monoclonal antibody was a purified unrelated IgG raised against ovalbumin in our laboratory.
Animals
Male Wistar or Nude-Rowett (athymic) rats, weighing 130–180 g, housed in temperature controlled rooms (22–25°C) with water and food ad libitum until use.
Results
Effect of IL-4 on hyperalgesic responses to carrageenin, bradykinin, TNFα, IL-1β, IL-8 and PGE2
Injection (in 100 μl, i.pl.) of carrageenin (100 μg), bradykinin (500 ng), TNFα (2.5 pg), IL-1β (0.5 pg), IL-8 (100 pg) and PGE2 (100 ng) into the hind-paws of Wistar rats evoked hyperalgesia, measured 3 h after injections. Treatment with IL-4 (2.5–10 ng, 50 μl, i.pl.), 30 min before carrageenin, bradykinin, or TNFα inhibited, in a dose-dependent manner, hyperalgesic responses to these agents. IL-4 (10 ng, i.pl.) inhibited the hyperalgesic responses to carrageenin, bradykinin and TNFα by 81, 84 and 86%, respectively. Hyperalgesic responses to IL-1β, IL-8 and PGE2 were not affected by IL-4 (Figure 1). Although IL-4 (10 ng, i.pl.) injected 2 h before IL-1β (0.5 pg, i.pl.) did not affect the response to IL-1β, when IL-4 was given 12 or 12+2 h before IL-1β the hyperalgesic response to this cytokine was inhibited by 30 and 74%, respectively (Figure 2A). The hyperalgesic response to IL-8 (100 pg, i.pl.) was not inhibited by IL-4 (10 ng, i.pl.) injected in any of the three schedules (2, 12 and 12+2 h before IL-8, Figure 2B). There was no visible paw inflammation following treatment with IL-4.
Figure 1.
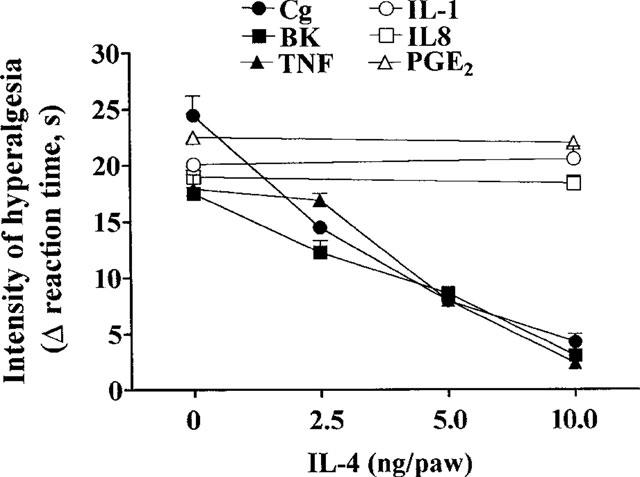
Effect of IL-4 (2.5–10 ng, 100 μl, i.pl.) on hyperalgesic response to injections (in 100 μl, i.pl.) of carrageenin (Cg, 100 μg), bradykinin (BK, 500 ng), TNFα (2.5 pg), IL-1β (0.5 pg), IL-8 (100 pg), and PGE2, (100 ng). IL-4 was injected into paws to be injected with a hyperalgesic agent, 30 min before the hyperalgesic agent. The intensity of hyperalgesia was measured 3 h after injection of hyperalgesic agents. Means±s.e.means in groups of five rats are shown.
Figure 2.
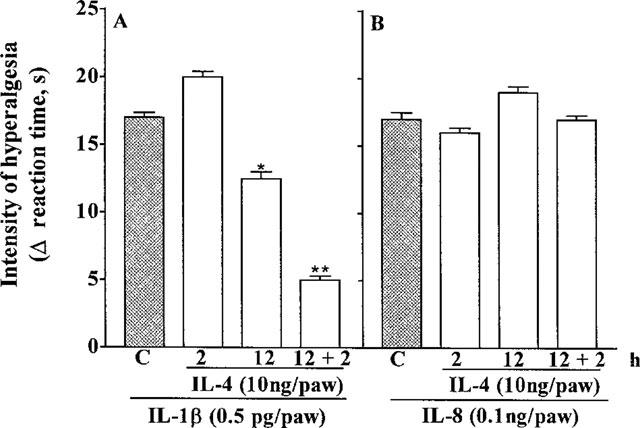
Effect of IL-4 (10 ng, 100 μl, i.pl.) on hyperalgesic responses to injections (in 100 μl, i.pl.) of IL-1β (0.5 pg, panel A) and IL-8 (100 pg, panel B). IL-4 was injected into paws to be injected with a hyperalgesic agent, 2, 12, or 2+12 h before the hyperalgesic agent. The intensity of hyperalgesia was measured 3 h after injection of hyperalgesic agents. Means±s.e.means in groups of five rats are shown; *P<0.001, **P<0.0001.
Effect of IL-4 on production of IL-1β and PGE2 by murine peritoneal macrophages stimulated with LPS
Murine peritoneal macrophages stimulated with LPS (3 μg ml−1 for 16 h) produced IL-1β and this production was not inhibited by addition to the macrophages of IL-4 (1.6 or 16 ng ml−1) for 12 h before stimulation with LPS. Addition to the macrophages of IL-4 (16 ng ml−1), for 2 h before stimulation with LPS partially inhibited the production of IL-1β (−40%, Figure 3A). Stimulation of the macrophages with LPS (3 μg ml−1) also evoked release of PGE2, and this release was inhibited by 40 and 70% by the treatment of the macrophages for 12 h with IL-4 at 1.6 and 16 ng ml−1, respectively. Treatment of macrophages with IL-4 (16 ng ml−1) for 2 h prior to stimulation with LPS (3 mg ml−1) did not inhibit LPS-induced release of PGE2 (Figure 3B).
Figure 3.
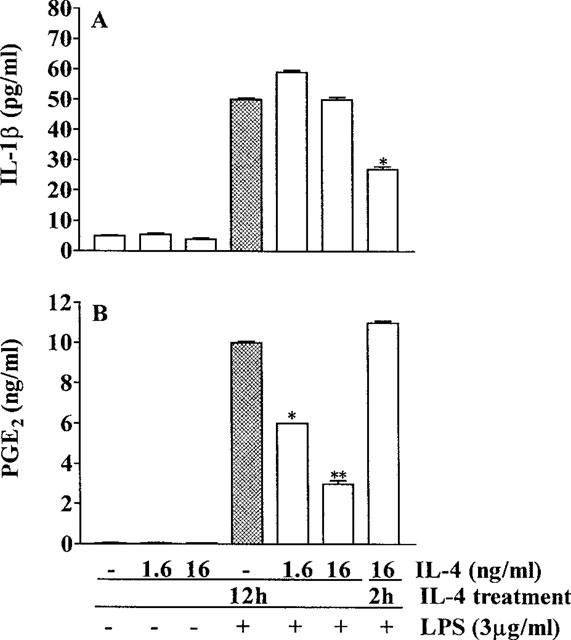
Effect of IL-4 on the production of IL-1β and PGE by murine macrophages stimulated with LPS (3 μg ml−1). The macrophages (106 cells well−1) were cultured with murine IL-4 (1.6 or 16 ng ml−1) in medium (+) or medium alone (−). Two h or 12 h later the macrophages were stimulated with LPS (3 μg ml−1) in medium (+) or with medium alone (−) for a further 16 h. At the end of this period the concentrations of IL-1β (panel A) and PGE2 (panel B) in the macrophage-conditioned media were measured. Means±s.e.means of triplicate wells are shown; *P<0.001, **P<0.0001.
Potentiation by monoclonal antibodies to IL-4 of hyperalgesic responses to carrageenin, bradykinin, TNFα, but not IL-1β and IL-8
Two MAbs to murine IL-4, 11B11 and BVDG (50 μg, i.pl.), but not a control MAb (50 μg, i.pl.), injected 30 min before injections (i.pl.) of carrageenin (10 μg), bradykinin (50 ng) and TNFα (0.25 pg) potentiated responses, measured 3 h after injection of these hyperalgesic agents (responses after 11B11: carrageenin=+74%, bradykinin=+117%, TNFα=+148%; responses after BVDG: carrageenin=+75%, bradykinin=+107%, TNFα=+204%). Hyperalgesic responses to IL-1β (0.05 pg, i.pl.) and IL-8 (10 pg, i.pl.) were not affected by the MAbs to murine IL-4 (Figure 4).
Figure 4.
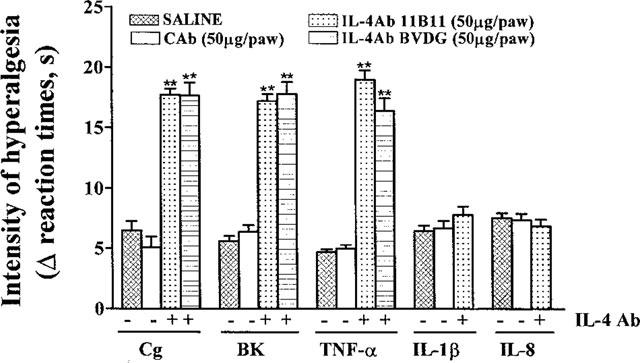
Effect of MAbs to IL-4, 11B11 and BVDG (50 μg in 50 μl, i.pl.), a control antibody and saline on hyperalgesic responses to injections (in 100 μl, i.pl.) of carrageenin (Cg, 10 μg), bradykinin (BK, 50 ng), TNFα (0.25 pg), IL-1β (0.05 pg), and IL-8 (10 pg). One of the MAbs or saline was injected into paws to be injected with a hyperalgesic agent, 30 min before the hyperalgesic agent. The intensity of hyperalgesia was measured 3 h after injection of hyperalgesic agents. Means±s.e.means in groups of five rats are shown. **P<0.0001.
Potentiation by a monoclonal antibody to IL-4 of hyperalgesic response to bradykinin of athymic (nude) rats
As in Wistar rats, in athymic Nude-Rowett rats BVDG (50 μg, i.pl.), but not a control antibody, potentiated by +101% hyperalgesic responses measured 3 h after injection of bradykinin (50 μg, i.pl., Figure 5A). The absence of the thymus in the Nude-Rowett rats was confirmed by histopathological analysis and in a splenocyte proliferation assay using Con-A. Proliferative responses (3H-thymidine incorporation, c.p.m.) to Con-A (5 μg ml−1) were for Wistar rats, culture medium alone=1,320±140 c.p.m., Con-A=12,316±940 c.p.m. and for Nude-Rowett rats, culture medium alone=1,050±128 c.p.m., Con-A=970±90 c.p.m.
Figure 5.
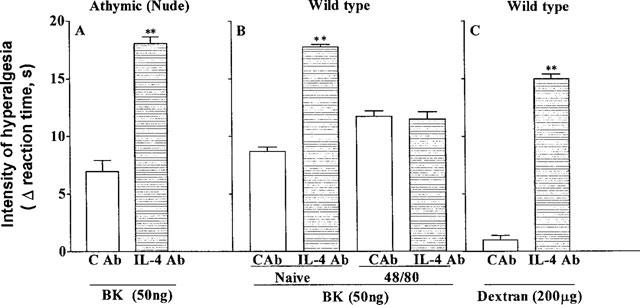
Effect of a MAb to IL-4, BVDG (50 μg in 50 μl, i.pl., horizontally hatched columns) and a control MAb (open columns) on hyperalgesic responses to injections (in 100 μl, i.pl.) of bradykinin (BK, 50 ng, panel A,B) and dextran (200 μg, panel C) in athymic (nude) rats (panel A), wild type rats (panel C) and wild type rats depleted of mast cells (panel B). The MAbs were injected into paws to be injected with a hyperalgesic agent, 30 min before the hyperalgesic agent. The intensity of hyperalgesia was measured 3 h after injection of hyperalgesic agents. Means±s.e.means in groups of five rats are shown; **P<0.0001.
Failure of a monoclonal antibody to IL-4 to potentiate the hyperalgesic response to bradykinin following mast cell depletion
Potentiation of the hyperalgesic response to bradykinin (50 ng, i.pl.), measured 3 h after its injection, by BVDG (50 μg, i.pl., given into paws to be injected, 30 min later, with bradykinin) was abolished by the depletion of mast cells by local treatment (i.pl.) with compound 48/80 (Figure 5B). No potentiation of the response to bradykinin by the MAb to IL-4 was observed in animals depleted of mast cells, compared with an increase in response of +104% in wild type (control) animals. There was no visible paw inflammation following treatment with compound 48/80.
Induction by a monoclonal antibody to IL-4 of a hyperalgesic response to dextran
Injection of dextran (200 μg, i.pl) into one hind-paw of Wistar rats did not evoke hyperalgesia, measured 3 h after injection. BVDG (50 μg), but not a control MAb, injected into paws to be injected, 30 min later, with dextran, rendered the paws sensitive to dextran, which now evoked a hyperalgesic response (Figure 5C).
Discussion
IL-4 has a number of biological activities, including B cell activation, proliferation of T cells and natural killer cells, upregulation of CD23 expression and stimulation of the expression of the Th1 phenotype by Th0 T helper cells. In addition, IL-4 has co-ordinated anti-inflammatory effects by suppressing the production of IL-1 (and also TNFα, IL-8 and IFNγ) while upregulation production of IL-1ra by LPS-stimulated monocytes (Vannier et al., 1992; Fenton et al., 1992). Other, essentially anti-inflammatory, effects of IL-4 include inhibition of the induction of the nitric oxide and synthase and COX-2 enzymes responsible for the production of nitric oxide and prostaglandins, respectively. In the present study the potential anti-hyperalgesic effect of IL-4 in a model of mechanical hyperalgesia was investigated.
Local administration of IL-4 inhibited, in a dose-dependent manner, hyperalgesic responses to carrageenin, bradykinin and TNFα. This anti-hyperalgesic effect of IL-4 was likely to have been a consequence of its capacity to inhibit the production of pro-inflammatory cytokines (namely TNFα, IL-1β and IL-8) since, in carrageenin-evoked hyperalgesia, bradykinin is generated and initiates the release of TNFα, which stimulates the release of IL-1 and IL-8 (Tiffany & Burch, 1989, Ferreira et al., 1993). Consistent with this notion, the release of IL-1β by activated murine macrophages was partially inhibited by treating the cells with IL-4 for 2 h. The absence of an effect of IL-4 on hyperalgesic responses to IL-8 and PGE2 was expected, since IL-8 stimulates the local production of sympathetic amines (Cunha et al., 1991), which sensitize nociceptors (Ferreira & Lorenzetti, 1981). IL-10, which has a profile of action similar to that of IL-4 in terms of its inhibitory actions on the production of cytokines generally regarded as pro- also inhibited hyperalgesic responses to carrageenin, bradykinin and TNFα, but not those to IL-8 and PGE2 (Poole et al., 1995). In contrast to IL-10, IL-4 injected 0.5 h or 2 h before IL-1β did not inhibit its hyperalgesic effect. Inhibition was observed only when IL-4 was injected either once, 12 h before, or twice, at 12+2 h before the administration of IL-1β. Since IL-1β-induced hyperalgesia is mediated by eicosanoid products (Ferreira et al., 1988; Seibert et al., 1994), the effect of IL-4 on PGE2 production by activated macrophages was investigated. Similar to its effect on IL-1β evoked hyperalgesia, IL-4 inhibited, in a dose-dependent manner, PGE2 production only when the macrophages were pre-treated with IL-4 for 12 h; treatment for 2 h before the challenge did not affect the production of the prostanoid. These results are consistent with IL-1β-induced hyperalgesia via the release of PGs (Ferreira et al., 1988; Seibert et al., 1994).
The results with murine macrophages are different from the rapid blockade by IL-4 of the release of PGE2 (60 min) in human monocytes and synoviocytes (Seitz et al., 1994, Endo et al., 1996). This difference might be a reflection of differences in the cell types studied or might be a species difference, or both. The lack of effect of IL-4 on the hyperalgesic response to IL-8 (given either 12 h before or 12+2 h before the IL-8), reinforces the suggestion that the hyperalgesia induced by IL-8 is subsequent to the release of mediators other than cyclo-oxygenase products (Cunha et al., 1991).
The finding that the hyperalgesic responses to carrageenin, bradykinin, and TNFα were enhanced by treatment of paws with two different MAbs to IL-4 strongly suggests that endogenous IL-4 has an important role in limiting inflammatory hyperalgesia. The failure of the MAbs to IL-4 to enhance the hyperalgesic effects of IL-1β and IL-8 suggests that the predominant effect of endogenous IL-4 in inflammatory hyperalgesia is to inhibit the release of cytokines, rather than the PGs which sensitize nociceptors (Ferreira & Lorenzetti, 1981; Nakamura & Ferreira, 1987).
IL-4 is released mainly by Th2 lymphocytes and by mast cells (Lord & Lamb, 1996; Tunon de Lara et al., 1994). To gain information about which cell types were releasing IL-4 during inflammatory hyperalgesia, the capacity of a MAb against IL-4 to enhance the hyperalgesic effect of bradykinin in athymic rats (deficient in T lymphocytes) and in rats depleted of mast cells (with compound 48/80, Di Rosa et al., 1971) was investigated. Histopathological analysis confirmed the absence of the thymus (in the athymic animals) and the effectiveness of the depletion of mast cells after compound 48/80. The absence of the thymus was also confirmed using a splenocyte proliferation assay. The finding that the MAb to IL-4 potentiated the hyperalgesic effect of bradykinin in athymic rats but not in animals depleted of mast cells strongly suggests that mast cells were the source of the IL-4 that limited the inflammatory hyperalgesia. Supporting evidence for mast cells as the source of the IL-4 was obtained in the experiment with dextran, which disrupts mast cells (Nishida et al., 1978; Nishida & Tomizawa, 1980). Thus, a MAb to IL-4 induced a hyperalgesic response to a substance, dextran, in (wild type) rats not previously sensitive to this agent.
In conclusion, the data described above provide evidence that IL-4, released by resident mast cells, limits the intensity and duration of inflammatory hyperalgesia. Consequently, a lack of IL-4 may contribute to the maintenance of chronic inflammatory pain. In the early phase of the hyperalgesic response, IL-4 appears to achieve its effects by inhibiting the production of hyperalgesic cytokines (e.g. TNFα and IL-1). In the later phase of the hyperalgesic response, the effect of IL-4 also may result also from inhibition of COX-2, since IL-4, given 12 h before IL-1β, inhibited the hyperalgesic effect of IL-1β and the release in vitro of PGE2 by murine macrophages stimulated with LPS. These interpretations are consistent with data from preliminary experiments in our laboratory with the relatively selective COX-2 inhibitor meloxicam.
Acknowledgments
The authors thank leda R. Santos for technical assistance and Dr Olga Célia Martinez Ibañez (Butantã Institute, Brazil) for providing the athymic rats. This work was supported by grants from CNPq and FAPESP (Brazil). This work utilized reagents generated with funding from the European Community Concerted Action Program Biomed I: ‘CYTOKINES IN THE BRAIN' (PL 931450).
Abbreviations
- BK
bradykinin
- Cg
carrageenin
- Con-A
concanavalin A
- o.p.m.
counts per minute
- °C
degrees centigrade
- h
hour
- IFNγ
interferon γ
- IL
interleukin
- IU
international unit
- IgG
immunoglobulin G
- i.pl.
intraplantar
- LPS
lipopolysaccharide
- μg
microgram
- μl
microlitre
- mg
milligram
- min
minutes
- MAb
monoclonal antibody
- ng
nanogram
- pg
picogram
- PGE2
prostaglandin E2
- TNFα
tumour necrosis factor α
- Th
T helper
- U
unit
- 3H
tritiated
- w/v
weight per volume.
References
- CALLARD R.E., MATTHEUS D.J., HIBBERT L. IL-4 and IL-13 receptors: are they one and the same. Immunol. Today. 1996;17:108–110. doi: 10.1016/0167-5699(96)80600-1. [DOI] [PubMed] [Google Scholar]
- CASSATELLA M.A., MEDA L., BONORA S., CESKA M., CONSTANTIN G. Interleukin-10 (IL-10) inhibits the release of proinflammatory cytokines from human polymorphonuclear leukocytes. Evidence for an autocrine role of tumour necrosis factor and IL-1β in mediating the production of IL-8 triggered by lipopolysaccharide. J. Exp. Med. 1993;178:2207–2211. doi: 10.1084/jem.178.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUNHA F.Q., POOLE S., LORENZETTI B.B., FERREIRA S.H. Interleukin-8 as a mediator of sympathetic pain. Br. J. Pharmacol. 1991;104:765–767. doi: 10.1111/j.1476-5381.1991.tb12502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUNHA F.Q., POOLE S., LORENZETTI B.B., FERREIRA S.H. The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. Br. J. Pharmacol. 1992;107:660–664. doi: 10.1111/j.1476-5381.1992.tb14503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUNHA F-Q., BOUKILI M.A., BRAGA D.A., MOTTA J.I., VARGAFTIG BB. &, FERREIRA S.H. Blockade by ferispiride of endotoxin-induced neutrophil migration in the rat. Eur. J. Pharmacol. 1993;238:47–52. doi: 10.1016/0014-2999(93)90503-a. [DOI] [PubMed] [Google Scholar]
- DINARELLO C.A., CANNON J.G., WOLFF S.M., BERNHEIM H.A., BEUTLER B., CERAMI A., FIGARI I.S., PALLADINO M.A., JR, O'CONNOR J.V. Tumour necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin 1. J. Exp. Med. 1986;163:1443–1449. doi: 10.1084/jem.163.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DINARELLO Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77:1627–1652. [PubMed] [Google Scholar]
- DI ROSA M., GIROUD J.P., WILLOUGHBY D.A. Studies of the mediators of the acute inflammatory response in rats in different sites by carrageenin and turpentine. J. Pathol. 1971;104:15–29. doi: 10.1002/path.1711040103. [DOI] [PubMed] [Google Scholar]
- ELIAS J.A., LENTZ V., CUMMINGS P.J. Transforming growth factor-β regulation of IL-6 production by unstimulated and IL-1-stimulated human fibroblasts. J. Immunol. 1991;146:3437–3443. [PubMed] [Google Scholar]
- ENDO T., OGUSHI F., SABURO S. LPS-dependent cyclooxygenase-2 induction in human monocytes is down-regulated by IL-13, but not by IFN-γ. J. Immunol. 1996;156:2240–2246. [PubMed] [Google Scholar]
- FACCIOLI L.H., SOUZA G.E.P., CUNHA F.Q., POOLE S., FERREIRA S.H. Recombinant interleukin-1 and tumour necrosis factor induce neutrophil migration in vivo by indirect mechanisms. Agents Actions. 1990;30:344–349. doi: 10.1007/BF01966298. [DOI] [PubMed] [Google Scholar]
- FENTON M.J., BURAS J.A., DONELLY R.P. IL-4 reciprocally regulates IL-1 and IL-1 receptor antagonist expression in human monocytes. J. Immunol. 1992;149:1283–1288. [PubMed] [Google Scholar]
- FERREIRA S.H., LORENZETTI B.B. Prostaglandin hyperalgesia, IV: a metabolic process. Prostaglandins. 1981;21:789–792. doi: 10.1016/0090-6980(81)90235-5. [DOI] [PubMed] [Google Scholar]
- FERREIRA S.H., LORENZETTI B.B., BRISTOW A.F., POOLE S. Interleukin-1β as a potent hyperalgesic agent antagonized by a tripeptide analogue. Nature. 1988;334:698–699. doi: 10.1038/334698a0. [DOI] [PubMed] [Google Scholar]
- FERREIRA S.H., LORENZETTI B.B., CORREA F.M.A. Central and peripheral antialgesic action of aspirin-like drugs. Eur. J. Pharmacol. 1978;53:39–48. doi: 10.1016/0014-2999(78)90265-0. [DOI] [PubMed] [Google Scholar]
- FERREIRA S.H., LORENZETTI B.B., POOLE S. Bradykinin initiates cytokine-mediated inflammatory hyperalgesia. Br. J. Pharmacol. 1993;110:1227–1231. doi: 10.1111/j.1476-5381.1993.tb13946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FIORENTINO D.F., ZLOTNIK A., MOSMANN T.R., HOWARD M., O'GARRA A. IL-10 inhibits cytokine production by activated macrophages. J. Immunol. 1991;147:3815–3822. [PubMed] [Google Scholar]
- HART P.R., VITTI G.F., BURGESS D.R., WHITTY G.A, , PICCOLI D.S., HAMILTON J.A. Potential antiinflammatory effects of interleukin 4: suppression of human monocyte tumor necrosis factor α, interleukin 1, and prostaglandin E2. Proc. Natl. Acad. Sci. U.S.A. 1989;86:3803–3807. doi: 10.1073/pnas.86.10.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LORD C.J.M., LAMB J.R. TH2 cells in allergic inflammation: a target of immunotherapy. Clin. Exp. Allergy. 1996;26:756–765. [PubMed] [Google Scholar]
- LUMPKIN M.D. The regulation of ACTH secretion by IL-1. Science. 1987;238:452–454. doi: 10.1126/science.2821618. [DOI] [PubMed] [Google Scholar]
- MORSTYN G., BURGESS A.W. Hemopoietic growth factors: a review. Cancer Res. 1988;48:5624–5637. [PubMed] [Google Scholar]
- NAKAMURA M., FERREIRA S.H. A peripheral sympathetic component in inflammatory hyperalgesia. Eur. J. Pharmacol. 1987;135:145–153. doi: 10.1016/0014-2999(87)90606-6. [DOI] [PubMed] [Google Scholar]
- NISHIDA S., TOMIZAWA S. Effects of compound 48/80 on dextran-induced paw edema and histamine content of inflammatory exudate. Bioch. Pharmacol. 1980;29:1073–1075. doi: 10.1016/0006-2952(80)90174-4. [DOI] [PubMed] [Google Scholar]
- NISHIDA S., WAGAWA K., TOMIZAWA S. Correlation between histamine content in exudate and degree of edema produced by dextran. Bioch. Pharmacol. 1978;27:2641–2646. doi: 10.1016/0006-2952(78)90340-4. [DOI] [PubMed] [Google Scholar]
- POOLE S., CUNHA F.Q., SELKIRK S., LORENZETTI B.B., FERREIRA S.H. Cytokine-mediated inflammatory hyperalgesia limited by interleukin-10. Br. J. Pharmacol. 1995;115:684–688. doi: 10.1111/j.1476-5381.1995.tb14987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RANKIN E.C., CHOY E.H., KASSIMOS D., KINGSLEY G.H., SOPWITH A.M., ISENBERG D.A., PANAYI G.S. The therapeutic effects of an engineered human anti-tumour necrosis factor alpha antibody (CDP571) in rheumatoid arthritis. Br. J. Rheumatol. 1995;34:334–342. doi: 10.1093/rheumatology/34.4.334. [DOI] [PubMed] [Google Scholar]
- ROCKEN M., RACKE M., SHEVACH E.M. IL-4-induced immune deviation as antigen-specific therapy for inflammatory autoimmune disease. Immunol. Today. 1996;17:225–231. doi: 10.1016/0167-5699(96)80556-1. [DOI] [PubMed] [Google Scholar]
- SEIBERT K., ZHANG Y., LEAHY K., HAUSER S., MASFERRER J., PERKINS W., LEE L., ISAKSON P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc. Natl. Acad. Sci. U.S.A. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEITZ M., LOETSCHER P., DEWALD B., TOWBIN H., CESKA M., BAGGIOLINI M. Production of interleukin-1 receptor antagonist, inflammatory chemotactic proteins, and prostaglandin E by rheumatoid and osteoarthritic synoviocytes-regulation by IFN-γ and IL-4. J. Immunol. 1994;152:2060–2065. [PubMed] [Google Scholar]
- STANDIFORD T.J., STRIETER R.M., CHENSUE S.W., WESTWICK J., KASAHARA K., KUNKEL S.L. IL-4 inhibits the expression of IL-8 from stimulated human monocytes. J. Immunol. 1990;145:1435–1439. [PubMed] [Google Scholar]
- STEFFERL A., HOPKINS S.J., ROTHWELL N.J., LUHESHI G.N. The role of TNF-alpha in fever: opposing actions of human and murine TNF-alpha and interactions with IL-beta in the rat. Br. J. Pharmacol. 1996;118:1919–1924. doi: 10.1111/j.1476-5381.1996.tb15625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIFFANY C.W., BURCH R.M. Bradykinin stimulates tumour necrosis factor and interleukin 1 release from macrophages. FEBS Lett. 1989;247:189–192. doi: 10.1016/0014-5793(89)81331-6. [DOI] [PubMed] [Google Scholar]
- TUNON DE LARA J.M., OKAYAMA Y., MCEUEN A.R., HEUSSER C.H., CHURCH M.K., WALLS A.F. Release and inactivation of interleukin-4 by mast cells. In: {it}Cells and cytokines in lung inflammation eds. Chignard, M., Pretolani, M., Renesto, P and Vargaftig, B.B. Ann. N. Y. Acad. Sci. 1994;725:50–58. doi: 10.1111/j.1749-6632.1994.tb39789.x. [DOI] [PubMed] [Google Scholar]
- VANNIER E., MILLER L.C., DINARELLO C.A. Co-ordinated anti-inflammatory effects of interleukin-4: Interleukin-4 suppresses interleukin-1 production but up-regulates gene expression and synthesis of interleukin-1 receptor antagonist. Proc. Natl. Acad. Sci. U.S.A. 1992;89:4076–4080. doi: 10.1073/pnas.89.9.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATKINS L.R., GOEHLER L.E., RELTON J. , BREWER M.T., MAIER S.F. Mechanisms of tumour necrosis factor-alpha (TNF-alpha) hyperalgesia. Brain-Res. 1995;692:244–250. doi: 10.1016/0006-8993(95)00715-3. [DOI] [PubMed] [Google Scholar]
- WATKINS L.R., WIERTELAK E.P., GOEHLER L.E., SMITH K.P., MARTIN D., MAIER S.F. Characterization of cytokine-induced hyperalgesia. Brain-Res. 1994;654:15–26. doi: 10.1016/0006-8993(94)91566-0. [DOI] [PubMed] [Google Scholar]