

Abstract
The effects of various inhibitors of the mitochondrial electron transport chain on the activity of ATP-sensitive K+ channels were examined in the Cambridge rat insulinoma G1 (CRI-G1) cell line using a combination of whole cell and single channel recording techniques.
Whole cell current clamp recordings, with 5 mM ATP in the pipette, demonstrate that the mitochondrial uncoupler sodium azide (3 mM) rapidly hyperpolarizes CRI-G1 cells with a concomitant increase in K+ conductance. This is due to activation of KATP channels as the sulphonylurea tolbutamide (100 μM) completely reversed the actions of azide. Other inhibitors of the mitochondrial electron transport chain, rotenone (10 μM) or oligomycin (2 μM) did not hyperpolarize CRI-G1 cells or increase K+ conductance.
In cell-attached recordings, bath application of 3 mM sodium azide (in the absence of glucose) resulted in a rapid increase in KATP channel activity, an action readily reversible by tolbutamide (100 μM). Application of sodium azide (3 mM), in the presence of Mg-ATP, to the intracellular surface of excised inside-out patches also increased KATP channel activity, in a reversible manner.
In contrast, rotenone (10 μM) or oligomycin (2 μM) did not increase KATP channel activity in either cell-attached, in the absence of glucose, or inside-out membrane patch recordings.
Addition of sodium azide (3 mM) to the intracellular surface of inside-out membrane patches in the presence of Mg-free ATP or the non-hydrolysable analogue 5′-adenylylimidodiphosphate (AMP-PNP) inhibited, rather than increased, KATP channel activity.
In conclusion, sodium azide, but not rotenone or oligomycin, directly activates KATP channels in CRI-G1 insulin secreting cells. This action of azide is similar to that reported previously for diazoxide.
Keywords: KATP channels, azide, mitochondrial inhibitors, rotenone, oligomycin
Introduction
It is well established that ATP-sensitive K+ (KATP) channels play a key role in the insulin secretory mechanism of pancreatic beta cells (Ashcroft & Rorsman, 1991). Closure of these channels results in depolarization of the cell membrane and Ca2+ influx via voltage-gated Ca2+ channels which in turn triggers exocytotic release of insulin (Ashford, 1990). Thus agents that modulate the activity of KATP channels are important therapeutic tools in the treatment of hyperglycaemia and hypoglycaemia.
In pancreatic beta cells (Trube et al., 1986; Dunne et al., 1987) and insulin secreting cells (Sturgess et al., 1988; Kozlowski et al., 1989), the hyperglycaemic agent diazoxide activates KATP channels, resulting in suppression of insulin secretion. Diazoxide-induced activation of KATP channels requires the presence of Mg2+ and ATP (Sturgess et al., 1988; Kozlowski et al., 1989; Dunne, 1989), suggesting that a phosphorylation-dependent process may underly this action of diazoxide. The KATP channel of the pancreatic beta cell comprises of two proteins: the sulphonylurea receptor, SUR1 and an inwardly rectifying K+ channel, Kir 6.2 (Inagaki et al., 1995; Sakura et al., 1995). Although the precise mode of action of diazoxide is not entirely clear it is thought to act directly on SUR1 (Inagaki et al., 1996; Tucker et al., 1997) rather than Kir 6.2. Recent studies also indicate that diazoxide may have actions in pancreatic beta cells, through inhibiting mitochondrial energy metabolism (Grimmsmann & Rustenbeck, 1998). However the functional significance of this, if any, in terms of KATP channel activity or stimulus secretion coupling is presently unknown.
In pancreatic beta cells, the activity of KATP channels is tightly regulated by the metabolic state of the cell, since a reduction in the cytosolic concentrations of ATP, by either an elevation in energy consumption or inhibition of energy metabolism, results in activation of KATP channels. Thus agents that interfere with ATP production via inhibition of mitochondrial energy metabolism, are commonly used to activate both native and cloned KATP channels (Ashcroft & Ashcroft, 1990). Indeed, metabolic inhibition by sodium azide (Misler et al., 1989), cyanide (Misler et al., 1986) and oligomycin (Niki et al., 1989) has been reported to activate KATP channels in pancreatic beta cells and insulin secreting cells. Cyanide and rotenone are also reported to activate KATP channels in rat substantia nigra dopaminergic neurones (Murphy & Greenfield, 1991; Roper & Ashcroft, 1995) and cyanide activates KATP channels in cardiac myocytes (Kakei & Noma, 1984; Ito et al., 1991) and skeletal muscle (Allard et al., 1995). Furthermore, in oocyte expression systems sodium azide is used routinely to activate KATP channel currents (Tucker et al., 1997; Gribble et al., 1997a). It is well known that sodium azide (Tsubaki & Yoshikawa, 1993) inhibits oxidative phosphorylation via inhibition of cytochrome oxidase, the final enzyme in the mitochondrial electron transport chain, thereby resulting in a rapid depletion of intracellular ATP. Indeed, the effects of sodium azide on KATP channels have been attributed to the resultant reduction in ATP levels (Gribble et al., 1997a). In contrast, rotenone and oligomycin deplete intracellular ATP levels via inhibition of the mitochondrial flavoprotein NADH dehydrogenase (Ragan & Racker, 1973) and the mitochondrial F1-ATPase (Penefsky, 1985), respectively.
In view of the recent report that diazoxide may also act to inhibit mitochondrial metabolism, an action presumably distinct from its activation of KATP channels, we have re-examined the effects of sodium azide, rotenone and oligomycin on KATP channels using the rat CRI-G1 insulinoma cell line. We now demonstrate that sodium azide, but not rotenone or oligomycin directly activate KATP channel currents even in the presence of high concentrations of ATP, suggesting that inhibition of mitochondrial energy metabolism may not underlie this effect. Furthermore, in a manner similar to diazoxide (Sturgess et al., 1988; Kozlowski et al., 1989), sodium azide-induced KATP channel activation requires the presence of MgATP.
Methods
Cell culture
Cells from the insulin-secreting cell line CRI-G1 were grown in Dulbecco's modified Eagle's medium with sodium pyruvate and glucose, supplemented with 10% (v/v) foetal calf serum and 1% penicillin and streptomycin at 37°C in a humidified atmosphere of 95% O2 and 5% CO2. Cells were passaged at 2–5 day intervals as described previously (Carrington et al., 1986), plated onto 3.5 cm petri dishes (Falcon 3001) and used 1–4 days after plating.
Electrophysiological recording and analysis
Experiments were performed using whole cell current- and voltage-clamp recording configurations to monitor membrane potential and macroscopic currents, respectively and cell-attached and inside-out configurations to monitor single channel responses, as described previously (Harvey et al., 1997). In voltage-clamp recordings the membrane potential was held at −50 mV and 10 mV steps of 100 ms duration were applied every 200 ms (range of voltages –120 mV to −30 mV). Current and voltage were measured using Axopatch 200B and List EPC-7 amplifiers, and currents evoked in response to the voltage-step protocol were analysed using pClamp 6.0 software (Axon Instruments, Foster City, CA, U.S.A). Single channel data were analysed for current amplitude (I) and average channel activity (Nf.Po) as described previously (Lee et al., 1995). Changes in Nf.Po as a result of drug effects are expressed as a percentage of control. Current clamp and single channel data were recorded onto digital audio tapes and replayed for illustration on a Gould TA 240 chart recorder.
Recording electrodes were pulled from borosilicate glass capillaries and had resistances of 1–5 MΩ for whole cell recordings and 8–12 MΩ for single channel experiments when filled with electrolyte solution. The pipette solution for whole cell recordings comprised (mM): KCl 140, MgCl2 0.6, CaCl2 2.73, Mg-ATP 5.0, HEPES 10.0, EGTA 10, pH 7.2 (free Ca2+ of 100 nM), whereas for cell-attached and single channel recordings it contained (mM): KCl 140, CaCl2 1, MgCl2 1, HEPES 10, pH 7.2. The bath solution for whole cell and cell-attached recordings comprised of normal saline (mM): NaCl 135, KCl 5, MgCl2 1, CaCl2 1, HEPES 10, pH 7.4, whereas for inside-out excised patches the bath solution contained either (mM): KCl 140, MgCl2 1, CaCl2 2, EGTA 10, HEPES 10, pH 7.2 (free Mg2+ of 0.6 mM and Ca2+ of <30 nM) or KCl 140, EDTA 10.0, CaCl2 4.6, HEPES 10, pH 7.2 (free Mg2+ and Ca2+ of <30 nM). The free Ca2+ and Mg2+ concentrations were calculated using the ‘METLIG' programme (P. England and R. Denton, University of Bristol, U.K.). In order to calculate these values it was necessary to assume that the salts used to prepare the ‘Mg2+-free' solution were contaminated with the trace levels of Mg2+ indicated by the manufacturer. All solution changes were achieved by superfusing the bath with a gravity feed system at a rate of 10 ml min−1 which allowed complete bath exchange within 2 min. All experiments were performed at room temperature (22–25°C).
Drugs
Sodium azide, rotenone, oligomycin, tolbutamide, Mg-AMP-PNP, K-ATP and Mg-ATP were obtained from Sigma, Poole, Dorset, U.K. Tolbutamide was made up as a 100 mM stock solution in dimethyl sulphoxide (DMSO). ATP (potassium and magnesium salts) was made up as a 100 mM stock solution in 10 mM HEPES at pH 7.2 and kept at −4°C until required, whereas Mg-AMP-PNP was stored at −4°C as a 10 mM stock solution in 10 mM HEPES.
Statistical analysis
All data are expressed as the mean±s.e.mean and statistical analyses were performed using unpaired student's t-test (unless otherwise stated).
Results
Sodium azide activates KATP channels
Under current clamp conditions with 5 mM ATP in the pipette solution to maintain KATP channels in the closed state, the mean resting membrane potential of CRI-G1 cells was −37.5±0.83 mV (n=19). Application of the metabolic inhibitor sodium azide (0.1–3 mM) under these conditions resulted in a rapid hyperpolarization of CRI-G1 cells to −66±1.6 mV (n=11; Figure 1a). The hyperpolarization was initiated within 0.5–1.0 min of application (including a dead space time of 10–15 s), reached a maximum 2–3 min later and was maintained in the continued presence of the agent. Examination of the voltage-clamped macroscopic currents indicate that the azide-induced hyperpolarization was accompanied by an increase in the slope conductance from a control value of 0.59±0.06 nS to 4.03±1.02 nS following exposure to 3 mM sodium azide (n=11; Figure 1b). The mean reversal potential (obtained from the point of intersection of the current-voltage relationships) associated with this increase in conductance was −78±0.64 mV (n=11) which is close to the calculated value for Ek of −84 mV under these conditions, indicating an increase in K+ conductance. The azide-induced hyperpolarization and increased K+ conductance were readily reversed on washout of this agent such that the membrane potential and slope conductance values following washout of azide were −38±1.3 mV and 0.44±0.09 nS (n=3). The sulphonylurea tolbutamide (100 μM) completely reversed the membrane hyperpolarization and increase in conductance to pre-azide levels (−36±1.8 mV and 0.39±0.08 nS) indicating that these actions of azide are due to activation of KATP channels (n=5; Figure 1). Lower concentrations of sodium azide were also examined, 1 mM (n=3) and 0.3 mM (n=2) induced similar levels of hyperpolarization and increased slope conductance. The threshold concentration for azide action was approximately 0.1 mM, where two out of five cells responded as described above.
Figure 1.
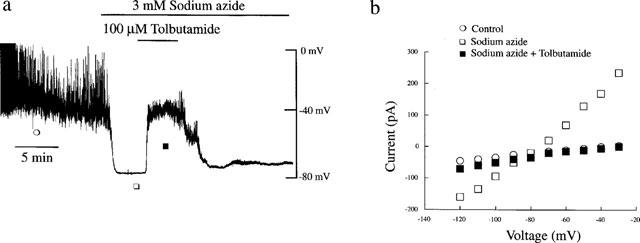
Sodium azide hyperpolarizes CRI-G1 cells via activation of KATP channel currents. (a) Whole cell current clamp trace of a CRI-G1 cell dialysed with 5 mM ATP. In this and subsequent figures, the trace begins approximately 5 min after obtaining the whole cell configuration. Application of sodium azide (3 mM) for the time indicated resulted in rapid hyperpolarization of the cell membrane from −38 mV to −78 mV, an action readily reversed by the sulphonylurea tolbutamide (100 μM). (b) The current-voltage relations for the currents obtained at the specified points in (a): control (○); sodium azide (□) and sodium azide and tolbutamide (▪). Sodium azide increased the membrane conductance relative to control and tolbutamide (100 μM) reversed the azide-induced increase in conductance with a reversal potential of −80 mV.
Effects of rotenone and oligomycin on whole cell currents and membrane potential
In order to determine whether the above effects of sodium azide on CRI-G1 cells were specific for this mitochondrial chain inhibitor, the actions of other mitochondrial inhibitors were examined. In particular, the effects of rotenone, a specific inhibitor of NADH dehydrogenase (Ragan & Racker, 1973) and oligomycin, which inhibits the mitochondrial F1-ATPase (Penefsky, 1985) on membrane potential and conductance were investigated. Under current clamp conditions with 5 mM ATP in the pipette solution, application of rotenone (10 μM) for at least 15–20 min failed to effect the membrane potential of CRI-G1 insulinoma cells (Figure 2a). Thus the mean resting membrane potential values obtained in the absence and following 15 min exposure to rotenone were −36±1.3 mV and −35±1.9 mV, respectively (n=5; P>0.05). In addition, rotenone had no effect on the slope conductance (obtained from the current voltage relationship) of CRI-G1 cells. The mean slope conductance value obtained in control conditions was 0.63±0.09 nS whereas in the presence of 10 μM rotenone it was 0.65±0.12 nS (n=5; P>0.05; Figure 2b).
Figure 2.
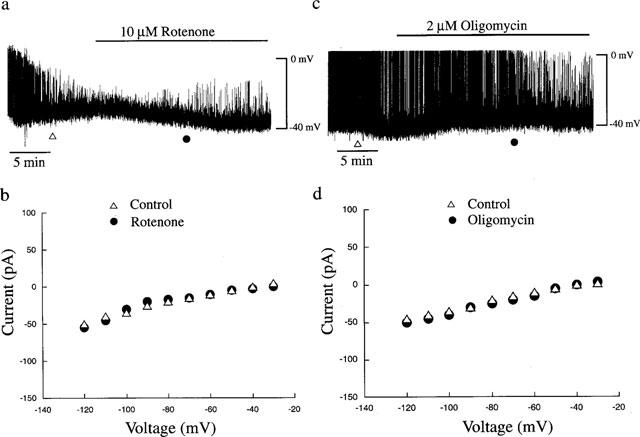
Effects of rotenone and oligomycin on membrane potential and whole cell currents. (a) The upper panel is a current clamp record of a cell dialysed with a 5 mM ATP-containing electrode solution. Application of rotenone (10 μM) for the time indicated had no effect on the resting membrane potential of CRI-G1 cells. (b) Current-voltage relations for the voltage clamped macroscopic currents at the times specified in a: control (▵) and 10 μM rotenone (•). Rotenone had no effect on the membrane conductance of CRI-G1 insulinoma cells. (c) Current clamp record of a CRI-G1 cell dialysed with a 5 mM ATP-containing electrode solution. Addition of oligomycin (2 μM) for the time indicated had no effect on the resting membrane potential of the cell. (d) Plot of the current-voltage relations for the voltage clamped currents obtained in c; control (▵) and oligomycin (•). Oligomycin failed to affect the slope conductance of CRI-G1 cells.
Similarly, application of oligomycin (2 μM) for at least 15–20 min, failed to effect either the membrane potential or slope conductance of CRI-G1 cells (Figure 2). Thus the membrane potential and slope conductance values obtained under control conditions were −35±1.5 mV and 0.54±0.02 nS, respectively, whereas following incubation with 2 μM oligomycin for at least 20 min the values obtained were −36±2.0 mV and 0.53±0.05 nS, respectively (n=3; P>0.05; Figure 2c,d). The lack of effect of rotenone and oligomycin on the electrical properties of CRI-G1 cells in the presence of excess intracellular ATP, is in keeping with their effects as metabolic inhibitors. However, it is unlikely that inhibition of mitochondrial energy metabolism per se underlies the azide-induced changes in membrane potential or conductance observed in CRI-G1 cells.
Sodium azide activates single K+ channel currents
Identification of KATP channels as the molecular target for the acute actions of azide in these cells was obtained from cell-attached and excised inside-out single channel recordings. Application of sodium azide (3 mM) to the bath during cell-attached recordings induced activation of KATP channel currents in six out of seven recordings (Figure 3b). Analysis of total channel activity (over a 60 min period), 1–2 min after formation of the cell-attached configuration, resulted in a mean channel activity (Nf.Po) of 0.05±0.02 and this increased to 0.22±0.07 (n=7; P<0.05) 6–7 min after the addition of 3 mM sodium azide to the bath. Identical control experiments in the absence of sodium azide resulted in no KATP channel activation with mean channel activity values of 0.05±0.02 and 0.04±0.02 (n=3; P>0.05) obtained at equivalent time periods (Figure 3a). The azide-induced increase in channel activity was sustained for the duration of recordings, in the continued presence of azide and could be inhibited reversibly by tolbutamide (100 μM; n=4; Figure 3b). Furthermore, following azide-induced increase in K+ channel activity cell-attached, excision of the patch into the inside-out configuration allowed unequivocal identification of the channel as KATP by inhibition by either 0.1 mM AMP-PNP (n=3; data not illustrated) or 1 mM ATP (n=3). Thus these data are consistent with KATP channels as the molecular target for sodium azide-induced hyperpolarization of CRI-G1 cells.
Figure 3.
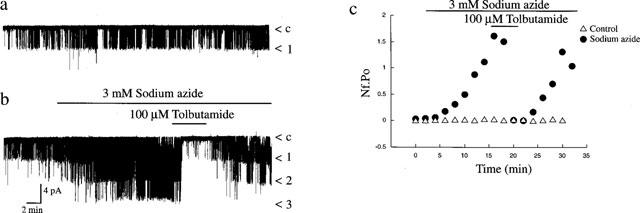
Sodium azide activates single potassium channels. (a) Cell attached recording from a single CRI-G1 cell, at 10 mV applied to the recording pipette. Continuous recording over a 20 min period showed that in the absence of any drugs there was no significant change in channel activity with time. Note that under these recording conditions channel openings are denoted as downward deflections (inward currents). In this and subsequent single channel recordings the symbol c refers to the closed state of the channel. (b) cell-attached recording of a CRI-G1 cell at 10 mV applied potential. Addition of sodium azide (3 mM) for the time indicated resulted in a rapid increase in channel activity, an action that was readily reversed by addition of tolbutamide (100 μM). (c) Graph of channel activity (Nf.Po) expressed as a function of time obtained from the cell-attached patches in a (▵) and b (•).
Dual effects of sodium azide in excised patches
In excised inside-out patches recordings were made in symmetrical (140 KCl in pipette and bath) K+-containing solution and the membrane potential was held at +40 mV. In the absence of MgATP in the bathing medium, application of sodium azide (3 mM) resulted in an inhibition of KATP channel activity on ten out of ten occasions (Figure 4a). This action of azide was reversible in only four of the ten patches. Run down of KATP channels during recordings is likely to underlie the lack of reversibility in the remaining six patches. The mean channel activity obtained in control conditions (in the absence of Mg2+), in the presence of 3 mM sodium azide and on washout were 1.25±0.29, 0.58±0.14 and 0.62±0.19, respectively (n=10). In contrast, in inside-out patches bathed in Mg2+-containing medium, addition of MgATP (0.1 mM) resulted in a 77.8±5.4% inhibition of KATP channel activity relative to control (n=10). Subsequent exposure to sodium azide (3 mM) in the continued presence of 0.1 mM MgATP resulted in an immediate (0.5–1 min) increase (270±76%) in KATP channel activity (n=10; Figure 4b). The corresponding mean levels of channel activity in the presence of MgATP (0.1 mM) and following application of sodium azide were 0.16±0.09 and 0.40±0.17, respectively (n=10; P<0.05). The azide-induced increase in channel activity was readily reversed on washout of azide, maintaining MgATP at 0.1 mM (0.03±0.01; n=4). These data indicate that in the absence of MgATP, sodium azide inhibited KATP channel activity, whereas in the presence of MgATP sodium azide resulted in rapid activation of these channels. These findings parallel identical dual actions reported for the hyperglycaemic agent, diazoxide on KATP channels in this cell line (Kozlowski et al., 1989).
Figure 4.
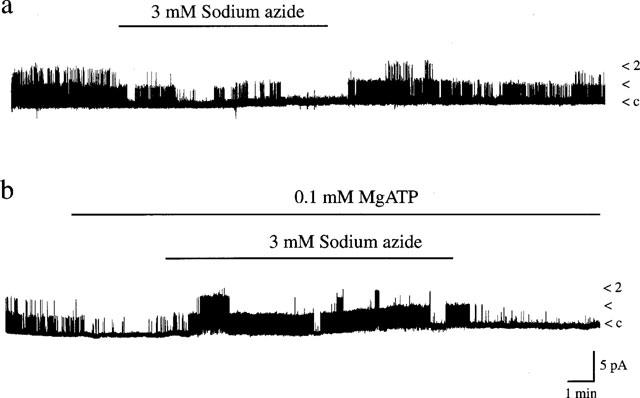
Effects of sodium azide on single channel currents. Continuous single channel currents recorded from inside-out membrane patches exposed to symmetrical 140 mM KCl at a membrane potential of +40 mV. Single channel openings are denoted by upward deflections. (a) Sodium azide (3 mM) inhibited KATP channels in the absence of Mg2+ in the bathing medium. This action of sodium azide was partially reversed on washout. The Nf.Po values obtained in control conditions, in the presence of sodium azide and following washout of azide were 1.06, 0.13 and 0.62, respectively. (b) In contrast, sodium azide (3 mM) activated KATP channels when 0.1 mM MgATP bathed the intracellular surface of inside out patches. Washout of azide partially reversed this action. The Nf.Po values were as follows: control 0.214; 0.1 mM MgATP 0.036; sodium azide and MgATP 0.600; wash 0.023.
In order to determine whether Mg2+ ions are a prerequisite for activation of KATP channels by sodium azide, the effects of sodium azide were examined in excised patches bathed in solution containing 0.1 mM ATP but in the absence of Mg2+ (by EDTA chelation and omission of MgCl2). Under these conditions, application of 0.1 mM ATP resulted in 97.3±1.5% inhibition of KATP channel activity (n=6, Figure 5), which was significantly greater (P<0.05) than that observed in the presence of MgATP as reported previously (Kozlowski et al., 1989). Subsequent addition of sodium azide (3 mM) in the presence of 0.1 mM ATP failed to activate KATP channel activity (n=6). The values of Nf.Po obtained in the presence of 0.1 mM ATP, sodium azide and ATP, and following washout of sodium azide but with ATP still present were 0.063±0.02, 0.039±0.008 and 0.016±0.005, respectively (n=6). These data are consistent with the absolute requirement of Mg2+ ions for sodium azide-induced activation of channel activity in the presence of ATP.
Figure 5.
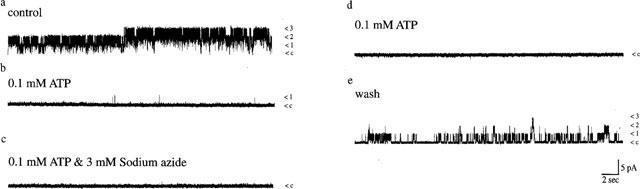
Sodium azide does not activate KATP channels in the absence of Mg2+ ions. Single channel currents recorded from an inside-out membrane patch exposed to symmetrical 140 KCl at a membrane potential of +40 mV. Single channel openings are denoted by upward deflections. Sodium azide (3 mM) inhibited, rather than activated KATP channels in patches bathed in 0.1 mM ATP and this was partially reversed on washout of azide. The Nf.Po values obtained in control conditions (a), in the presence of 0.1 mM ATP (b), in the combined presence of 0.1 mM ATP and sodium azide (3 mM; c), washout of azide (d) and washout of 0.1 mM ATP (e) were 2.25, 0.025, 0.024, 0.032 and 0.27, respectively.
Rotenone and oligomycin do not activate single KATP channel currents
It is possible that in the whole cell configuration some intracellular component critical for the effects of rotenone and oligomycin may be dialysed from the cell interior. Thus in order to investigate this likelihood the effects of these mitochondrial inhibitors were also examined in the cell-attached recording mode. Application of rotenone (10 μM; Figure 6a) or oligomycin (2 μM; Figure 6b) to the bathing medium, in the absence of extracellular glucose, did not induce activation of KATP channel currents in cell-attached recordings. Thus, the mean channel activity obtained in control conditions were 0.16±0.09 (n=5; rotenone) and 0.03±0.01 (n=8; oligomycin) which were not significantly altered following at least 10–15 min exposure to rotenone (0.13±0.05; n=5; Figure 6c) or oligomycin (0.03±0.01; n=8; P>0.05; Figure 6c), respectively. Furthermore, addition of either rotenone (10 μM; Figure 7) or oligomycin (2 μM; data not illustrated) to the intracellular surface of excised inside-out membrane patches bathed in 0.1 mM MgATP failed to activate KATP channel currents. Thus the mean channel activity obtained in control patches (exposed to 0.1 mM MgATP) were 0.08±0.04 (rotenone) and 0.09±0.03 (oligomycin), whereas following application of these inhibitors the mean currents were 0.07±0.04 (n=4; rotenone; P>0.05) and 0.04±0.01 (n=5; oligomycin; P>0.05). These data clearly demonstrate that, in cells exposed to zero glucose, rotenone or oligomycin, in contrast to sodium azide, have no effect on KATP channel activity, indicating that there is no correlation between their capacity to inhibit mitochondrial function and KATP channel activation.
Figure 6.
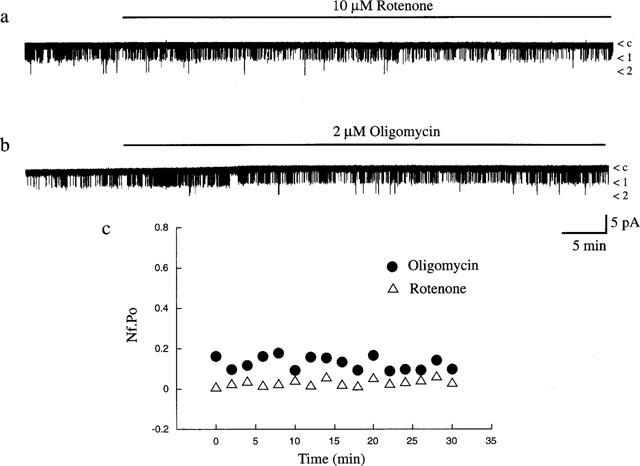
Rotenone and oligomycin do not activate KATP channels. Cell-attached recording from a single CRI-G1 cell at 10 mV applied potential. Application of rotenone (10 μM; a) or oligomycin (2 μM; b) for the time indicated failed to have any effect on channel activity over the time period of recording. (c) Graph of channel activity (Nf.Po) expressed as a function of time obtained from the cell attached patches in a (▵) and b (•).
Figure 7.
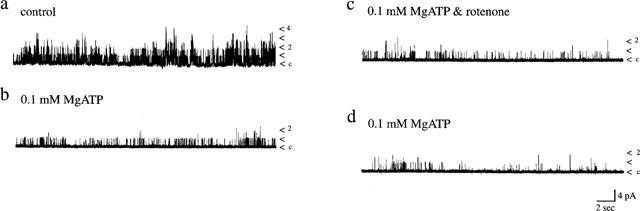
Effects of rotenone on single channel currents. Single channel currents recorded from inside-out membrane patches exposed to symmetrical 140 mM KCl at a membrane potential of +40 mV. Rotenone (10 μM) failed to activate KATP channels in the presence of 0.1 mM MgATP. The Nf.Po values obtained in control conditions, following application of 0.1 mM MgATP, in the combined presence of 0.1 mM MgATP and rotenone and following washout of rotenone were 0.265, 0.096, 0.072 and 0.045, respectively.
AMP-PNP prevents sodium azide activation of KATP channels
The sodium azide-induced activation of KATP channels appears to be dependent upon the presence of MgATP, suggesting that nucleotide hydrolysis or a phosphorylation dependent process is required for KATP channel activation. Therefore, the effects of the non-hydrolysable ATP analogue, Mg-AMP-PNP were investigated on azide-induced activation of KATP channels. In whole cell recordings, 5 mM Mg-ATP was substituted with 5 mM Mg-AMP-PNP in the pipette solution and under current clamp recordings conditions, the mean resting membrane potential and slope conductance values were −32.4±1.25 mV and 0.49±0.04 nS, respectively (n=7; Figure 8a). After at least 10 min following formation of the whole cell configuration (to allow complete dialysis of Mg-AMP-PNP into the cell interior), sodium azide (3 mM) was applied to the bath. In contrast to its actions in control conditions (following dialysis with an electrode solution containing 5 mM Mg-ATP), azide failed to result in hyperpolarization of the membrane of CRI-G1 cells (n=5; Figure 8a). Thus the membrane potential and slope conductance values obtained after 5 min exposure to azide were −32.2±2.3 mV and 0.44±0.06 nS, respectively (n=5; P>0.05). However, on two separate occasions, Mg-AMP-PNP failed to prevent azide-induced hyperpolarization and increased K+ conductance which may be due to insufficient dialysis of Mg-AMP-PNP.
Figure 8.
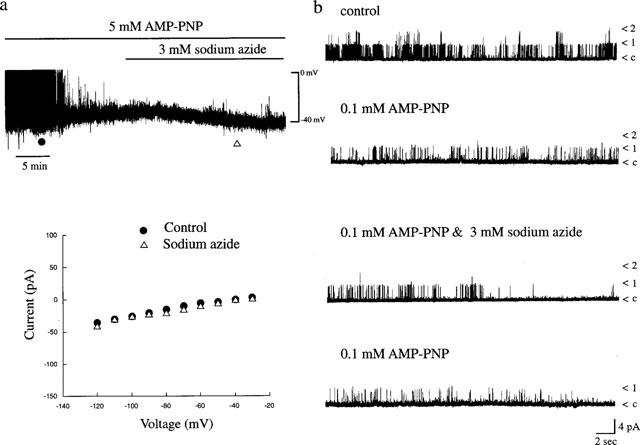
(a) Sodium azide activation of KATP channels is blocked by AMP-PNP. (a) The upper panel is a whole cell current clamp trace of cell dialysed with 5 mM AMP-PNP. Application of sodium azide (3 mM) for the time indicated failed to hyperpolarize CRI-G1 cells. The lower panel is a plot of the current-voltage relations for the voltage clamped currents obtained at the times specified in a: control (•) and sodium azide (▵). Following dialysis with AMP-PNP, sodium azide failed to effect the membrane conductance of CRI-G1 cells. (b). Single channel currents recorded from an inside-out membrane patch exposed to symmetrical 140 mM KCl at a membrane potential of +40 mV. Following application of 0.1 mM AMP-PNP which itself inhibited KATP channels, subsequent addition of sodium azide (3 mM) failed to increase KATP channel activity. The Nf.Po values were as follows: control 0.207; 0.1 mM AMP-PNP 0.087; 0.1 mM AMP-PNP and sodium azide 0.065; washout of azide 0.044.
In excised inside-out membrane patches, Mg-AMP-PNP was substituted for ATP in the bath solution and the effects of sodium azide examined (Figure 8b). In a manner similar to Mg-ATP, addition of Mg-AMP-PNP (0.1 mM) to the intracellular surface of the patch resulted in 71.5±1.3% inhibition of KATP channel activity (n=4). Subsequent addition of sodium azide (3 mM) in the continued presence of Mg-AMP-PNP (0.1 mM) failed to increase the activity of KATP channel currents in four out of four patches (Figure 8b). The mean channel activity (Nf.Po) obtained following addition of 0.1 mM Mg-AMP-PNP, in the combined presence of Mg-AMP-PNP and sodium azide, and following washout of sodium azide were 0.048±0.005, 0.025±0.007 and 0.008±0.003, respectively (n=4).
Discussion
The present study shows, using the whole cell recording configuration with 5 mM Mg-ATP in the electrode solution to maintain KATP channels closed, that the mitochondrial inhibitor sodium azide results in rapid hyperpolarization of CRI-G1 insulin-secreting cells, with an associated increase in K+ conductance. This was due to activation of KATP channels as the sulphonylurea tolbutamide, at concentrations reported to inhibit maximally KATP channels in this cell line (Sturgess et al., 1988; Lee et al., 1994), completely reversed the effects of sodium azide. In contrast, rotenone and oligomycin, inhibitors of NADH dehydrogenase and F1-ATPase, respectively, failed to affect either the membrane potential or slope conductance of these cells, suggesting that acute inhibition of mitochondrial energy metabolism is unlikely to explain the azide-induced activation of KATP channel currents in CRI-G1 insulin-secreting cells.
Definitive identification of KATP channels as the molecular target for sodium azide-induced hyperpolarization was obtained from single channel studies. In cell-attached recordings KATP channels were opened by sodium azide when applied to the bath solution. Furthermore, in excised inside-out membrane patches bathed in a MgATP-containing solution, sodium azide induced a rapid increase in KATP channel activity that was readily reversed by the sulphonylurea tolbutamide. In contrast, rotenone and oligomycin both failed to increase the activity of KATP channels in cell-attached recordings and in excised inside-out patches in the presence of MgATP. The lack of effect of rotenone and oligomycin in excised patches in comparison to sodium azide under all recording conditions indicate that acute inhibition of mitochondrial function is unlikely to induce soluble mediators (other than decreased ATP) capable of altering KATP channel function.
The failure of rotenone and oligomycin to activate KATP channels in cell-attached recordings in this study contrasts with previous reports showing that these agents activate KATP channels in insulin secreting cells (Niki et al., 1989) and dopaminergic substantia nigra neurones (Roper & Ashcroft, 1995) within 5–7 min. This is perhaps surprising since rotenone and oligomycin are expected ultimately to cause depletion of intracellular ATP levels even though they inhibit mitochondrial function by distinct and different mechanisms. Thus inhibition of electron flow via the electron transport chain (by either sodium azide or rotenone) or inhibition of the ATP synthase (by oligomycin), ultimately results in depletion of intracellular ATP levels as the processes of electron transfer and ATP synthesis are obligatorily coupled in mitochondria (Senior, 1988).
Another possible explanation for the lack of effect of rotenone and oligomycin on cell-attached KATP channel activity is that these compounds are less effective at depleting intracellular levels of ATP than sodium azide. However, this seems unlikely as rotenone, oligomycin and sodium azide all produce comparable reductions in intracellular ATP levels (Niki et al., 1989; Rustenbeck et al., 1997; Gribble et al., 1997a). It should be noted that recent evidence indicates that oligomycin fails to activate KATP channels in pancreatic beta cells even though it significantly reduces ATP production (Rustenbeck et al., 1997). Alternatively the metabolic source and/or site of ATP production may be important in CRI-G1 insulinoma cells. Indeed, in guinea-pig cardiac myocytes (Weiss & Lamp, 1987) ATP produced by glycolysis is more effective than mitochondrial oxidative phosphorylation at inhibiting KATP channels, and in the present experiments there was a complete absence of glucose during cell-attached recordings. Regardless of these considerations it is clear that the effects of sodium azide observed in the present study are unrelated to inhibition of mitochondrial function and subsequent ATP depletion. Indeed the rapid reversibility of sodium azide-induced KATP channel activation in CRI-G1 cells is not consistent with the involvement of metabolic poisoning in this process. The ability of sodium azide to either inhibit or activate KATP channels, depending on the presence or absence of MgATP respectively adds further support to the notion that these actions of sodium azide are unrelated to mitochondrial function.
Previous studies have shown that the ability of diazoxide to activate KATP channels depends upon the presence and concentration of Mg-ATP in the intracellular environment (Trube et al., 1986; Dunne et al., 1987; Sturgess et al., 1988). Thus KATP channels are not activated by diazoxide either in the absence of Mg-ATP or if high concentrations of Mg-ATP are present (Dunne et al., 1987). Furthermore, a phosphorylation process is thought to underlie diazoxide-induced KATP channel activation as the presence of Mg2+ ions is required for activation (Sturgess et al., 1988) and diazoxide is ineffective when Mg-ATP is replaced by the non hydrolysable ATP analogue, Mg-AMP-PNP (Kozlowski et al., 1989; Dunne, 1989). Similarly, in this study sodium azide-induced KATP channel activation required the presence of intracellular Mg-ATP, such that sodium azide failed to activate KATP channels in inside-out patches in the absence of Mg-ATP. Mg2+ ions were also required for the actions of sodium azide, as activation of the channels was only observed when Mg-ATP-containing solution bathed the intracellular surface of inside-out patches. In addition, there was no activation of KATP channels by sodium azide when ATP (in the absence of Mg2+ ions) or Mg-AMP-PNP was present to inhibit the KATP channels. Together these data indicate that, like diazoxide, ATP hydrolysis or a phosphorylation process is most likely required for KATP channel activation by sodium azide in CRI-G1 insulinoma cells, although we cannot rule out the possibility of a more direct interaction with a nucleotide binding site associated with the KATP channel complex (Babenko et al., 1998).
In addition to KATP channel activation, the present data indicate that in the absence of Mg-ATP, sodium azide inhibits KATP channel activity in excised patches. Diazoxide is also reported to inhibit KATP channels under similar conditions in this cell line (Kozlowski et al., 1989) and this has been attributed to diazoxide-induced enhancement of the rate of channel run down. However, further experiments are required to establish whether a change in the kinetics of KATP channel run down underlies this inhibitory action of sodium azide. Interestingly, sodium azide is also reported to inhibit cloned KATP channels in oocyte expression studies (Gribble et al., 1997a), an action that has been attributed to a direct action of azide rather than some product of metabolic inhibition.
The characteristics of sodium azide-induced activation of KATP channels in CRI-G1 cells are identical, in most respects to those of diazoxide, suggesting that a similar mechanism or site of action may underly the actions of these agents. For instance, the membrane hyperpolarization induced by sodium azide during whole cell recordings occurred over the same rapid time course observed with diazoxide (Trube et al., 1986; Sturgess et al., 1988). The time course of sodium azide-induced KATP channel activation in excised patches also parallels that of diazoxide. The inhibitory effects of sodium azide observed in this study and the requirement for MgATP for KATP channel activation are also characteristic effects of diazoxide in this cell line (Kozlowski et al., 1989). Diazoxide is reported to evoke a small but significant depletion in ATP levels that has been attributed to inhibition of mitochondrial function (Grimmsmann & Rustenbeck, 1998). However, in view of the lack of effect of rotenone and oligomycin in this study, diazoxide-induced activation of KATP channels in CRI-G1 cells is unlikely to be due to inhibition of mitochondrial energy metabolism.
Recent studies suggest that diazoxide may act directly on the sulphonylurea receptor, SUR1 (Inagaki et al., 1996; Tucker et al., 1997). In particular the nucleotide binding folds (NBF1 and NBF2) appear to be important for the stimulatory actions of diazoxide as mutations in these regions abolish diazoxide-induced activation of the cloned KATP channel (Nichols, 1996; Shyng et al., 1997; Gribble et al., 1997b). Thus even though sodium azide and diazoxide exhibit no similarities in structure, it is possible that sodium azide, like diazoxide acts directly on SUR1. However, in the absence of comparing the actions of sodium azide on Kir 6.2/SUR1 and the truncated form of Kir 6.2 (Tucker et al., 1997) or on SUR1 with mutations at NBF2 and/or NBF1 heterologously expressed in oocytes, the precise site of sodium azide action is unclear. Inhibition of KATP channels by ATP is thought to occur at a site distinct from the stimulatory MgADP and diazoxide sites on SUR1 as mutations that block activation of KATP channels by these agents do not prevent the inhibitory actions of ATP (Gribble et al., 1997b; Shyng et al., 1997). The ability of AMP-PNP and ATP to inhibit KATP channels activated by sodium azide in the present study also supports the notion that the inhibitory and stimulatory nucleotide sites are distinct.
In conclusion, these data indicate that in CRI-G1 insulin secreting cells, sodium azide either activates or inhibits KATP channels depending on whether Mg-ATP is present. These actions of sodium azide are unlikely to be due to a fall in intracellular ATP levels as the Mg-ATP concentration was clamped during whole cell recording and as KATP activation was not evident with oligomycin or rotenone during cell-attached recordings. Consequently, this complication should be considered when using this agent for metabolic inhibition and subsequent activation of KATP channels.
Abbreviations
- CRI-G1
Cambridge rat insulinoma G1
- DMSO
Dimethyl sulphoxide
- AMP-PNP
5′-adenylylimidodiphosphate.
References
- ALLARD B., LAZDUNSKI M., ROUGIER O. Activation of ATP-dependent K+ channels by metabolic poisoning in adult mouse skeletal muscle: role of intracellular Mg2+ and pH. J. Physiol. 1995;485:283–296. doi: 10.1113/jphysiol.1995.sp020730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASHCROFT S.J.H., ASHCROFT F.M. Properties and functions of ATP-sensitive K+ channels. Cell. Signalling. 1990;2:197–214. doi: 10.1016/0898-6568(90)90048-f. [DOI] [PubMed] [Google Scholar]
- ASHCROFT F.M., RORSMAN P. Electrophysiology of the pancreatic β cell. Prog. Biophys. Mol. Biol. 1991;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- ASHFORD M.L.J.Potassium channels and modulation of insulin secretion Potassium Channels: Structure, Classification, Function and Therapeutic Potential 1990Chichester: Ellis Horwood Limited; 300–325.Cook, N.S. (ed). pp [Google Scholar]
- BABENKO A.P., AGUILAR-BRYANT L., BRYAN J. A view of SUR/6.x, KATP channels. Annu. Rev. Physiol. 1998;60:667–687. doi: 10.1146/annurev.physiol.60.1.667. [DOI] [PubMed] [Google Scholar]
- CARRINGTON C.A, , RUBERY E.D, , PEARSON E.C., HALES C.N. Five new insulin-producing cell lines with differing secretory properties. J. Endocrinol. 1986;109:193–200. doi: 10.1677/joe.0.1090193. [DOI] [PubMed] [Google Scholar]
- DUNNE M.J., ILLOT M.C., PETERSEN O.H. Interaction of diazoxide, tolbutamide and ATP4− on nucleotide-dependent K+ channels in an insulin-secreting cell line. J. Membr. Biol. 1987;99:215–224. doi: 10.1007/BF01995702. [DOI] [PubMed] [Google Scholar]
- DUNNE M.J. Protein phosphorylation is required for diazoxide to open ATP-sensitive potassium channels in insulin (RIN m5F) secreting cells. FEBS Lett. 1989;208:262–266. doi: 10.1016/0014-5793(89)80734-3. [DOI] [PubMed] [Google Scholar]
- GRIBBLE F.M., ASHFIELD R., AMMALA C., ASHCROFT F.M. Properties of cloned ATP-sensitive K+ currents expressed in Xenopus oocytes. J. Physiol. 1997a;498:87–98. doi: 10.1113/jphysiol.1997.sp021843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIBBLE F.M., TUCKER S.J., ASHCROFT F.M. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J. 1997b;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIMMSMANN T., RUSTENBECK I. Direct effects of diazoxide on mitochondrial in pancreatic β-cells and on isolated liver mitochondria. Br. J. Pharmacol. 1998;123:781–788. doi: 10.1038/sj.bjp.0701663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARVEY J., MCKENNA F., HERSON P.S., SPANSWICK D., ASHFORD M.L.J. Leptin activates ATP-sensitive potassium channels in the rat insulin-secreting cell line, CRI-G1. J. Physiol. 1997;504:527–535. doi: 10.1111/j.1469-7793.1997.527bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT J.P., NAMBA N., INAZAWA J., GONZALEZ G., AGUILAR-BRYAN L., SEINO S., BRYAN J. Reconstitution of IKATP: an inward rectifier subunit plus the sulphonylurea receptor. Science. 1995;270:1166–1169. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT J.P., WANG C.Z., AGUILAR-BRYAN L., BRYAN J., SEINO S. A family of sulphonylurea receptors determines the sensitivity of the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- ITO H., NAKAJIMA T., TAKIKAWA R., HAMADA E., IGUCHI M., SUGIMOTO T., KURACHI Y. Coenzyme Q10 attenuates cyanide-activation of the ATP-sensitive K+ channel current in single cardiac myocytes of the guinea-pig. Arch. Pharmacol. 1991;344:133–136. doi: 10.1007/BF00167394. [DOI] [PubMed] [Google Scholar]
- KAKEI M., NOMA A. Adenosine 5′-triphosphate-sensitive single potassium channel in the atrio ventricular node cell of the rabbit heart. J. Physiol. 1984;352:265–284. doi: 10.1113/jphysiol.1984.sp015290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOZLOWSKI R.Z., HALES C.N., ASHFORD M.L.J. Dual effects of diazoxide on ATP-K+ currents recorded from an insulin-secreting cell line. Br. J. Pharmacol. 1989;97:1039–1050. doi: 10.1111/j.1476-5381.1989.tb12560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE K., OZANNE S.E., ROWE I.C.M., HALES C.N., ASHFORD M.L.J. The effects of trypsin of ATP-sensitive potassium channel properties and sulphonylurea receptors in the CRI-G1 insulin secreting cell line. Mol. Pharmacol. 1994;45:176–185. [PubMed] [Google Scholar]
- LEE K., ROWE I.C.M., ASHFORD M.L.J. Characterisation of an ATP-modulated large conductance Ca2+-activated K+ channel present in rat cortical neurones. J. Physiol. 1995;488:319–337. doi: 10.1113/jphysiol.1995.sp020969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MISLER S., FALKE L.C., GILLIS K., MCDANIEL M.L. A metabolic-regulated potassium channel in rat pancreatic beta cells. Proc. Natl. Acad. Sci. U.S.A. 1986;83:7119–7123. doi: 10.1073/pnas.83.18.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MISLER S., GEE W.M., GILLIS K.D., SCHARP D.W., FALKE L.C. Metabolic-regulated ATP-sensitive K+ channel in human pancreatic islets. Diabetes. 1989;38:422–427. doi: 10.2337/diab.38.4.422. [DOI] [PubMed] [Google Scholar]
- MURPHY K.P.S.J., GREENFIELD S.A. ATP-sensitive potassium channels counteract anoxia in neurones of the substantia nigra. Exp. Brain. Res. 1991;84:355–358. doi: 10.1007/BF00231456. [DOI] [PubMed] [Google Scholar]
- NICHOLS C.G., SHYNG S.-L., NESTOROWICZ A., GLASER B., CLEMENT J., GONZALEZ G., AGUILAR-BRYAN L., PERMUTT A.M., BRYAN J.P. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- NIKI I., ASHCROFT F.M., ASHCROFT S.J. The dependence on intracellular ATP concentration of ATP-sensitive K-channels and of Na,K-ATPase in intact HIT-T15 beta cells. FEBS Lett. 1989;257:361–364. doi: 10.1016/0014-5793(89)81572-8. [DOI] [PubMed] [Google Scholar]
- PENEFSKY H.S. Mechanism of inhibition of mitochondrial adenosine triphosphate by dicyclohexylcarbodiimide and oligomycin: relationship to ATP synthesis. Proc. Natl. Acad. Sci. U.S.A. 1985;82:1589–1593. doi: 10.1073/pnas.82.6.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAGAN C.I., RACKER E. Resolution and reconstitution of the mitochondrial electron transport system IV. The reconstitution of the rotenone-sensitive reduced nicotinamide adenine dinucleotide-ubiquinone reductase from reduced nicotinamide adenine dinucleotide dehydrogenase and phospholipids. J. Biol. Chem. 1973;248:6876–6884. [PubMed] [Google Scholar]
- ROPER J., ASHCROFT F.M. Metabolic inhibition and low internal ATP activate K-ATP channels in rat dopaminergic substantia nigra neurones. Pflugers Arch. Eur. J. Physiol. 1995;430:44–54. doi: 10.1007/BF00373838. [DOI] [PubMed] [Google Scholar]
- RUSTENBECK I., HERRMENN C., GRIMMSMANN T. Energetic requirement of insulin secretion distal to calcium influx. Diabetes. 1997;46:1305–1311. doi: 10.2337/diab.46.8.1305. [DOI] [PubMed] [Google Scholar]
- SAKURA H., AMMALA C., SMITH P.A., ASHCROFT F.M. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel expressed in pancreatic beta cells, brain, heart and skeletal muscle. FEBS Lett. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- SENIOR A.E. ATP synthesis by oxidative phosphorylation. Physiol. Rev. 1988;68:177–231. doi: 10.1152/physrev.1988.68.1.177. [DOI] [PubMed] [Google Scholar]
- SHYNG S.-L., FERRIGNI T., NICHOLS C.G. Regulation of KATP channel activity by diazoxide and MgADP; Distinct functions of the two nucleotide binding folds of the sulphonylurea receptor. J. Gen. Physiol. 1997;110:643–654. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STURGESS N.C., KOZLOWSKI R.Z., CARRINGTON C.A., HALES C.N., ASHFORD M.L.J. Effects of sulphonylureas and diazoxide on insulin secretion and nucleotide-sensitive channels in an insulin-secreting cell line. Br. J. Pharmacol. 1988;95:83–94. doi: 10.1111/j.1476-5381.1988.tb16551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRUBE G., RORSMAN P., OHNO-SHOSAKU T. Opposite effects of tolbutamide and diazoxide on the ATP-sensitive K+ channel in mouse pancreatic beta cells. Pflugers Arch. 1986;407:493–499. doi: 10.1007/BF00657506. [DOI] [PubMed] [Google Scholar]
- TSUBAKI M., YOSHIKAWA S. Fourier-transform infra-red study of azide binding to the Fe3-CuB binuclear site of bovine cytochrome oxidase. Biochem. 1993;32:174–182. doi: 10.1021/bi00052a023. [DOI] [PubMed] [Google Scholar]
- TUCKER S.J., GRIBBLE F.M., ZHAO C., TRAPP S., ASHCROFT F.M. Truncation of Kir6.2 produces ATP-sensitive K+channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- WEISS J.N., LAMP S.T. Glycolysis preferentially inhibits ATP-sensitive K+ channels in isolated guinea pig cardiac myocytes. Science. 1987;238:67–69. doi: 10.1126/science.2443972. [DOI] [PubMed] [Google Scholar]